
Abstract
Purpose of Review
Advances in molecular genetics have improved our understanding of primary ciliary dyskinesia. The purpose of this review is to describe the integration of genetics into clinical practice.
Recent Findings
This review describes > 50 genes which have been identified to cause multiple motile ciliopathies. Known genotype–phenotype relationships are explored, including genes associated with worse prognosis (CCDC39, CCDC40, CCNO). Features which indicate referral for genetic testing such as a family history, situs defects and lifelong chronic upper and lower respiratory tract disease are described along with how genetics fits into current guidelines for diagnostic algorithms, and the potential challenges and advantages.
Summary
As we move forward, the growing genomic knowledge about primary ciliary dyskinesia will aid diagnosis, understanding of prognosis and the establishment of future therapeutic trials.
Similar content being viewed by others

Introduction
Primary ciliary dyskinesia is a lifelong chronic condition of mucociliary clearance dysfunction. It was first described by Swiss physician Manes Kartagener in 1933 as a triad of bronchiectasis, sinusitis and situs inversus [1]. In the past decade, next generation sequencing has resulted in rapid gene discovery in this condition. More than 2000 pathogenic variants in 50 genes have been described to cause PCD, and it is estimated currently that up to 90% PCD cases can be confirmed through genetic testing. The majority of patients have private mutations, meaning the variant has never been previously detected in another individual. This genetic heterogeneity expands our knowledge of the motile ciliopathy phenotype and presents unique challenges for accurate integration of complex genetic information into diagnosis and management of this chronic respiratory condition. There is the opportunity for integration of genomic sequencing into diagnostic pathways, development of personalising treatment plans based on genotype–phenotype relationships and ultimately, development of PCD-specific gene–based therapies. In this review, we describe the current knowledge of PCD genetics and discuss whom to refer for genetic testing and the advantages and challenges of the integration of genotyping into clinical practice.
Motile cilia play a key role in the respiratory tract in clearing mucus, pollutants and pathogens from the airway. When the cilia are ineffective in PCD, patients suffer from persistent productive cough, develop respiratory infections and exacerbations throughout childhood, and nearly all patients with PCD have developed bronchiectasis by adulthood [2]. A common first feature of PCD is neonatal respiratory distress, often requiring hospitalisation [3]. Length of hospital stay in neonates correlates with lung function later in life [4]. Most PCD infants have a daily wet cough and cilia dysfunction in the upper airways and Eustacean tubes which can result in loss of sense of smell and rhinosinusitis as well as conductive hearing loss from otitis media (glue ear) [5]. Due to the involvement of cilia in the reproductive organs (the fallopian tubes in females and efferent duct in males), PCD patients can be infertile. Male patients with PCD may be infertile due to shared defects between respiratory cilia and sperm flagella, which share many axonemal proteins [6] Furthermore, as cilia are important in the left–right symmetry during development, almost half of PCD patients have situs inversus, and 8–12% have complex congenital heart disease [7]. Recurrent respiratory infections and the development of bronchiectasis lead to progressive lung function decline through childhood. A large international registry suggested that lung function impairment at ages 6–9 was similar between PCD and cystic fibrosis (CF), but that lung function was substantially more impaired in young adults with CF compared to PCD [8, 9]. A description of these different clinical characteristics that present in PCD patients at different life stages is shown in Table 1.
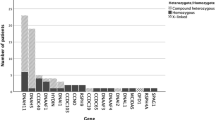
The most recent investigation into the international prevalence of PCD based on large genomic databases found it to affect an estimated 1 in 7554 people [10]. In the majority of PCD cases, inheritance is autosomal recessive, but there are exceptions which are dominant or X-linked [11]. Pathogenic variants in 1 of 50 genes (Table 2) affect the development, structure and/or the axonemal motors of the cilia [2].
Normal Cilia Structure and Role in Disease
The cilium structure is approximately 6–7 µm long and is made up of an axoneme consisting of a central pair of single microtubules which are surrounded by nine outer doublet microtubules. These outer microtubules are connected to each other by the nexin–dynein regulatory complex, and each outer pair is joined to the central pair by radial spokes. The cilium is attached to the cell at a point at the base of the cilium called the basal body [12]. To allow the cilia to beat, the dynein motor proteins hydrolyse ATP to then apply force onto the microtubules which stimulate the bending of the cilium. The cilia beat in a motion with a forward effective stroke which enables them to move the mucus and then return to their original position with a recovery stroke. In a healthy individual, the cilia beat at a frequency of between 7 and 16 Hz [13]. Known PCD genes and how they correspond to abnormalities in the cilia ultrastructure are shown in Fig. 1, a cross-section diagram of cilia ultrastructure.

