
Abstract
Sustainable food production for a growing world population will pose a central challenge in the coming decades. Organic farming is among the feasible approaches to achieving this goal if the yield gap to conventional farming can be decreased. However, uncertainties exist to which extend—and for which phenotypes in particular—organic and conventional agro-ecosystems require differentiated breeding strategies. To answer this question, a heterogeneous spring barley population was established between a wild barley and an elite cultivar to examine this question. This initial population was divided into two sets and sown one in organic and the other in conventional managed agro-ecosystems, without any artificial selection for two decades. A fraction of seeds harvested each year was sown the following year. Various generations, up to the 23th were whole-genome pool-sequenced to identify adaptation patterns towards ecosystem and climate conditions in the allele frequency shifts. Additionally, a meta-data analysis was conducted to link genomic regions’ increased fitness to agronomically related traits. This long-term experiment highlights for the first time that allele frequency pattern difference between the conventional and organic populations grew with subsequent generations. Further, the organic-adapted population showed a higher genetic heterogeneity. The data indicate that adaptations towards new environments happen in few generations. Drastic interannual changes in climate are manifested in significant allele frequency changes. Particular wild form alleles were positively selected in both environments. Clustering these revealed an increased fitness associated with biotic stress resistance, yield physiology, and yield components in both systems. Additionally, the introduced wild alleles showed increased fitness related to root morphology, developmental processes, and abiotic stress responses in the organic agro-ecosystem. Concluding the genetic analysis, we demonstrate that breeding of organically adapted varieties should be conducted in an organically managed agro-ecosystem, focusing on root-related traits, to close the yield gap towards conventional farming.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The cultivation of barley (Hordeum vulgare L.) and human development have been closely associated over the last 10,000 years (Zohary et al. 2012). Due to this tight bond, barley has accompanied human migration across almost all continents (von Bothmer et al. 2003; Malysheva-Otto et al. 2006). This movement led to the adaptation of genetic material to a wide range of local environments forming a highly diverse gene pool across the globe (von Bothmer et al. 2003; Morrell and Clegg 2007). Compared to this passive process, modern breeding strategies and the development of new farming technologies have shaped the allele frequencies of current high-yielding varieties at a considerable pace and extent (Khush 2001). As a result, these varieties have been adapted to applying agrochemicals and modern management strategies and harvesting processes (Hedden 2003). Furthermore, the demand for substantial yields will continue to increase in the coming decades (Ray et al. 2012). Moreover, the focus on yield maximization in conventional farming practices results in ecological damage. Modern societies are increasingly less willing to accept this one-sided practice of food production (Tilman 1998). The reduction of biodiversity, an increase in pollution, and climate change are relevant to the future development of strategies for farmers and policymakers. Based on shifts in priorities, organic farming has become more relevant over the past few years (Willer et al. 2008). Despite the benefit of ecosystem services, organic farming results in a yield reduction of 20–30% compared to conventional farming (Reganold and Wachter 2016). Therefore, it is imperative to counteract this reduction by increasing yields, as the predicted demand for food will increase by ~ 70% until the year 2050 (Ray et al. 2012, 2013). As the genetic material used in organic farming is derived from old varieties or varieties developed for conventional agriculture, the yield gap can be reduced by developing organically adapted varieties (Murphy et al. 2007). Previous studies have highlighted this effect and pointed out the demand for selection towards local adaptation to account for environmental interactions (Simmonds 1991; Ceccarelli 1994). Besides, conventional agriculture is also facing severe challenges and changes caused by the energy shortage and, in particular, mineral fertilizer (Randive et al. 2021).
However, conventionally adapted germplasm seems unsuitable for use as a single source of variation to achieve this goal (Murphy et al. 2007; Le Campion et al. 2020). Hence, integrating wild form genetic resources, which have continuously undergone natural evolutionary adaptation, could reverse the depletion of genetic diversity (Nevo 1998) and enrich alleles suitable for organic farming and nutrient efficiency. The value of landraces and wild material has been demonstrated in evolution and physiology experiments before (Grando and Ceccarelli 1995; Raggi et al. 2022).
Over the last two decades, a long-term evolutionary adaptation experiment has been ongoing to identify relevant alleles for organic agriculture in a spring barley backcross population. Two backcrosses of the former elite barley cultivar Golf (recurrent) and the wild form of barley ISR42-8 (donor) were performed to establish the starting population BC2F3. These seeds were sown in two different agro-ecological environments (organic and conventional management practice, Fig. 1, Supplementary Table 1). The organic and conventional populations were harvested separately for each following year, and seeds were used to establish the next generation. As no artificial selection occurred over 20 generations, mainly natural evolutionary adaptation processes towards conventionally and organically characterized agro-ecosystems will have altered the composition of these two wild form introgressed populations. The present study aimed to observe and compare these spring barley populations for molecular genetic adaptation footprints towards the agro-ecosystems.

Workflow overview. A Crossing scheme for the establishment of the initial spring barley population. B Field experiments with generations and treatments. Seeds from previous years were used to establish the next generation. The picture in the center is a drone shoot of the field with the barley plots in their defined crop rotation (unblurred row, rotating in these plots only, no shift to any other row). Yellow boxes indicate the conventional (left) and organic populations (right) in any exemplary year. The gray arrows indicate which crops were fertilized with manure in the organic agro-ecosystem. The treatments for both agro-ecosystems are indicated on top and below. C Selected populations and sampling strategy.
2 Material and methods
2.1 Plant material and population construction
More than 25 years ago, a long-term adaptation experiment was launched using barley as a model. The crossing scheme used in this study is described elsewhere (Cox 1984). The population was derived from the initial cross of the cultivar Golf (H. vulgare L. ssp. vulgare) and wild form ISR42-8 (Hordeum vulgare L. ssp. spontaneum) as the recurrent and donor parents. Golf was selected as an elite cultivar and used as the maternal plant in the initial barley cross, while the wild form was used as the donor to increase the genetic diversity of the population (Fig. 1A). Golf is a spring barley variety developed by Nickerson in the UK and registered under the ID “RPB 822-77” on the 23.01.1984 at the Bundessortenamt in Germany. No GMO or DH techniques have been applied in the development of this line variety. A single F1 plant was backcrossed to the cultivar to retain only a single wild form allele per locus, and six BC1F1 plants were randomly selected and further backcrossed. Two plants of each BC2F1, in a total of 12 sublines, were used to produce BC2F2 plants. All crossings were performed in a greenhouse under controlled conditions to avoid admixtures. An equal number of obtained seeds from each (sub)cross in BC2F2 were then sown to ensure that all the BC2F3 introgression lines had equal contributions for the following evolutionary adaptation process.
2.2 Agro-environmental evolutionary adaptation experiments
Cultivation occurred under organic and conventional cropping conditions for 22 generations (from 1997 to 2019) at two field stations managed by the University of Bonn: Dikopshof (1997 to 2010; 50° 48′ 22.6″N 6° 57′ 05.5″E) and Klein–Altendorf (2009 to 2019; 50° 36′ 47.2″N 6° 59′ 39.3″E). The organically farmed system employed a seven-field crop rotation without any application of pesticides or growth regulators. In contrast, the conventionally farmed system was based on a three-field crop rotation, complemented by on-demand plant and fungicide control following professional practice standards (Fig. 1B). In addition, 80–100 kg/ha of nitrogen (calcium ammonium nitrate) fertilizer was applied at the booting and flowering stages. Cattle manure (20 t/ha) was applied to two crops in the seven-field rotation of the organic system (Fig. 1B, gray arrows).
Further details of the crop management strategy are presented in Supplementary Table 1. The pH value was adjusted to 7, and the organic matter content of both systems ranged from 1.5 to 2% across the entire duration of the experiment. The sowing density was 320 and 360 grains/m2 for the conventional and organic systems, respectively. Based on a plot size of 9 × 15 m, the annual population size was approximately 45,000 individual plants. The populations rotated in defined plots (Fig. 1B) to ensure that selection pressure is caused only by diseases, availability/lack of nutrients and minerals, crop rotation, and weather conditions. The plants were harvested using a plot combine harvester after an average cropping period of 137 days. In order to avoid admixture, the seeds for the following generation were harvested from the center of the plots.
Climate data of the experimental fields were collected from local field-based weather stations. Radiation, wind speed, humidity, precipitation, and temperature were measured every 10 min at 1 m above ground level. As there were deviations in the measured average of the two sites (Dikopshof and Klein–Altendorf), annual average values were calculated for the two data sets separately. The weather data was aggregated to mean and summary values (Suppl. Figure 1). The potential evapotranspiration during the barley vegetation period was estimated by calculating the reference evapotranspiration value using the Penman-Monteith equation (Allen et al. 1998). In addition, the barley crop evapotranspiration (ETC) value (Allen et al. 1998; Zotarelli and Dukes 2010) was calculated, and the soil–water balance was determined to estimate periods with water deficits (ETA) (Meyer and Green 1980; Minasny and McBratney 2018). The limit of thermal time for the initial, crop development, mid, and end stages was set at 220, 650, 1300, and 1650 growing degree days, respectively, using 0 °C as the base temperature. The sowing date was set as starting point for these calculations. The water balance was based on a maximal root depth of 75 cm. When the usable field capacity decreased to less than 30%, the evapotranspiration value was corrected as described previously (Meyer and Green 1980). The correct estimation of ETC by the FAO Penman-Monteith equation was illustrated in Pozníková et al. (2014).
The texture of the soil, which was composed predominantly of loam, was characterized by a high-field capacity of approximately 25%. The soil type is Luvisol. The soil type at both field stations was a Luvisol with a cation exchange capacity of 210 and 240, with a high usable filed capacity of 191 and 198 mm in the top 110 cm soil, respectively. Both field stations were located in plane areas with no risks for water erosion.
2.3 Sample preparation for genotyping
Pooled samples for five offspring generations (BC2F3 [initial offspring generation grown in a greenhouse], BC2F12, BC2F16, BC2F22, and BC2F23) from both the conventional and organic environments were generated (18 pool samples in total, Fig. 1D). For every generation, seeds have been stored in a cold chamber under controlled humidity and temperature conditions up to the point of sampling (4 °C, 50% humidity). The required sample size for a representative allele frequency estimation in each population was estimated by applying Cochran’s formula (Cochran 1977) with an aimed precision level of p < 0.05 and a narrow confidence interval of p > 0.996 to determine the correct allele frequency with a maximum deviation of 5% from the true value. Based on a theoretically expected allele frequency of infinite population size—12.5% (Cox 1984) of the donor wild form alleles, a sample size of 296 genotypes per generation and environment was required to estimate the total genetic diversity of the population. Based on these estimates, 300 genotypes were pooled. Pool samples at the leaf tissue level were constructed by harvesting equal amounts of leaf discs per genotype utilizing a hole puncher, which were then pooled. To avoid any bias, tissue materials were vigorously homogenized using a TissueLyser II ball mill (Qiagen GmbH, Hilden, Germany). Total genomic DNA was extracted using the Plant DNA Mini kit (VWR International GmbH, Darmstadt, Germany) in accordance with manufacturer’s instructions. Two samples of each generation and environment with 300 genotypes were pooled, creating a test set of 600 genotypes per population-environment combination.
2.4 Genotyping of parents and populations
The genotyping of the populations was performed by following the method proposed by Schneider et al. (2022). WGS (Novogene Co., Ltd., Beijing, China) (Belkadi et al. 2015) with a moderate depth of 10 reads per locus was applied. Similarly, the two parents, cultivar Golf and wild form ISR42-8, were sequenced. The seeds used to genotype the parental lines origin from the same batches from which the population has been established in the 1990s. The sequence quality was obtained with FastQC (Andrews 2010). As the quality was sufficiently high, no trimming was performed. The alignment to the Hordeum vulgare reference genome toplevel v2 (downloaded 30.05.2019 (Mascher et al. 2017)) was executed using bwa mem (version 0.7.1 (Li 2013)). Aligned reads were strictly filtered by sambamba view (Tarasov et al. 2015) (version 0.6.6), while only retaining single mapped reads with a mapping quality higher than 30 and removing unpaired reads:
Duplicates were removed using sambamba markdup. Variant calling was performed with SAMtools mpileup and BCFtools call (version 1.8 (Li et al. 2009)) with a minimum variant base quality of 30 and overall read quality of 25 for parents and pool progeny samples in one run. Indels were omitted. Additionally, the two flags -t AD and --positions were specified. AD, the allelic depth format, generates information about the read calls for each allele and sample. The position flag was used to restrict the pileup to sites where the parents were homozygote, variant from one another, and the position was reported as variant in the Ensembl (Cunningham et al. 2019) variant reference database. The BCFtools options -v and -m were used to generate VCF file. As described previously (Yao et al. 2020), the proposed method is the best routine for large plant genome variant calling with superior specificity and sensitivity.
Afterward, haplotypes were constructed based on the cultivar and wild form alleles. The SNPs located within or close to an annotated gene were aggregated into one single haplotype, providing a haplotype allele frequency (haplotype allele frequency). More than 20 million variants were identified between the cultivar and wild form donor. Variant detection of next-generation sequencing data is exceptionally error-prone (Nielsen et al. 2011; Olson et al. 2015); therefore, variant loci not reported in the Ensembl online database were removed (Cunningham et al. 2019). This approach retained about 3.6 million SNPs from pooled SNPs calling.
2.5 Genetic diversity determination
The allele frequency for each SNP was calculated as \({AF}_{Parent1} = {RD}_{Allele\, Parent1}/({RD}_{Allele\, Parent1} +{ RD}_{Allele\, Parent2})\,\mathrm{ and }\,{AF}_{Parent2} = 1 - {AF}_{Parent1}\), where RD is the read depth. The allele frequency and the read depth were further used to calculate the haplotype allele frequency (haplotype allele frequency). As the SNP’s allele frequency was corrected by the read coverage for haplotype allele frequency estimation, a minimum cutoff of coverage per SNP was not set, and all SNPs with coverage of ≥ 1 were included for analysis. Further details are described in Schneider et al. (2022).
Based on the fact that the population has two founders (Golf and ISR42-8) and their haplotypes are known, this information can be incorporated into the pooled sequencing of the population. Gene annotation derived from IPK database (Anonymous IPK Galaxy Blast Suite n.d.) with positional annotation for high-confidence (HC) genes was used as an anchor to generate haplotypes directly linked to a function.
HC gene-derived haplotypes’ associated SNP allele frequency scores were used to perform pairwise statistical comparisons of generations and systems. More detailed, to identify any significant variations, pairwise gene-derived haplotype comparisons of the SNP allele frequency distributions were compared between generations and environments. Prior to this step, the two samples per generation and system were merged to achieve higher confidence. Haplotype allele frequency calls were smoothed using a loess function with a span of 0.03 to accommodate for potential false-positive allele frequencies based on expected LD patterns. As the SNP density and SNP allele frequency distribution per gene-anchored haplotype varied widely, a dynamic algorithm selected the appropriate test statistic.
Furthermore, commonly utilized approaches to identify consistent differences between populations lack accuracy and result in many false positives (Wiberg et al. 2017). Exemplarily for our data set, some haplotypes were characterized by relatively few SNPs, some by many SNPs with 0% haplotype allele frequency, and others by mixtures of distributions. Therefore, an algorithm was developed to choose the appropriate test statistic based on the haplotype’s SNP allele frequency distribution pattern. The Mann–Whitney U test was applied if the number of SNPs per haplotype was smaller than 30 (15 SNPs per haplotype and sample), as a proper estimate of the distribution could not be completed, or if one of both test samples consisted of all zero values. If less than 15% of all values were zero or one test sample did not have any zero AF entries, a generalized linear model, based on a negative binomial distribution, was used to test the statistical significance. A zero-inflated, negative binomial distribution characterized the largest haplotype fraction. These haplotypes were analyzed with the Pscl R package (Zeileis et al. 2008) implementation in Julia if less than 75% of all SNPs had a wild form haplotype allele frequency of 0%. Lastly, a zero-inflated Poisson-based linear model was applied if the number of SNPs with no wild form haplotype allele frequency detected exceeded 75%. Pscl package separately calculates two test statistics for Poisson/negative binomial and zero-inflated statistics. Therefore, both p values were extracted and saved in the statistics summary table together with the information of the used test statistic; the average haplotype allele frequency; and variation, read depth, SNP, and zero value count. Significant candidate haplotypes were based on p values of < 0.05 for the negative binomial distribution and an additional filter based on the zero-inflated distribution if calculated (p < 0.05). These analyses were combined with a meta-analysis of priorly published QTL loci and were further applied to identify candidate genes, as described below.
2.6 Overall genetic structure analysis
The potential influence of genetic drift was simulated in a customized Julia (Bezanson et al. 2017) script related to the R function sim.drift (Sharma 2019). The following settings were applied: population size, 10,000 (equals the center part of the field plot used for the establishment of next year’s population); ploidy level, 2; the number of generations, 21; as initial allele frequency, 0.1; the number of loci, 34,237; and iterations, 1000. Additionally, the wild form haplotype allele frequency of the BC2F3 was used as a starting point to calculate the variation based on drift effects for the following generations. Over the 1000 iterations, a median value and the 95% confidence interval were calculated and compared against the gene-based haplotype allele frequencies of the organic and conventional populations. Corresponding generations were tested against one another. A negative binomial linear model was applied to calculate the probability (p) value.
Besides, we tested how many haplotypes did not show evidence of any wild form alleles being present. This “introgression rate” was assessed by counting those haplotypes with less than 0.005 wild form haplotype allele frequency across all generations.