Genotype–phenotype associations in PCD. PCD genes (black italics) and their structural and functional consequence. Blue text indicates known genotype and clinical phenotype associations. Less than (^) sign indicates normal ultrastructure and asterisk (*) cases with normal nasal nitric oxide have been described
Genotype–Phenotype Relationships
Gene discovery has improved the understanding of genotype–phenotype relationships. The burden of PCD can depend on the causative genotypes. Patients are at higher risk of severe disease, measured by lung function, when ciliogenesis is severely impaired (e.g. defects in CCNO or MCIDAS) or in defects of microtubular stabilisation (CCDC39 and CCDC40 genes) [14]. These associations have been shown across multiple large international studies although the mechanism behind this worsened disease severity has not yet been fully elucidated. Conversely, some genes or specific mutations are associated with reduced risk of severe disease. Mutations in the outer dynein arm heavy chain gene DNAH11 are associated with the preservation of lung function when compared to other genotypes. A reduced incidence of neonatal distress is also reported in patients with DNAH11 genotypes which could be linked to the subsequent preservation of lung function [15]. RSPH1, MNS1, DNAH9, and the His154Pro mutation in CCDC103 have also been described as associated with milder disease [16,17,18,19]. Of note, some patients with these genotypes will have severe disease, and several important factors beyond genotype are likely to play a part, for example, age at diagnosis, socio-economic factors, access and adherence to treatment, chronic infections and exacerbations.
Further genotype–phenotype relationships have been observed in PCD based on the subtle difference in the proteins involved in ciliary structure and function in different cell types. For example, some genotypes are associated with preserved fertility (CCDC151) or situs solitus (HYDIN, RSPH4A, RSPH9, RSPH1, RSPH3, DRC1, DRC2, DRC3, CCNO, MCIDAS) due to the redundant function of these proteins in the fallopian tubes or embryonic development, respectively [20, 21]. Other PCD genes are associated with the presence of hydrocephalus due to dysfunction of motile cilia in the brain ventricles (FOXJ1, CCNO, MCIDAS) [22,23,24]. Understanding these genotype–phenotype relationships may allow the counselling of patients with increased accuracy regarding prognosis and put in place personalised management plans factoring in genotype. These may include, for example, the relevance of referral to colleagues in specialties such as ENT, fertility clinics and cardiology.
In recent years, genetic testing has reduced in cost and become more accessible which has aided PCD diagnosis. Testing can be conducted through PCD-specific gene panels, whole exome or whole genome sequencing. Each comes with advantages and disadvantages in terms of cost, sequence coverage, complexity of data analysis and potential for incidental findings and multiple variants of unknown significance. Gene panels may present the option with lowest cost and least complexity but at the expense of worst coverage, while whole genome sequencing is costly but provides additional information [25, 26]. Whole genome sequencing in healthcare has recently been trialled alongside clinical practice in large government funded projects in the UK and USA [27, 28]. Some commercially available ‘panels’ will conduct whole exome sequencing but only report on requested genes, giving the opportunity to revisit a diagnosis as gene discovery continues to progress. The UK National Health Service 100,000 Genome Project was funded by the UK government and included subpopulations with specific diseases of interest including early onset or aggressive bronchiectasis. This study revealed underdiagnosis of PCD in an adult bronchiectasis cohort. The project found 17 out of 142 (12%) bronchiectasis patients had disease-causing mutations in known PCD genes [27]. This highlights a high frequency of undiagnosed PCD in early onset or aggressive bronchiectasis and indicates the need for more genetic testing for PCD in adults with severe unexplained bronchiectasis.