A principal component analysis (PCA) was performed with the R Bioconductor package PCAtools (Blighe and Lun 2019) to identify genetic structures and variations between generations and environments. The explanatory effect and significance of each principal component (PC) on the generations, replicates, and agro-ecosystems were calculated by the stats (R Core Team 2020) cor.test function, based on a Pearson correlation test. Samples were categorized by the number of replicates, environments, and generations. The diversity estimation within each environment and generation was tested based on the variation between the two samples. The distance of the samples was calculated for each PC separately and multiplied with the explanatory weight of the PC. Based on these values, a single value over the first five components was summarized and compared between the environments. A dendrogram of genetic distances based on the HC gene haplotype allele frequency was created with the pvclust (Suzuki et al. 2019) package with the ward.D2 agglomeration and canberra distance method. Haplotypes with a read depth above 100 were used for this calculation. The FST value was calculated using VCFtools (Danecek et al. 2011) with default settings and VCF and BAM files as input.
Abrupt changes in haplotype frequency were regarded as a combination of crossing over (crossing-overs) events combined with strong fitness differences between both sides of the crossing-overs region. These regions were manually identified after a loess adjustment of the haplotype allele frequency with a span of 0.03. We considered all regions as potential crossing-overs/adaptive selection regions when a change of the haplotype allele frequency (Δ > 0.1) from one genomic segment to another was observed. Each segment needed to be longer than 5 MB, defined by a homogeneous haplotype allele frequency in the segment.
2.7 Predictions of coding and putative amino acid sequences
The splice variants of the coding DNA reference from the Ensembl Plants genome-centric portal for plant species (Cunningham et al. 2019) were filtered for the presence of start and stop codons and used to map the obtained reads. The longest was used as a reference if multiple splice variants had a start and stop codon. Information of the physical location of the gene was utilized to extract a consensus sequence for the cultivar and wild form donor from BAM files using SAMtools and BCFtools (Li et al. 2009). If necessary, the reverse complement strand was assessed with the FASTX-toolkit (Gordon and Hannon 2010). The obtained consensus sequences of the two parents were aligned to the selected coding DNA splice variant by MEGACC muscle (Edgar 2004) alignment implementation and mafft (Katoh and Standley 2013) alignment using default settings. All steps were implemented in a custom Julia script to process 127,149 gene splice variants. Introns, as well as up- and downstream start and stop codons, were removed. The presence of start and stop codons in the parental coding DNA alignment was tested, and deletions were counted. Additionally, the DNA sequence was translated into a putative sequence of amino acids with Bio.Seq (Ward 2020) to estimate mismatches and stop codons. The most similar alignment of muscle and mufft to the coding DNA was selected. Some genes characterized by poor alignment quality were reprocessed on the splice variant level. All splice variants of these genes were aligned to the coding DNA, and the most similar splice variant (compared to the reference coding DNA) was selected and added to the coding sequence list. In total, 28,601 complete CDS alignments were utilized to identify and select candidate genes based on structural variations like insertions, deletions, preliminary stop codons, or SNPs in the exons.
2.8 Evolutionary adaptation signature and candidate gene selection
A meta-analysis of published QTL loci was performed. Several QTL studies have observed marker-trait associations for numerous traits, including drought, yield physiology, resistance to biotic stressors, and yield components. Many of these studies employed the 9K iSelect genotyping chip. Therefore, each haplotype was aligned on this genetic map (Fig. 3). The NCBI BLASTn algorithm (Altschul et al. 1990) and the bwa mem option were used to retrieve the unique physical position of all markers in the 9K iSelect chip on the barley reference genome V2. Markers with multiple loci were omitted. The same procedure was followed for other genetic maps and specifically designed markers reported in previous publications. Potential candidate genes related to QTL were selected based on significant (p < 0.001) haplotype allele frequency variations between the environments or generations, the functional prediction (Anonymous IPK Galaxy Blast Suite n.d.), and the coding DNA variation. The identified candidate loci were clustered in the six agronomically relevant categories: root morphology, biotic resistance, nutrient related, drought tolerance, yield components, and yield physiology. The haplotype allele frequency was compared generation-wise for significant variations between the environments using an unpaired Wilcoxon test, using the ggpubr R package (Kassambara 2020).
3 Results
3.1 Characterization of genetic diversity between the parental lines
The two parents were genotyped by whole-genome re-sequencing (WGS). Five progeny generations (BC2F3, BC2F12, BC2F16, BC2F22, and BC2F23) with two replicates each were separately genotyped for both agro-ecosystems by pooled WGS, which resulted in 400 million high-quality paired-end reads for each library. The genome-wide coverage was uniform, with an average of nine reads per base (Supplementary Figure 2). Overall, 3,991,259 high-confidence single-nucleotide polymorphisms (SNPs) and 621,434 indels were identified. As many as 3,632,910 SNPs were annotated to 34,343 high-confidence genes, according to the haplotyping approach presented by Schneider et al. (2022), which led to a median enrichment of 55 SNPs per gene and a median haplotype size of 71,585 base pairs. The calculated allele frequencies of all SNPs related to the same gene were aggregated to one haplotype allele frequency (haplotype allele frequency) with a median coverage of 434 reads per haplotype.
The coding sequences with annotated start and stop codons were aligned to the parental haplotypes to predict the functional effects of SNP variants and identify candidate genes that might cause haplotype allele frequency changes in linkage blocks. A total of 28,601 complete coding DNA alignments were generated, and 640,611 amino acid variants in 16,570 predicted protein sequences were identified between the parents. Compared to the reference coding DNA, 3831 and 3305 genes in the wild form and the cultivar genomes, respectively, showed evidence of deletions or insertions. A high degree of variation was observed at the DNA and the predicted amino acid level.
3.2 Agro-ecosystem-induced genetic variation
The observed donor genome-wide allele frequency (genome-wide allele frequency) of the BC2F3 generation was 10.07%, a deviation of 0.5% from the expected frequency for a population of the applied population size, according to Cox (1984). The genome-wide allele frequency was continuously reduced to 6.95% in the BC2F23 generation in the conventionally evolving population. In the organic system, the donor genome-wide allele frequency decreased to 8.96% in the 12th generation but increased to 10.57% in the 23rd generation, which was 0.5% greater than in the initial BC2F3 (Supplementary Figure 3, green line).
One potential cause for these allele frequency changes across generations might be the drift. So, the potential drift effect on these populations was estimated by simulations. One thousand iterations on (I) the average genome-wide allele frequency and (II) on all gene haplotypes individually were performed to estimate the drift effect in the real observations. Both I and II were compared to the observed haplotype allele frequency in the 12th, 16th, 22nd, and 23rd generations. Compared to the drift simulation, ten times larger 95% confidence intervals and a deviating average genome-wide allele frequency in the conventional and organic systems were observed (Supplementary Figure 4), indicating significant discrepancies between the drift effect simulations and the generation-wise conventional and organic detected haplotype allele frequency (Supplementary Table 3). Therefore, we conclude that drift alone cannot explain most allele frequency changes.
Additionally, the introgression rate was assessed based on the haplotype allele frequency across all generations. Of 34,344 HC genes, 531 (1.55%) had a wild form haplotype allele frequency < 0.005 and, thus, were not expected to be introgressed into the population. Most of these genes were located on chromosome 2H as well as on the short arms of chromosomes 3H and 4H.
In the conventional BC2F23, 7115 wild form and 15,124 cultivar-derived haplotypes showed a higher haplotype allele frequency compared to the BC2F3. Three thousand sixty-six wild form haplotypes were no longer found in the BC2F23 generation of the conventional system, of which 524 were already missing by the 12th generation. In contrast, only 547 haplotype positions in the organic system were found to no longer carry wild form haplotypes (Supplementary Table 4, Supplementary Figure 3).
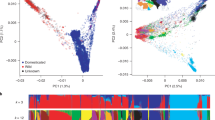
A genetic diversity analysis estimated the genomic variation between the generations of both populations in both agro-ecosystems to dissect an agro-environmental evolutionary adaptation process. The principal component analysis (PCA) revealed large genetic differences between the generations and the two agro-ecological environments. The first principal component (PC) described the significant (p < 0.001) differences between the organic and conventional environments, while the second PC separated the cumulative effects of different years and generations (Fig. 2).

Principal component analysis of the haplotype allele frequency in respect to generation and agro-ecosystem. Per combination of generation (12th, 16th, 22nd, and 23rd) and agro-ecosystem (organic, conventional), two samples were drawn (replication). The generations are presented in different colors, while different shapes indicate the different agro-ecosystems (A). The explanatory effect of the principal components is illustrated in B, where the first PC (PC1) explains the majority of variation between the farming systems, and the second PC explains the variation attributed to the generation effect. The replications did not affect the clustering of the samples. Stars indicate level of significance. A The majority of variation is explained by the first PC (51.24%), while the second PC explains 7.74%. The first five PCs explain 71.06 % of the total variance.
Furthermore, the diversity between the two pool samples prepared for each generation and environment was compared to estimate the within-generation genetic diversity between the two systems. This analysis indicated a three to 13-fold higher genetic variation over the first five PCs in the organic environment for the 12th, 16th, and 23rd generations (Table 1, Supplementary Table 2).