There are few studies describing the specificity of PCD genetic testing as it is predominantly conducted on individuals with a clinical or pathology-based diagnosis. Screening larger, broader populations will present some challenges due to the size of the genes and number of genes involved. Approximately 1 in 20 people are carriers of a PCD mutation, and variants of unknown significance in one or more of the PCD genes are identified in most of the population without disease. The Hannah et al. study, which describes PCD prevalence of 1:7500, used pathogenic or likely pathogenic mutations in 26 of the known 50 genes for their prediction model, and therefore, this is likely to be an underestimate. A second analysis including variants of unknown significance in the prediction models resulted in a prevalence estimation of 1 in 200. This suggests at least 1 in 200 people have 2 variants of unknown significance in a PCD gene. Therefore, results of PCD genetic screening in general populations with just one clinical feature would be unreliable [10]. However, over the next few years, understanding of disease-causing mutations may improve with prediction modelling and databases of known variants. Therefore, genetic screening will become increasingly possible in populations where the prevalence of PCD is predicted to be high. For example, genetic testing may be achievable in any child born with situs inversus, one in four of which are predicted to have PCD, or term infants with unexplained neonatal respiratory distress [29]. Currently, careful referral for genetic testing and confirmation of findings using pathology-based tests remains crucial.
Whom to Refer for Genetic Testing
Whom to refer for PCD testing may depend on the age of the patient and the availability of local testing facilities. In the UK, the genomics medicine testing directory recommends (1) neonatal presentation with at least one of the following: (a) situs inversus plus lower airway or nasal symptoms OR (b) persistent respiratory distress where other causes have been excluded OR (c) persistent rhinorrhoea and cough where other causes have been excluded OR (2) testing in childhood with at least one of the following: (a) Persistent life-long wet cough (where CF has been excluded), (b) unexplained bronchiectasis (CF excluded) and (c) serous otitis media in association with lower and upper airway symptoms and (3) testing in adults who have had symptoms as above since early childhood, often associated with infertility or subfertility [30].
There have been tools developed for the identification of people suitable for PCD testing; these include the PICADAR tool which is a predictive score with seven simple questions to predict the likelihood of having PCD. It can be used in any patient with chronic respiratory symptoms starting in early childhood [31]. The seven questions and associated points are the following: was the patient born preterm or full term (2 points), did the patient experience chest symptoms in the neonatal period (2 points), was the patient admitted to a neonatal unit (2 points), does the patient have a situs abnormality (4 points), does the patient have a congenital heart defect (2 points), does the patient have persistent perennial rhinitis (1 point), does the patient experience chronic ear or hearing symptoms (1 point)? It is recommended to test anyone with a score of 4 or above. The emphasis on the neonatal period means this is difficult to translate to adults and older children and the referral may need to be more pragmatic. Most PCD patients will have a life-long history of upper and lower respiratory disease. In patients with bronchiectasis, some findings on HRCT can give a suggestion that PCD might be the causative factor. Patients tend to have bilateral disease concentrated in the lower and middle lobes, as these are least effective at clearing with cough alone. There may also be widespread mucus plugging, but neither feature is uncommon in other causes of adult bronchiectasis [32, 33]. In general, adults with PCD will have more severe disease with lower lung function, are younger than their counterparts with idiopathic disease and be more likely to isolate a pathogen growth from sputum and often Pseudomonas aeruginosa [32, 33].
PCD diagnosis is usually a process which involves more than one testing modality, and in Europe, genetics remains at the end of this pathway once a number of pathology-based tests have been conducted. There are European Respiratory Society (ERS) and American Thoracic Society (ATS) guidelines for PCD diagnosis as shown in Fig. 2. The ERS guidelines recommend initial testing with nasal nitric oxide (nasal NO). Nasal NO measures the concentration of nitric oxide gas in the sinuses using a gas analyser. Most PCD patients have significantly reduced nasal NO compared to healthy controls [34]. A cut-off of less than 77 nl/min is commonly used to identify patients with possible PCD [35]. However, the test is not 100% sensitive or 100% specific. A systematic review and meta-analysis using EM or genetic-based diagnosis as the gold standard suggested a sensitivity of 97.6% and specificity of 96% in patients with a high clinical suspicion of disease [36]. Cilia function can be directly analysed from nasal epithelial tissue using high-speed video microscopy (HSVM). Analysis of cilia beat frequency and more importantly beat pattern can be analysed by light microscopy [37]. To confirm a diagnosis, transmission electron microscopy (TEM) can be used to visualise the ultrastructure of the cilia. Genetic testing may finally be used to confirm the diagnosis. ERS guidelines state that a hallmark defect detected TEM, and/or biallelic pathogenic mutations in a known PCD gene can confirm PCD, while repeatedly abnormal HSVM make a diagnosis highly likely. [38].