The population differentiation fixation index (FST) increased with each subsequent generation between the agro-ecosystems from 0.012 (BC2F12) to 0.113 (BC2F23) (BC2F16 = 0.043; BC2F22 = 0.052). Similar observations were made based on genetic distance clustering (Supplementary Figure 5).
When the initial generation BC2F3 was included in the PCA, the first PC described the difference between the starting generation (not adapted) and the offspring’s. The second PC splits the organic from the conventional environment and indicates adaptation patterns towards the agro-ecosystems (Supplementary Figure 6). Meanwhile, based on the first PC, the conventionally farmed population only evolved until the 12th generation, and after that changed little.
Besides the system effect, the generation, and with this also the climate and weather had the second-highest effect on the micro-evolutionary processes (Fig. 2B). The weather was highly volatile during the annual vegetation period from 1998 to 2019. So, the collected weather data were examined to identify conditions that might have caused allele frequency changes (Fig. 1C). The approximation of barley’s actual evapotranspiration (ETa, Supplementary Figure 7) revealed the presence of different periods with varying selection pressure. The years 1998, 2001, 2002, 2005, 2007, 2008, 2009, 2012, and 2013 were characterized by sufficient water availability for plants throughout the growth period. High air humidity levels in the anthesis stage were observed in 2007, 2012, 2016, and 2018. The ratio of days with precipitation to days without in the anthesis stage was found to be above average (0.3) for 2007 (0.41), 2012 (0.39), and 2016 (0.38). Based on evapotranspiration calculations (ETc), drought stress periods occurred in 1999, 2000, 2006, 2011, 2015, 2017, 2018, and 2019, highlighting the other end of the spectrum. Multiple consecutive days in 2011, 2017, 2018, and 2019 were characterized by an actual ETa/ETC ratio of < 0.7.
3.3 Identification of chromosomal hotspot region undergoing (micro)evolutionary adaptation
Haplotype allele frequency variations between the initial and offspring generations, as well as both agro-ecological environments, were observed on all seven chromosomes (Fig. 3). Many loci in the telomeric regions indicated increasing wild form haplotype allele frequency, while the wild form haplotype allele frequency was reduced in most pericentromeric regions in both environments. A strong increase in wild form haplotype allele frequency was observed in both agro-ecosystems on the short arm of chromosome 1H (locus = 0–26 Mbp; wild form haplotype allele frequency: F3 = 0.169, O23 = 0.392, C23 = 0.391; mean haplotype allele frequency in environment organic (O) and conventional (C) at generation X23), the long arms of 2H (locus = 645–690 Mbp; haplotype allele frequency: F3 = 0.173, O23 = 0.219, C23 = 0.371), 3H (locus = 630–647 Mbp; haplotype allele frequency: F3 = 0.064, O23 = 0.486, C23 = 0.462), and 6H (locus = 530–590 Mbp; haplotype allele frequency: F3 = 0.155, O23 = 0.279, C23 = 0.1972) (Further details can be obtained from https://michaelschneiderrun.shinyapps.io/allelefreqvariation/).

Genome-wide wild form haplotype allele frequency. Different facets show different chromosomes (chromosome name is indicated in the top left), while colors differentiate the generations and line types and shapes the agro-ecosystems (solid line = conventional; dashed-dot line = original population BC2F3; dashed line = organic populations). Lines represent a loess adjusted wild form haplotype allele frequency estimate. The wild form haplotype allele frequency (y-axis) is shown across the chromosome by physical position (x-axis). The gray area presents the peri-centromere of each chromosome (Mascher et al. 2017). A Chromosome 1H; B = 2H; C = 3H; D = 4H; E = 5H; F = 6H, G = 7H, H = unknown.
Besides, the two pericentromeric regions on chromosome 3H and 5H, characterized by large linkage blocks, were positively selected for the wild form alleles (3H, 210–450 Mbp; 5H, 370–490 Mbp). The increased wild form haplotype allele frequency on chromosome 5H was associated with QGL5H/QGW5H (Fig. 4, Supplementary Table 5), a quantitative trait loci (QTL) for grain length and width (Watt et al. 2019). The search for candidate genes for this region resulted in several hits with a wild form haplotype allele frequency of ~ 0.5 for both environments in the late generations (Supplementary Table 6). Remarkably, the average wild form haplotype allele frequency of the entire region on chromosome 5H was low in the BC2F3 (0.0009), but increased to 0.372 in the conventional and 0.284 in the organic populations over successive generations (values for F23). The below-expected allele frequency in the initial generation might be associated with the close physical bound of QGL5H/QGW5H with the seed dormancy locus 1 (SD1, Fig. 4). The tight linkage between the agronomically favorable allele of ear length and the unfavorable allele of extensive dormancy has caused an unintended selection during seed multiplication against both these wild form alleles. Still, the wild form haplotype allele frequency of QGL5H/QGW5H increased to ≥ 0.45 in both agro-ecosystems, while the wild form haplotype allele frequency of SD1 decreased to < 0.01.

Zoomed image of the 5H chromosome with emphasis on the region beginning at 350 to 510 Mb. Colors differentiate the generations and line types and shapes the agro-ecosystems (solid line = conventional; dashed-dot line = initial BC2F3 population; dashed line = organic populations). Lines represent a loess adjusted wild form haplotype allele frequency estimate, while the blurred points in the background show the actual measured wild form haplotype allele frequency of each block, separately. The wild form haplotype allele frequency (y-axis) is shown across the chromosome by physical position (x-axis). The figure illustrates the wild form allele frequency development across multiple generations and farming systems on the entire 5H chromosome. The region in the black square is magnified to visualize with an increased resolution (gray box above). Arrows indicate the physical position of adaptational relevant alleles.
This development can be concluded that those few genotypes in the BC2F3 generation with a crossing-over event between these two genes must have had an adaptational (fitness) advantage compared to the genotypes with the QGL5H/QGW5H Golf allele.
A similar pattern of lower than the average wild form haplotype allele frequency values in the initial population was observed in the peri-centromere of chromosome 2H (250–400 Mbp; haplotype allele frequency: F3 = 0.027). However, unlike the region on 5H, no substantial increase of the wild form haplotype allele frequency was observed in either system (O23 = 0.004, K23 = 0.004).
Compared to the linkage block in the peri-centromere on 5H, the peri-centromere of chromosome 3H revealed a major differentiation between the organic and conventional populations (Fig. 5). The wild form haplotype allele frequency reached 0.18 in the organic system but decreased below 0.02 in the conventional one (F3 = 0.059). In multiple QTL studies, this region was characterized by enriched loci for various traits, including plant height (Marzec and Alqudah 2018), root morphology (Reinert et al. 2016; Oyiga et al. 2020), and flowering time (Guan et al. 2012; Xu et al. 2017) (Supplementary Table 5).

Zoomed image of the 3H chromosome with emphasis on the region beginning at 35 to 160 Mb. Colors differentiate the generations and line types and shapes the agro-ecosystems (solid line = conventional; dashed-dot line = initial population; dashed line = organic population). Lines represent a loess adjusted wild form haplotype allele frequency estimate, while the blurred points in the background show the actual measured wild form haplotype allele frequency of each block, separately. The wild form haplotype allele frequency (y-axis) is shown across the chromosome by physical position (x-axis). The figure illustrates the wild form allele frequency development across multiple generations and farming systems on the entire 3H chromosome. The region in the black square is magnified to show an increased resolution (gray box above). Arrows indicate the physical position of adaptational relevant alleles in this region.
The microevolutionary adaptation processes on chromosome 3H also allowed exemplifying recombination events in the telomeres ((sub-) Fig. 5). The brittleness loci of brt1 and brt2 are located on the short arm (Komatsuda et al. 2004), and the wild form carries the alleles for ear brittleness (brt1 and brt2), but not the modern cultivars (Brt1 and Brt2). Both brt1 and brt2 are negatively selected by machine harvesting and showed reduced wild form haplotype allele frequency of 0 and 0.005 in the conventional and organic adapted BC2F12 and following, respectively (compared to BC2F3 = 0.06, Fig. 5). In addition, this region was characterized by linkage blocks undergoing increased (four) and decreased (four) wild form haplotype allele frequency changes in the organic population. The transition from down- to up-selected regions became more distinct with each subsequent generation, indicating these haplotype allele frequency patterns are not likely to be by chance. Contrasting, the wild form haplotype allele frequency in the entire region was reduced to 0.003 in the conventional populations.
3.3.1 Adaptation in the context of linkage disequilibrium
The reported haplotype allele frequency changes are presumably the result of adaptational selection and crossing-overs (crossing-overs). Approximately equal numbers of crossing-overs were present in both agro-ecosystems from the start of the experiment, but the crossing-overs number in the BC2F3 is unknown as crossing-overs changes allele combinations in the gametes but not the allele frequencies. However, differences in the fitness of alleles near crossing-overs visualize former crossing-overs events in later generations through abrupt changes of haplotype frequency. Our goal was to trace the remaining crossing-overs/fitness combinations in the BC2F23. Therefore, the loess-adjusted wild form haplotype allele frequency of the 23rd generation in both organic and conventional agro-ecosystems was examined to identify underlying agronomically relevant crossing-over events (Fig. 6).