© ERS 2023: European Respiratory Journal 54 (3) 1901066; https://doi.org/10.1183/13993003.01066-2019 Published 5 September 2019
Summary of a European Respiratory Society and b American Thoracic Society guidelines on the techniques and steps to confirm a PCD diagnosis. For further details including meaning of superscript symbols please refer to the source publication. Reproduced with permission of the
The ATS guidelines place greater emphasis of clinical parameters, nasal NO testing and genetics rather than functional tests. The ATS recommendations require at least two of the four clinical features of PCD before undertaking nasal NO testing. If nasal NO testing is available and levels of NO are low (< 77 nl/min) (and if cystic fibrosis (CF) is excluded), then PCD diagnosis is considered to be confirmed. If nasal NO testing is not available, then genetic testing is carried out to confirm a PCD diagnosis. If a biallelic pathogenic variant associated with PCD is not present, then ATS guidelines recommend that TEM analysis is also carried out [39].
It is important to note that none of the diagnostic tests is fully sensitive or specific. PCD mutations can have unique diagnostic phenotypes, so a patient could present with a normal nasal NO but have abnormalities in the cilia ultrastructure by TEM (e.g. RSPH1 mutations) or a patient could have abnormal nasal NO but normal TEM (e.g. DNAH11 mutations). Up to 30% of people with PCD have normal cilia ultrastructure when examined with TEM [40] The poor sensitivity or specificity of all the tests is the reason that both guidelines recommend an approach combining multiple tests.
A technique that may be used to aid genetic diagnosis, especially in the confirmation of a variant of unknown significance, and that is quicker and perhaps more readily available than other methods is immunofluorescence staining. This is achieved by staining cells that have been dried onto slides with primary and secondary antibodies and then visualising them with a confocal microscope. This was shown by a study in the UK to be highly specific and in a diagnostic cohort identified nearly 90% of the patients with already confirmed PCD and confirmed normal staining for the rest of the cohort who were considered ‘highly unlikely’ to have PCD [41]. Subsequently, additional antibodies have been added as a potential for immunofluorescence which may uplift its usefulness above that of TEM. Specifically, a group used immunofluorescence staining to identify individuals with the HYDIN mutation using a SPEF2 antibody as SPEF2 is associated with the central pair projections which are absent in people with pathogenic mutations in this gene [42]. DNAH11 antibodies can also be used with immunofluorescent staining to successfully recognise defects in this large gene where TEM results are usually normal. Immunofluorescence can confirm this defect in the majority of patients, although some with intronic variants continue to display normal staining patterns, suggesting DNAH11 still reaches the axoneme but does not function properly [43].
Use of Genetics in Isolation and Pitfalls
Despite its many advantages, it is also important to consider the limitations of diagnosing PCD by genetics. Firstly, genetic screening cannot be used in isolation, and frequently, results require confirmation with another technique such as TEM or HSVM or IF. Importantly genetic testing misses approximately 30% of PCD patients, so a negative result on a gene panel does not mean that the clinician can exclude PCD as a diagnosis [2].
There are over 200 proteins which make up a cilium and 700 involved in its biogenesis. Mutations in only 50 genes have so far been implicated in PCD. There is a steady gene discovery with 2 or 3 new genes identified each year. In addition, genes that are known about are not always tested. HYDIN, for example, has a pseudogene HYDIN2 which makes it very difficult to reliably align sequences and detect mutations in these. HYDIN is therefore not included in many commercially available PCD genetic panel. HYDIN mutations in PCD patients can present with abnormal HSVM as they have a reduced beating amplitude as well as showing subtle central complex defects when analysed with electron microscope tomography [44]. Recently, advances in genomics techniques have facilitated improved the diagnosis of HYDIN using digital masking, long read sequencing and detection of SPEF immunofluorescent techniques. Where these techniques have been used HYDIN has been shown to be the 4th/5th largest causative gene accounting for ~ 8% of cohorts [45]. The majority of mutations in PCD genes are nonsense mutations or deletions which therefore truncate the protein. However, about 30% are missense mutations in which only a single amino acid is changed, and thus, it is harder to define whether they are contributing towards the disease. Many of the mutations associated with PCD are private mutations, meaning they have not been previously seen in another patient or family [46]. The majority of these private missense mutations are assessed by clinical geneticists as variants of unknown significance. Meaning, it is not known whether they are disease-causing or not [10]. This emphasises the need for complementary functional tests to confirm PCD diagnosis and complement information from genetic testing, particularly where clinical features are non-classical.