Detection of the crossing-over events combined with strong fitness differences between both sides of the crossing-overs region in the 23rd generation of the organic and conventional populations. Gray = conventional; orange = organic adapted population. A Count of crossing overs which became visible by adaptational-driven changes of the allele frequency by chromosome, separated by agro-ecosystem. B The loess corrected wild form haplotype allele frequency across the genome (span = 0.03). The facet show different chromosomes. Running numbers (red, organic; blue, conventional) present the position and count of crossing overs across the genome in the populations. Wherever two lines become visible, the two pool samples (replicates) differed in wild form haplotype allele frequency. The gray background presents the peri-centromere. Dots above the allele frequency curves indicate prominent plant physiology genes—significant differences between the agro-ecosystems are shown for HvPpDH1, HCElf2, and PpDH2.
As expected, more recombinations occurred in the telomeres. No chromosome showed increased counts of crossing-overs/fitness combinations, but comparing the agro-ecosystems highlighted 131 genome-wide crossing-overs in the organic but only 48 crossing-overs in the conventional agro-ecosystem. This unequal decline in detectable crossing-overs/fitness combinations could result from a more targeted selection in the conventional agro-ecosystem. Assuming equal numbers of crossing-overs per genotype, this might indicate that the organic BC2F23 population has a 273% higher genotype diversity than the conventional BC2F23.
Aligning eight loci varying between spring and winter barley (Mascher et al. 2017) highlighted significant differences between the agro-ecosystems for HvPpDH1, HCElf2, and HvPpDH2 (p < 0.05) in terms of wild form haplotype allele frequency between the agro-ecosystems (Fig. 6).
3.4 Changes in allele frequency as alternative QTL for key trait categories
In the following analysis, we aimed to transfer the genome-wide patterns onto single traits and phenotypes. To do so, a meta-data analysis of reported QTL data was performed for various agronomically important traits from the following six categories: nutrient uptake, biotic stress, drought tolerance, yield physiology, yield components, and root morphology. Many of these QTL alleles were detected in crosses where the wild form ISR42-8 was used as a parental line. The wild form haplotype allele frequency of the QTL regions was assessed (Supplementary Table 5) and compared between the two agro-ecosystems (selection force of interest) and among generations (Fig. 7A). All reported QTLs were clustered into the six trait categories mentioned above. The wild form haplotype allele frequency of the yield components (F3 = 0.0728, O23 = 0.149, C23 = 0.116—mean wild form haplotype allele frequency for the category across associated QTLs, Fig. 7B), the biotic stress (F3 = 0.097, O23 = 0.205, C23 = 0.196), and yield physiology (F3 = 0.0755, O23 = 0.1489, C23 = 0.104) categories were significantly increased in both agro-ecosystems above the levels of the BC2F3 generation (p > 0.01).

Combination of (meta-)QTLs and haplotype allele frequency patterns. A The wild form allele frequency relative to the physical position across the seven chromosomes. The yellow line represents the organic, while the gray line shows the conventionally adapted BC2F23 population. Whenever two lines become visible, the two replicates of each population differentiate from each other. The gray background illustrated the peri-centromere. Dots above the allele frequency lines present QTLs identified in a meta-analysis, clustered in the same six groups as aggregated in sub-figure B. The QTLs were clustered into six groups: biotic stress (resistance genes), drought tolerance (reaction to drought), nutrients (QTLs or genes identified and related to the transport or uptake of nutrients), yield physiology (e.g., plant height and heading dates), root morphological traits (e.g., root length), and yield components (e.g., kernel size). Many of the analyzed studies used ISR42-8 as donor parent to create introgression lines. The square and triangle indicate if the allele frequencies equal or differentiate between both agro-ecosystem adapted populations after 20 generations in field. B The wild donor allele frequencies in the QTL regions (based on Sup. Table 5) were clustered in six groups and plotted for each generation and environment. The y-axis illustrates the wild form allele frequency, while the generations are plotted on the x-axis. The farming systems are separated by shape and color (organic, yellow diamond; conventional, gray star). The gray and yellow lines give the standard deviation for the respective population, trait, and generation across all considered QTLs. Stars indicate significant difference levels between the organic and conventional populations.
Significant differences in drought tolerance (po23/c230.0019, comparing the 23rd generation of organic [po23] and conventional [pc23] populations; allele frequency F3 = 0.113, O23 = 0.099, C23 = 0.035), yield physiology (po23/c23 = 0.0031; F3 = 0.076, O23 = 0.149, C23 = 0.104), and root morphology (po23/c23 < 0.0001; F3 = 0.071, O23 = 0.162, C23 = 0.071) between the two agro-ecosystems were observed. Even though the wild form haplotype allele frequency of drought tolerance decreased in the 12th generation (O12 = 0.0652, F3 = 0.113), the process reversed in the organic system in the following generations (O23 = 0.0989), analogous to the more frequent occurrence of drought stress in the subsequent generations (Fig. 1C). In contrast, wild form haplotype allele frequency associated with drought tolerance-related genomic regions was further reduced in the conventional system (C23 = 0.0351). Root-related traits demonstrated a marked difference between the agro-ecosystems (O23 = 0.1621, C23 = 0.0707). While the wild form haplotype allele frequency followed a linear increase in the organic system across generations, there was no difference in the conventionally adapted generations compared to the initial generation.
Remarkably, the wild form haplotype allele frequency of all six clusters increased in the organic agro-ecosystem across generations. At the same time, the conventionally farmed population benefited from the wild form donor alleles, particularly in biotic stress resistance, yield physiology, and yield components.
4 Discussion
4.1 Evaluation of the methods for populations characterization
We used whole-genome sequencing of pooled samples to locate allele frequency trajectories in a biparental cross derived from an elite cultivar and wild barley. Parental haplotypes were tracked down in the populations based on biparental origin. Unlike other species, such as Arabidopsis thaliana, barley’s genome size and structure result in pronounced challenges in terms of sequencing costs. Pool sequencing of crops with exceptionally large genomes can overcome these problems and has been shown to provide reliable allele frequency estimates (Schneider et al. 2022). Additionally, haplotype-based analysis can successfully compensate for low sequencing coverage per genotype (0.03×), as reported previously (Schneider et al. 2022; Tilk et al. 2019). Still, deviations between the true and observed haplotype allele frequency might occur, which could be attributed to either methodical issues or genomic attributes. Such likely false-positive haplotype allele frequency calls can be identified using expected LD patterns or canceled using a loess adjustment of the haplotype allele frequency. Single gene-anchored haplotype blocks with a strongly divagating haplotype allele frequency to neighboring blocks will likely provide false information. Such deviations could result from undetected copy number variants, where one parent inherits more copies of the same gene than the other (Muñoz-Amatriaín et al. 2013), and due to a potential low resolution of repetitive sequences of the used reference genome mapped against in this region (Mascher et al. 2017). Such variations might result in unexpected wild form haplotype allele frequency patterns. Another potential source of unexpected wild form haplotype allele frequency might be the inadequate linear alignment of the reference genome for wild form alignments (Beier et al. 2017). The genomes of cultivated barley and its wild form might differ in this background, and therefore, haplotype blocks might be mismatched or broken apart. We reduced the bias of such abnormal patterns by defining potential candidate regions under adaptation to have a minimum number of haplotype blocks with a similar wild form haplotype allele frequency and smoothing these deviations by using a loess function.
4.2 Genome-wide adaptation patterns
This study’s results highlight that organically and conventionally farmed populations can benefit from the genetic diversity of distinct wild form alleles (Supplementary Table 5, 6). ISR42-8, which was used as donor, was reported to have an expanded root system (Naz et al. 2014), increased drought tolerance (Sayed et al. 2012; Honsdorf et al. 2014, 2017), increased seed and ear length and kernel weight (Zahn et al. 2020), reduced thresh ability (Schmalenbach et al. 2011), reduced plant height (Naz et al. 2014), late flowering (Wang et al. 2010), enhanced biotic stressor resistance genes (Von Korff et al. 2005; Schmalenbach et al. 2008; Dragan et al. 2013), and ear brittleness (Schmalenbach et al. 2011).
The highest magnitude of wild form haplotype allele frequency changes was noticed until the BC2F12. This indicates that few major alleles, like Denso (Fig. 5), seemed to have dominated the evolutionary adaptation process in the first generations towards the broad-sense environment (climate, soil type, photoperiod, light intensity, precipitation intensity). The climate characteristics of the broad-sense environment are illustrated by an average daily temperature of 14.5 °C, an average daily radiation of 2,250 W/m2, and a total average evapotranspiration of 300 mm and 250 mm of precipitation in the growth period (April to July) and are complemented by a highly fertile Luvisol growth substrate.
These humid growth conditions strongly deviate from the natural habitat the wild form has adapted to. Consequently, most wild form alleles attributed to ISR42-8 natural habitat, like flowering time delay, negatively affect individual reproduction and fitness. Although late flowering was hypothesized to be unbeneficial in the cropping environment (sown in spring), we observed an increase of wild form haplotype allele frequency at the HvPpdH2 locus. As a QTL for drought tolerance was identified in the same genomic window, the positive selection of the wild form allele of HvPpdH2 might only be a coincidence based on linkage. Nevertheless, it was observed before that winter wheat produces significantly more root biomass than spring types (Thorup-Kristensen et al. 2009). We hypothesize from this aspect that the wild form allele of HvPpdH2 might cause increased root development, which might ultimately result in higher drought tolerance.
Contrasting to the wild form, the second parent Golf, an elite breeding variety, was developed for such environmental conditions.
With this genetic background in mind, we see an initial adaptation phase towards the new environment. Consequently, allelic variations between the organic and conventional agro-ecosystems are not distinguishable until the 12th generation. After these initial adaptations reached an equilibrium state, a cumulative and linear differentiation of the organically from the conventionally adapted populations was detected across the generations. While the conventionally farmed population seemed to have reached a state of equilibrium in the generations following the 12th, the organic population showed higher within-population diversity and genetic variation across generations (Fig. 2A). We can only speculate on the implications this might have—for instance, that the increased heterogeneity is a state of equilibrium itself and is beneficial in buffering varying environmental factors. Besides, alleles derived from the wild form were observed at higher rates in the organic population. Regardless, we have to point out that the experimental site was shifted from one location (Dikopshof) to another (Klein-Altendorf) after the 12th generation. This could have some implications on the composition of the populations, too. Especially the organic population and its enhanced interaction with the soil microbiome might be affected, which could also result in a changed allele frequency pattern (Hawkes et al. 2020; Sandrini et al. 2022). Considering the transition phase of 3 years and almost identical soils, we suspect this effect to be minor.
4.3 Detection of sharp regional linkage disequilibrium changes caused by crossing-overs and adaptational selection
Across the genome, sharp transitions in allele frequencies between regions were noted. These changes were not coincidental, as such changes also became more and more distinct across generations in this self-pollinating crop (Figs. 3 and 6). Allele frequency changes depend on genetic drift, mutation, migration, and/or selection (Wright 1990). While genetic drift was estimated as a minor effect in the present study (Supplementary Figure 4, Supplementary Table 2), mutations were, due to their rarity and the few generations in which they might become significant, negligible, and migration was avoided by the experimental design. These often-small segments of sharp transitions may have resulted from breeding behavior (backcrossing followed by self-pollinating) combined with evolutionary adaptation (selection). Backcrossing and selfing will reduce the length of heterozygous chromosome segments, which are the only source of recombination between nearby genes (Hanson 1959). In this self-pollinating crop, the following evolutionary adaptation will address the remaining segment rather than the responding allele itself. This will result in the observed haplotype allele frequency transitions and selection of entire genome regions (Fig. 6). The unequal number of observed crossing-overs/fitness combinations in the 23rd generation between agro-ecosystems can hint that higher diversity is promoted in the organic environment.
4.4 Dissection of candidate regions and the wild form haplotype allele frequency patterns
For the given population size and backcrossing level, the expected wild form haplotype allele frequency in the BC2F3 was 10.5% (Cox 1984). However, only a minor deviation of 0.43% was detected. This underlines the power of the conducted crossing scheme as well as the pooling and sequencing strategy. This deviation was most likely associated with the reduced allele frequency in the peri-centromeres of chromosomes 2H and 5H (Fig. 3). An incomplete representation of the ISR42-8 alleles in the population on 5H was observed earlier (Schmalenbach et al. 2011). Interestingly, the wild form haplotype allele frequency of this region on chromosome 5H exceeded > 0.3 in both agro-ecosystems after less than ten generations. Considering that a yield-related QTL (Watt et al. 2019) and the seed dormancy gene SD1 (Pourkheirandish and Komatsuda 2007) are located in this region, this leads to the assumption that the linkage between these loci had a substantial impact on the genetic composition (Fig. 4). Once a linkage is broken, evolutionary selection processes are speedy, and even rare haplotypes can be up-selected to the major haplotypes within a few generations—if the fitness effect of this allele (or linkage block) is high. Similar patterns were observed for the brittleness genes brt1 and brt2 (Komatsuda et al. 2004), responsible for a vital domestication trait (von Bothmer et al. 2003) (Fig. 5). The wild form donor carried both unfavorable alleles of ear brittleness (brt1 and brt2), which could explain the adverse evolutionary adaptation in both environments. However, the selection of Brt1 and Brt2 alleles may have occurred due to an artificial influence because predominantly genotypes with stable ears lose their kernels less easily, which is why their grains are harvested more often by a combine harvester.
Although the populations endured varying weather conditions throughout the experimental period, the climatic background of a generally humid, maritime, and mild climate with a seasonal photoperiod remained similar for both agro-ecosystems (Suppl. Figure 1, max day length 16.5 h). While most wild form alleles were negatively selected in this period, the genomic region surrounding the Denso locus showed a high increase in the wild form haplotype allele frequency (Fig. 5). The semi-dwarf growth habit of ISR 42-8, likely associated with this locus, was beneficial in terms of genetic fitness, presumably because the dwarfed growth type of ISR42-8 might contribute in resistance to lodging and nutrient use efficiency.
Besides these examples presented, many more regions have shown distinct and linear adaptation patterns. In all these regions, single or multiple genes might be under selection, which makes the presented data an alternative approach to identify candidate genes (compared to GWAS or QTL mapping). Similar approaches based on the allele frequency exist for the Fst calculation (Berner 2019) and the identification of common variants (Park et al. 2011).
4.5 Allele frequency patterns for phenotype approximations
Furthermore, we were interested in special adaptations towards biotic and abiotic stresses. The populations endured varying weather conditions, imposing water and heat stress and biotic attack (Fig. 1C). A positive selection of resistance alleles towards biotic stressors was observed. Still, contrasting to our hypothesis and common sense, those resistance genes present in the gene pool of this population towards biotic stress showed no statistical difference between organic and conventional environments (po/c = 0.8, F23) (Fig. 7). Additionally, the allele frequency equilibrium of resistance genes was below 0.3 for all inspected candidate genes, indicating a non-persistent adaptation to biotic stress. This finding is in line with a report by Allard (1988), highlighting that the resistance alleles present in the populations only had an additive but no significant singular fitness effect.
The most prevalent variation between the agro-ecosystems was detected in root-associated genes. Due to expected nutrient-deficient growing conditions, a different root system may have been a critical trait in the organic compared to the conventional environment. While other trait-associated allele frequencies have risen above 0.5 for the wild form allele, most root-related alleles were characterized by intermediate fitness (Supplementary Table 5, 6), suggesting high variability in the root morphology within the organic population. A comparative experiment of root phenotypes between the 24th generation of the organic and conventional population supported our genotype-based observations and identified longer roots and increased root phenotypic variability in the organic population (Siddiqui et al. 2022). Similarly, Bhaskar et al. (2019) reported winter wheat composite cross populations having developed fewer seminal roots, thicker roots, and a heavier root system after 11 generations of adaptation towards organic farming. In a comparison between modern varieties, landraces and H. spontaneum, the latter was found to have less seminal roots than both modern varieties and landraces and thinner seminal roots than modern varieties (Grando and Ceccarelli 1995). Lower and thinner seminal roots are expected to increase root hydraulic resistance, which is of advantage in crops growing on storage water (Passioura 1972). These observations made in previous studies support our genomic-based assumptions.
Additionally, the allele frequency of the agronomically relevant candidate loci was clustered in the clusters’ yield components and physiology, nutrient uptake and allocation, and drought tolerance. However, the nutrient and yield component cluster did not show significant variations. Nevertheless, it has been shown by Raggi et al. (2022) that diverse populations established from high-yielding varieties can also quickly gain specific adaptations towards nutrient limited environments. Reasons for these observations could be (I) the allelic diversity of the two parents was not wide enough; (II) the period was too short for differentiation; (III) too few loci were grouped in these classes; (IV) epigenetic changes, which have not been reviewed in this study, was well as (V) the holobiont of plant-soil microbe interactions. Nevertheless, as described before, the ear length alleles, introduced into the population by the wild form, were positively selected across the generations and highlighted a favorable yield allele.
Concluding on the candidate QTLs, matched with observed allele frequency variation patterns, should serve as a potential explanation for which agronomic effects could have caused all those changes. Nevertheless, as already mentioned, we could not match all loci under selection with a corresponding phenotype. Therefore, the presented variations and similarities between the agro-ecosystems might not persist when more candidate genes are included in this approximation.