Why a Diagnosis Is Important and Advantages of Genetic Testing
Many patients receive a diagnosis of PCD on clinical grounds without diagnostic testing, some patients receive a confirmed diagnosis based on diagnostic testing without genetics and worldwide, it is likely only a small minority of patients receive a genetic diagnosis of PCD.
Making the diagnosis of PCD is essential, and the most accurate diagnosis possible should be made using the combination of diagnostic tests recommended by international guidelines to avoid under- and over-diagnosis. A confirmed diagnosis of PCD in a child without bronchiectasis enables protocolised care aimed at preserving lung function and delaying or preventing the development of irreversible pulmonary and non-pulmonary pathology. In those with established bronchiectasis, a diagnosis of PCD is still important as it alters management. Additional measures following a diagnosis of PCD may include genetic counselling, counselling regarding fertility, intensified treatment of upper airway disease, cardiac investigations and intensified airway clearance and additional measures to treat pulmonary disease.
There are a range of different management strategies for PCD patients, most addressing symptoms of disease, and there are currently no licensed PCD-specific therapies. For the pulmonary symptoms, it is strongly advised that PCD patients practice airway clearance techniques to improve mucociliary clearance [47]. In the case of pulmonary exacerbations then bronchiectasis or cystic fibrosis management guidelines are usually followed. Patients with PCD are at high risk of exacerbations and are frequently chronically infected with pathogens, most commonly P. aeruginosa and H. influenzae. In adulthood, Pseudomonas aeruginosa becomes the most common infecting pathogen. Other treatments for improving pulmonary function such as hypertonic saline and recombinant human deoxyribonuclease I (rhDNase) have been investigated for PCD treatment but have not been confirmed to be beneficial [48, 49]. Nevertheless, hypertonic saline is widely used to aid mucus clearance. rDNAse is more controversial as, although effective in CF, it was found to be ineffective in non-CF bronchiectasis.
A multicentre randomised trial of prophylactic azithromycin found that the treatment reduced exacerbations compared to placebo by approximately 50% in patients with PCD, mirroring results observed in a broader bronchiectasis population. Inhaled antibiotics are also frequently used in the PCD population, particularly when P. aeruginosa infection is present [50]. An otolaryngologist can help assist with symptoms associated with the upper airways and ears using techniques such as sinonasal rinsing and nasal steroids [51]. A multidisciplinary team is required for each patient, and thus, for other aspects of the condition, genetic counselling and psychological support may also be beneficial.
Beyond making an accurate diagnosis of PCD, which is crucial, there are added benefits of a genetic diagnosis. Knowledge regarding genotype/phenotype relationships can assist in prognostication and screening for complications. PCD is an area of intense research, and future therapies targeted at specific genetic mutations are emerging. Genetic testing can therefore enable patients to participate in clinical trials.
Novel treatment options for PCD patients are being developed using gene or transcript therapies. A study used a lentivirus vector to correct a DNAI1 mutation in cells, and they were able to demonstrate the transduced DNAI1-rectified outer arm defect seen in this mutation. This partially improved the mucociliary clearance, but not all of the cells were transduced [52]. Another study used the same technique in a mouse model and found that they successfully transduced cells in the airway epithelium but did not significantly change the cilia beat frequency [53]. Gene editing using clustered regularly interspaced short palindromic repeats (CRISPR) has the potential to be used in PCD, but there are yet to be any studies published. Another study has been published using transcription-activator-like effector nucleases (TALEN) in which the authors were able to partially reinstate cilia function in ex vivo epithelial cells which had the DNAI1 mutation [54]. The concept of using RNA to develop treatments is also being explored by research scientists and also has the potential to be beneficial as it is considered less likely to have harmful side effects than direct DNA therapies [49].