5 Conclusion
The observations made in this study can be concluded as (I) an adaptation to the habitat (climate zone) takes several generations, and (II) a follow-up period of multiple generations was required to adjust the allele frequency towards the ecological niche, defined by the agro-ecosystem. Furthermore, (III) the allele (and with that likely also the genotype) diversity is higher in the organically compared to the conventionally adapted populations, which might also result in (IV) wild form alleles being more likely to be positively selected in the organic environment. Lastly, in the context of climate change and agronomical transformations, (V) wild forms contributed beneficial alleles to the population and, therefore, might be a valuable source for modern plant breeding. Based on an innovative pool genotyping approach, linked with a long-term natural selection trial, this work highlights for the first time the potential of wild alleles to close the yield gap between organic and conventional agro-systems. We encourage organic breeders to focus on traits related to root morphology, developmental processes, and abiotic stress responses selected within an organic agro-ecosystem.
Data availability
Raw variant calling data and processed data can be found under https://doi.org/10.5281/zenodo.4304046https://doi.org/10.5281/zenodo.4304046.
Code availability
Code used to perform the analysis can be found at https://github.com/mischn-dev/haplotype allele frequencycall.
References
Afsharyan NP, Sannemann W, Léon J, Ballvora A (2020) Effect of epistasis and environment on flowering time in barley reveals a novel flowering-delaying QTL allele. J Exp Bot 71:893–906. https://doi.org/10.1093/jxb/erz477
Allard RW (1988) Genetic changes associated with the evolution of adaptedness in cultivated plants and their wild progenitors. J Hered 79:225–238. https://doi.org/10.1093/oxfordjournals.jhered.a110503
Allen R, Pereira L, Raes D, Smith M (1998) Evapotranspiration guidelines for computing crop water requirements: guidelines for computing crop water requirements (FAO Irrigation and Drainage Paper No.56). FAO M56
Alqudah AM, Koppolu R, Wolde GM et al (2016) The genetic architecture of barley plant stature. Front Genet 7:117. https://doi.org/10.3389/fgene.2016.00117
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Andrews S (2010) FastQC - A quality control tool for high throughput sequence data. Babraham Bioinforma. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 6 May 2019
Anonymous IPK Galaxy Blast Suite (n.d.). https://webblast.ipk-gatersleben.de/barley_ibsc/downloads/. Accessed 2 Apr 2020
Bedawy IMA, Dehne HW, Léon J, Naz AA (2018) Mining the global diversity of barley for Fusarium resistance using leaf and spike inoculations. Euphytica 214:18. https://doi.org/10.1007/s10681-017-2103-1
Beier S, Himmelbach A, Colmsee C et al (2017) Construction of a map-based reference genome sequence for barley, Hordeum vulgare L. Sci Data 4:1–24. https://doi.org/10.1038/sdata.2017.44
Belkadi A, Bolze A, Itan Y et al (2015) Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A 112:5473–5478. https://doi.org/10.1073/pnas.1418631112
Berner D (2019) Allele frequency difference AFD-an intuitive alternative to FST for quantifying genetic population differentiation. Genes 10(4):308. https://doi.org/10.3390/genes10040308
Bezanson J, Edelman A, Karpinski S, Shah VB (2017) Julia: a fresh approach to numerical computing. SIAM Rev 59:65–98. https://doi.org/10.1137/141000671
Bhaskar V, Weedon OD, Finckh MR (2019) Exploring the differences between organic and conventional breeding in early vigour traits of winter wheat. Eur J Agron 105:86–95. https://doi.org/10.1016/j.eja.2019.01.008
Blighe K, Lun A (2019) PCAtools: PCAtools: everything principal components analysis. https://github.com/kevinblighe/PCAtools. Accessed 28 Mar 2019
Ceccarelli S (1994) Specific adaptation and breeding for marginal conditions. Euphytica 77:205–219. https://doi.org/10.1007/BF02262633
Cochran WG (1977) Sampling Techniques, 3rd edn. John Wiley and Sons
Cox TS (1984) Expectations of means and genetic variances in backcross populations. Theor Appl Genet 68:35–41. https://doi.org/10.1007/BF00252308
Cunningham F, Achuthan P, Akanni W et al (2019) Ensembl 2019. Nucleic Acids Res 47:D745–D751. https://doi.org/10.1093/nar/gky1113
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Dragan P, Kopahnke D, Steffenson BJ et al (2013) Genetic fine mapping of a novel leaf rust resistance gene and a barley yellow dwarf virus tolerance (BYDV) Introgressed from Hordeum bulbosum by the Use of the 9K iSelect Chip. Adv Barley Sci 11:269–284. https://doi.org/10.1007/978-94-007-4682-4_23
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Gordon A, Hannon G (2010) Fastx-Toolkit. http://hannonlab.cshl.edu/fastx_toolkit/. Accessed 24 Jun 2020
Grando S, Ceccarelli S (1995) Seminal root morphology and coleoptile length in wild (Hordeum vulgare ssp. spontaneum) and cultivated (Hordeum vulgare ssp. vulgare) barley. Euphytica 86:73–80. https://doi.org/10.1007/BF00035941
Guan JC, Koch KE, Suzuki M et al (2012) Diverse roles of strigolactone signaling in maize architecture and the uncoupling of a branching-specific subnetwork. Plant Physiol 160:1303–1317. https://doi.org/10.1104/pp.112.204503
Hanson WD (1959) Early generation analysis of lengths of heterozygous chromosome segments around a locus held heterozygous with backcrossing or selfing. Genetics 44:833–837. https://doi.org/10.1093/genetics/44.5.833
Hawkes CV, Bull JJ, Lau JA (2020) Symbiosis and stress: how plant microbiomes affect host evolution. Phil Trans R Soc B 375:20190590. https://doi.org/10.1098/RSTB.2019.0590
Hedden P (2003) The genes of the Green Revolution. Trends Genet 19:5–9. https://doi.org/10.1016/S0168-9525(02)00009-4
Honsdorf N, March TJ, Berger B, Tester M, Pillen K (2014) High-throughput phenotyping to detect drought tolerance QTL in Wild Barley IntrogressionLines. PLoS ONE 9(5):e97047. https://doi.org/10.1371/journal.pone.0097047
Honsdorf N, March TJ, Pillen K (2017) QTL controlling grain filling under terminal drought stress in a set of wild barley introgression lines. PLoS ONE 12(10):e0185983. https://doi.org/10.1371/journal.pone.0185983
Itoh H, Tatsumi T, Sakamoto T et al (2004) A rice semi-dwarf gene, Tan-Ginbozu (D35), encodes the gibberellin biosynthesis enzyme, ent-kaurene oxidase. Plant Mol Biol 54:533–547. https://doi.org/10.1023/B:PLAN.0000038261.21060.47
Kassambara A (2020) ‘ggpubr’: “ggplot2” Based Publication Ready Plots. R Packag. version 0.2.5. https://cran.r-project.org/package=ggpubr. Accessed 1 May 2020
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Khush GS (2001) Green Revolution: the way forward. Nat. Rev. Genet. 2:815–822. https://doi.org/10.1038/35093585
Komatsuda T, Maxim P, Senthil N, Mano Y (2004) High-density AFLP map of nonbrittle rachis 1 (btr1) and 2 (btr2) genes in barley (Hordeum vulgare L.). Theor Appl Genet 109:986–995. https://doi.org/10.1007/s00122-004-1710-0
Le Campion A, Oury FX, Heumez E, Rolland B (2020) Conventional versus organic farming systems: dissecting comparisons to improve cereal organic breeding strategies. Org Agric 10:63–74. https://doi.org/10.1007/s13165-019-00249-3
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/MAP format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 00:1–3. ArXiv ID 1303.3997v2. https://doi.org/10.48550/arXiv.1303.3997
Liller CB, Walla A, Boer MP et al (2017) Fine mapping of a major QTL for awn length in barley using a multiparent mapping population. Theor Appl Genet 130:269–281. https://doi.org/10.1007/s00122-016-2807-y
Lüpken T, Stein N, Perovic D et al (2014) High-resolution mapping of the barley Ryd3 locus controlling tolerance to BYDV. Mol Breed 33:477–488. https://doi.org/10.1007/s11032-013-9966-1
Malysheva-Otto LV, Ganal MW, Röder MS (2006) Analysis of molecular diversity, population structure and linkage disequilibrium in a worldwide survey of cultivated barley germplasm (Hordeum vulgare L.). BMC Genet 7:6. https://doi.org/10.1186/1471-2156-7-6
Marzec M, Alqudah AM (2018) Key hormonal components regulate agronomically important traits in barley. Int J Mol Sci 19:1–12. https://doi.org/10.3390/ijms19030795
Mascher M, Gundlach H, Himmelbach A et al (2017) A chromosome conformation capture ordered sequence of the barley genome. Nature 544:427–433. https://doi.org/10.1038/nature22043