Conclusion
Research genetics have improved our understanding of multiple motile ciliopathies. Now, the challenge is to integrate genetics into clinical practice for diagnosis of these conditions. We have highlighted whom to refer, how genetics fits into current guidelines for diagnostic algorithms and the potential challenges and advantages, as well as techniques, which could be incorporated into future guidelines for diagnosis and therapies that are currently being developed. As we move forward, the growing genomic knowledge about this condition will aid the establishment of future clinical trials.
References
Kartagener M. Zur Pathogenese der Bronchiektasien: II. Mitteilung: Familiäres Vorkommen von Bronchiektasien. Beiträge zur Klinik der Tuberkulose und spezifischen Tuberkulose-Forschung. 1933;84:73–85.
Lucas JS, Davis SD, Omran H, Shoemark A. Primary ciliary dyskinesia in the genomics age. Lancet Respir Med. 2020;8(2):202–16.
Goutaki M, Meier AB, Halbeisen FS, Lucas JS, Dell SD, Maurer E, et al. Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur Respir J. 2016;48(4):1081–95.
Wee WB, Leigh MW, Davis SD, Rosenfeld M, Sullivan KM, Sawras MG, et al. Association of neonatal hospital length of stay with lung function in primary ciliary dyskinesia. Ann Am Thorac Soc. 2022;19(11):1865–70.
Pifferi M, Bush A, Rizzo M, Tonacci A, Di Cicco M, Piras M, et al. Olfactory dysfunction is worse in primary ciliary dyskinesia compared with other causes of chronic sinusitis in children. Thorax. 2018;73(10):980–2.
Sironen A, Shoemark A, Patel M, Loebinger MR, Mitchison HM. Sperm defects in primary ciliary dyskinesia and related causes of male infertility. Cell Mol Life Sci. 2020;77:2029–48.
Turner JP, Corkey CW, Lee JV, Levison H, Sturgess J. Clinical expressions of immotile cilia syndrome. Pediatrics. 1981;67(6):805–10.
Halbeisen FS, Goutaki M, Spycher BD, Amirav I, Behan L, Boon M, et al. Lung function in patients with primary ciliary dyskinesia: an iPCD Cohort study. Eur Respir J. 2018;52(2).
Roehmel JF, Doerfler FJ, Koerner-Rettberg C, Brinkmann F, Schlegtendal A, Wetzke M, et al. Comparison of the lung clearance index in preschool children with primary ciliary dyskinesia and cystic fibrosis. Chest. 2022;162(3):534–42.
Hannah WB, Seifert BA, Truty R, Zariwala MA, Ameel K, Zhao Y, et al. The global prevalence and ethnic heterogeneity of primary ciliary dyskinesia gene variants: a genetic database analysis. Lancet Respir Med. 2022;10(5):459–68.
Paff T, Loges NT, Aprea I, Wu K, Bakey Z, Haarman EG, et al. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am J Hum Genet. 2017;100(1):160–8.
Satir P, Christensen ST. Structure and function of mammalian cilia. Histochem Cell Biol. 2008;129(6):687–93.
Chilvers MA, Rutman A, O’Callaghan C. Functional analysis of cilia and ciliated epithelial ultrastructure in healthy children and young adults. Thorax. 2003;58(4):333–8.
Davis SD, Rosenfeld M, Lee H-S, Ferkol TW, Sagel SD, Dell SD, et al. Primary ciliary dyskinesia: longitudinal study of lung disease by ultrastructure defect and genotype. Am J Respir Crit Care Med. 2019;199(2):190–8.
Shoemark A, Rubbo B, Legendre M, Fassad MR, Haarman EG, Best S, et al. Topological data analysis reveals genotype–phenotype relationships in primary ciliary dyskinesia. Eur Respir J. 2021;58(2).
Shoemark A, Moya E, Hirst RA, Patel MP, Robson EA, Hayward J, et al. High prevalence of CCDC103 p. His154Pro mutation causing primary ciliary dyskinesia disrupts protein oligomerisation and is associated with normal diagnostic investigations. Thorax. 2018;73(2):157–66.
Fassad MR, Shoemark A, Legendre M, Hirst RA, Koll F, Le Borgne P, et al. Mutations in outer dynein arm heavy chain DNAH9 cause motile cilia defects and situs inversus. Am J Hum Genet. 2018;103(6):984–94.
Knowles MR, Ostrowski LE, Leigh MW, Sears PR, Davis SD, Wolf WE, et al. Mutations in RSPH1 cause primary ciliary dyskinesia with a unique clinical and ciliary phenotype. Am J Respir Crit Care Med. 2014;189(6):707–17.
Ta-Shma A, Hjeij R, Perles Z, Dougherty GW, Abu Zahira I, Letteboer SJ, et al. Homozygous loss-of-function mutations in MNS1 cause laterality defects and likely male infertility. PLoS Genet. 2018;14(8):e1007602.
Paff T, Onoufriadis A, Anthony D, Shoemark A, Micha D, Kuyt B, et al. Mutation in the CCDC114 gene causes primary ciliary dyskinesia with normal fertility in the isolated Volendam population. Tijdschrift Voor Kindergeneeskunde. 2013;81:92-.
Best S, Shoemark A, Rubbo B, Patel MP, Fassad MR, Dixon M, et al. Risk factors for situs defects and congenital heart disease in primary ciliary dyskinesia. Thorax. 2019;74(2):203–5.
Wallmeier J, Frank D, Shoemark A, Nöthe-Menchen T, Cindric S, Olbrich H, et al. De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am J Hum Genet. 2019;105(5):1030–9.
Wallmeier J, Al-Mutairi DA, Chen C-T, Loges NT, Pennekamp P, Menchen T, et al. Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Genet. 2014;46(6):646–51.
Boon M, Wallmeier J, Ma L, Loges NT, Jaspers M, Olbrich H, et al. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Nat Commun. 2014;5(1):4418.
Baz-Redón N, Rovira-Amigo S, Paramonov I, Castillo-Corullón S, Cols-Roig M, Antolín M, et al. Implementation of a gene panel for genetic diagnosis of primary ciliary dyskinesia. Arch Bronconeumol (English Edition). 2021;57(3):186–94.
Fassad MR, Patel MP, Shoemark A, Cullup T, Hayward J, Dixon M, et al. Clinical utility of NGS diagnosis and disease stratification in a multiethnic primary ciliary dyskinesia cohort. J Med Genet. 2020;57(5):322–30.
Shoemark A, Griffin H, Wheway G, Hogg C, Lucas JS, Camps C, et al. Genome sequencing reveals underdiagnosis of primary ciliary dyskinesia in bronchiectasis. Eur Respir J. 2022;60(5).
AoURP I. The “All of Us” research program. N Engl J Med. 2019;381(7):668–76.
Postema MC, Carrion-Castillo A, Fisher SE, Vingerhoets G, Francks C. The genetics of situs inversus without primary ciliary dyskinesia. Sci Rep. 2020;10(1):3677.
England N. National Genomic Test Directory. Testing criteria for rare and inherited disease. 2020.
Behan L, Dimitrov BD, Kuehni CE, Hogg C, Carroll M, Evans HJ, et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J. 2016;47(4):1103–12.
Shah A, Shoemark A, MacNeill SJ, Bhaludin B, Rogers A, Bilton D, et al. A longitudinal study characterising a large adult primary ciliary dyskinesia population. Eur Respir J. 2016;48(2):441–50.
Shoemark A, Polverino E, Blasi F, Ringshausen FC, De Soyza A, Vendrell M, et al. Primary ciliary dyskinesia in adults with bronchiectasis: data from the Embarc registry. Eur Respiratory Soc. 2018.
Walker WT, Jackson CL, Lackie PM, Hogg C, Lucas JS. Nitric oxide in primary ciliary dyskinesia. Eur Respir J. 2012;40(4):1024–32.
Beydon N, Kouis P, Marthin JK, Latzin P, Colas M, Davis SD, et al. Nasal nitric oxide measurement in children for the diagnosis of primary ciliary dyskinesia: European Respiratory Society technical standard. Eur Respir J. 2023;61(4).
Shapiro AJ, Josephson M, Rosenfeld M, Yilmaz O, Davis SD, Polineni D, et al. Accuracy of nasal nitric oxide measurement as a diagnostic test for primary ciliary dyskinesia. A systematic review and meta-analysis. Ann Am Thorac Soc. 2017;14(7):1184–96.
Rubbo B, Shoemark A, Jackson CL, Hirst R, Thompson J, Hayes J, et al. Accuracy of high-speed video analysis to diagnose primary ciliary dyskinesia. Chest. 2019;155(5):1008–17.
Lucas JS, Barbato A, Collins SA, Goutaki M, Behan L, Caudri D, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J. 2017;49(1).
Shapiro AJ, Davis SD, Polineni D, Manion M, Rosenfeld M, Dell SD, et al. Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2018;197(12):e24–39.
Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013;188(8):913–22.
Shoemark A, Frost E, Dixon M, Ollosson S, Kilpin K, Patel M, et al. Accuracy of immunofluorescence in the diagnosis of primary ciliary dyskinesia. Am J Respir Crit Care Med. 2017;196(1):94–101.
Cindrić S, Dougherty GW, Olbrich H, Hjeij R, Loges NT, Amirav I, et al. SPEF2-and HYDIN-mutant cilia lack the central pair–associated protein SPEF2, aiding primary ciliary dyskinesia diagnostics. Am J Respir Cell Mol Biol. 2020;62(3):382–96.
Dougherty GW, Loges NT, Klinkenbusch JA, Olbrich H, Pennekamp P, Menchen T, et al. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am J Respir Cell Mol Biol. 2016;55(2):213–24.
Olbrich H, Schmidts M, Werner C, Onoufriadis A, Loges NT, Raidt J, et al. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am J Hum Genet. 2012;91(4):672–84.
Shapiro AJ, Sillon G, D’Agostino D, Baret L, López-Giráldez F, Mane S, et al. HYDIN variants are a common cause of primary ciliary dyskinesia in French Canadians. Ann Am Thorac Soc. 2023;20(1):140–4.
Horani A, Ferkol TW, Dutcher SK, Brody SL. Genetics and biology of primary ciliary dyskinesia. Paediatr Respir Rev. 2016;18:18–24.
Schofield LM, Duff A, Brennan C. Airway clearance techniques for primary ciliary dyskinesia; is the cystic fibrosis literature portable? Paediatr Respir Rev. 2018;25:73–7.
Paff T, Daniels JM, Weersink EJ, Lutter R, Noordegraaf AV, Haarman EG. A randomised controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia. Eur Respir J. 2017;49(2).
Paff T, Omran H, Nielsen KG, Haarman EG. Current and future treatments in primary ciliary dyskinesia. Int J Mol Sci. 2021;22(18):9834.
Kobbernagel HE, Buchvald FF, Haarman EG, Casaulta C, Collins SA, Hogg C, et al. Efficacy and safety of azithromycin maintenance therapy in primary ciliary dyskinesia (BESTCILIA): a multicentre, double-blind, randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2020;8(5):493–505.
Strippoli M-PF, Frischer T, Barbato A, Snijders D, Maurer E, Lucas JS, et al. Management of primary ciliary dyskinesia in European children: recommendations and clinical practice. Eur Respir J. 2012;39(6):1482–91.
Chhin B, Negre D, Merrot O, Pham J, Tourneur Y, Ressnikoff D, et al. Ciliary beating recovery in deficient human airway epithelial cells after lentivirus ex vivo gene therapy. PLoS Genet. 2009;5(3):e1000422.
Ostrowski LE, Yin W, Patel M, Sechelski J, Rogers T, Burns K, et al. Restoring ciliary function to differentiated primary ciliary dyskinesia cells with a lentiviral vector. Gene Ther. 2014;21(3):253–61.
Lai M, Pifferi M, Bush A, Piras M, Michelucci A, Di Cicco M, et al. Gene editing of DNAH11 restores normal cilia motility in primary ciliary dyskinesia. J Med Genet. 2016;53(4):242–9.
Author information
Authors and Affiliations
Contributions
E.C, A.S and J.D.C wrote the main article text, prepared figures and approved the final version.
Corresponding author
Ethics declarations
Conflict of Interest
AS has received grants, contracts or consulting fees from TranslateBio, Insmed, Spirovant and ReCode Therapeutics; JDC has received grants, contracts or consulting fees from Astrazeneca, Chiesi, GlaxoSmithKline, Insmed, Grifols, Novartis, Boehringer Ingelheim, Pfizer, Janssen, Antabio and Zambon.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cant, E., Shoemark, A. & Chalmers, J.D. Primary Ciliary Dyskinesia: Integrating Genetics into Clinical Practice. Curr Pulmonol Rep 13, 57–66 (2024). https://doi.org/10.1007/s13665-023-00332-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13665-023-00332-x