Meyer WS, Green GC (1980) Water use by wheat and plant indicators of available soil water 1. Agron J 72:253–257. https://doi.org/10.2134/agronj1980.00021962007200020002x
Minasny B, McBratney AB (2018) Limited effect of organic matter on soil available water capacity. Eur J Soil Sci 69:39–47. https://doi.org/10.1111/ejss.12475
Morrell PL, Clegg MT (2007) Genetic evidence for a second domestication of barley (Hordeum vulgare) east of the Fertile Crescent. Proc Natl Acad Sci U S A 104:3289–3294. https://doi.org/10.1073/pnas.0611377104
Mulki MA, Bi X, von Korff M (2018) Flowering locus T3 controls spikelet initiation but not floral development. Plant Physiol 178:1170–1186. https://doi.org/10.1104/pp.18.00236
Muñoz-Amatriaín M, Eichten SR, Wicker T et al (2013) Distribution, functional impact, and origin mechanisms of copy number variation in the barley genome. Genome Biol 14:1–17. https://doi.org/10.1186/gb-2013-14-6-r58
Murphy KM, Campbell KG, Lyon SR, Jones SS (2007) Evidence of varietal adaptation to organic farming systems. F Crop Res 102:172–177. https://doi.org/10.1016/j.fcr.2007.03.011
Muzammil S, Shrestha A, Dadshani S et al (2018) An ancestral allele of pyrroline-5-carboxylate synthase1 promotes proline accumulation and drought adaptation in cultivated barley. Plant Physiol 178:771–782. https://doi.org/10.1104/pp.18.00169
Naz AA, Arifuzzaman M, Muzammil S et al (2014) Wild barley introgression lines revealed novel QTL alleles for root and related shoot traits in the cultivated barley (Hordeum vulgare L.). BMC Genet 15:1–12. https://doi.org/10.1186/s12863-014-0107-6
Nevo E (1998) Genetic diversity in wild cereals: regional and local studies and their bearing on conservation ex situ and in situ. Genet Resour Crop Evol 45:355–370. https://doi.org/10.1023/A:1008689304103
Nielsen R, Paul JS, Albrechtsen A, Song YS (2011) Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet 12:443–451. https://doi.org/10.1038/nrg2986
Olson ND, Lund SP, Colman RE, Foster JT, Sahl JW, Schupp JM, Keim P, Morrow JB, Salit ML, Zook JM (2015) Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front Genet 6:235. https://doi.org/10.3389/fgene.2015.00235
Oyiga BC, Palczak J, Wojciechowski T et al (2020) Genetic components of root architecture and anatomy adjustments to water-deficit stress in spring barley. Plant Cell Environ 43:692–711. https://doi.org/10.1111/pce.13683
Park JH, Gail MH, Weinberg CR et al (2011) Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc Natl Acad Sci U S A 108:18026–18031. https://doi.org/10.1073/pnas.1114759108
Passioura JB (1972) The effect of root geometry on the yield of wheat growing on stored water. Aust J Agric Res 23:745–752. https://doi.org/10.1071/AR9720745
Pourkheirandish M, Komatsuda T (2007) The importance of barley genetics and domestication in a global perspective. Ann Bot 100:999–1008. https://doi.org/10.1093/aob/mcm139
Pozníková G, Fischer M, Pohanková E, Trnka M (2014) Analyses of spring barley evapotranspiration rates based on gradient measurements and dual crop coefficient model. Acta Univ Agric Silvic Mendelianae Brun 62:1079–1086. https://doi.org/10.11118/actaun201462051079
R Core Team (2020) The R Project for Statistical Computing. https://www.r-project.org/. Accessed 22 May 2021
Raggi L, Ceccarelli S, Negri V (2022) Genomics of a barley population evolved on-farm under different environmental conditions. Agroecol Sustain Food Syst 46:1330–1359. https://doi.org/10.1080/21683565.2022.2106011
Randive K, Raut T, Jawadand S (2021) An overview of the global fertilizer trends and India’s position in 2020. Miner Econ 34:371–384. https://doi.org/10.1007/s13563-020-00246-z
Ray DK, Ramankutty N, Mueller ND et al (2012) Recent patterns of crop yield growth and stagnation. Nat Commun 3:1–7. https://doi.org/10.1038/ncomms2296
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8:66428. https://doi.org/10.1371/journal.pone.0066428
Reganold JP, Wachter JM (2016) Organic agriculture in the twenty-first century. Nat Plants. https://doi.org/10.1038/nplants.2015.221
Reinert S, Kortz A, Léon J, Naz AA (2016) Genome-wide association mapping in the global diversity set reveals new QTL controlling root system and related shoot variation in Barley. Front Plant Sci 7:1061. https://doi.org/10.3389/fpls.2016.01061
Sandrini M, Moffa L, Velasco R, Balestrini R, Chitarra W, Nerva L (2022) Microbe-assisted crop improvement: a sustainable weapon to restore holobiont functionality and resilience. Hortic Res 9:uhac160. https://doi.org/10.1093/HR/UHAC160
Sato K, Yamane M, Yamaji N et al (2016) Alanine aminotransferase controls seed dormancy in barley. Nat Commun 7:1–9. https://doi.org/10.1038/ncomms11625
Sayed MA, Schumann H, Pillen K et al (2012) AB-QTL analysis reveals new alleles associated to proline accumulation and leaf wilting under drought stress conditions in barley (Hordeum vulgare L.). BMC Genet 13:1–12. https://doi.org/10.1186/1471-2156-13-61
Schmalenbach I, Körber N, Pillen K (2008) Selecting a set of wild barley introgression lines and verification of QTL effects for resistance to powdery mildew and leaf rust. Theor Appl Genet 117:1093–1106. https://doi.org/10.1007/s00122-008-0847-7
Schmalenbach I, March TJ, Pillen K et al (2011) High-resolution genotyping of wild barley introgression lines and fine-mapping of the threshability locus thresh-1 using the illumina goldengate assay. G3 Genes. Genomes, Genet 1:187–196. https://doi.org/10.1534/g3.111.000182
Schneider M, Shrestha A, Ballvora A, Léon J (2022) High-throughput estimation of allele frequencies using combined pooled-population sequencing and haplotype-based data processing. Plant Methods 18:1–18. https://doi.org/10.1186/S13007-022-00852-8
Siddiqui MN, Schneider M, Barbosa MB, Léon J, Ballvora A (2022) Natural selection under conventional and organic cropping systems affect root architecture in spring Barley. Sci Rep 12:20095. https://doi.org/10.1038/s41598-022-23298-3
Sharma A (2019) R/sim.drift.R. https://rdrr.io/github/rknx/drift/src/R/sim.drift.R. Accessed 30 Nov 2020
Simmonds NW (1991) Selection for local adaptation in a plant breeding programme. Theor Appl Genet 82:363–367. https://doi.org/10.1007/BF02190624
Suzuki R, Terada Y, Shimodaira H (2019) pvclust: Hierarchical clustering with p-values via multiscale bootstrap resampling. https://cran.r-project.org/package=pvclust. Accessed 8 May 2019
Takahashi R, Bashir K, Ishimaru Y, Nishizawa NK, Nakanishi H (2012) The role of heavy-metal ATPases, HMAs, in zinc and cadmium transport in rice. Plant Signal Behav 7(12):1605–1607. https://doi.org/10.4161/psb.22454
Tarasov A, Vilella AJ, Cuppen E et al (2015) Sambamba: fast processing of NGS alignment formats. Bioinformatics 31:2032–2034. https://doi.org/10.1093/bioinformatics/btv098
Thorup-Kristensen K, Cortasa MS, Loges R (2009) Winter wheat roots grow twice as deep as spring wheat roots, is this important for N uptake and N leaching losses? Plant Soil 322:101–114. https://doi.org/10.1007/s11104-009-9898-z
Tilk S, Bergland A, Goodman A et al (2019) Accurate allele frequencies from ultra-low coverage pool-seq samples in evolve-and-resequence experiments. G3 Genes. Genomes, Genet 9:4159–4168. https://doi.org/10.1534/g3.119.400755
Tilman D (1998) The greening of the green revolution. Nature 396:211–212. https://doi.org/10.1038/24254
von Bothmer R, van Hintum T, Knüpffer H, Sato K (2003) Diversity in Barley (Hordeum vulgare) 1st edn, vol 7, Elsevier
Von Korff M, Wang H, Léon J, Pillen K (2005) AB-QTL analysis in spring barley. I. Detection of resistance genes against powdery mildew, leaf rust and scald introgressed from wild barley. Theor Appl Genet 111:583–590. https://doi.org/10.1007/s00122-005-2049-x
Wang G, Schmalenbach I, von Korff M et al (2010) Association of barley photoperiod and vernalization genes with QTLs for flowering time and agronomic traits in a BC2DH population and a set of wild barley introgression lines. Theor Appl Genet 120:1559–1574. https://doi.org/10.1007/s00122-010-1276-y
Ward B (2020) Bio.Seq Julia Package. https://github.com/BioJulia/Bio.jl/. Accessed 30 Jul 2020
Watt C, Zhou G, McFawn LA et al (2019) Fine mapping of qGL5H, a major grain length locus in barley (Hordeum vulgare L.). Theor Appl Genet 132:883–893. https://doi.org/10.1007/s00122-018-3243-y
Wiberg RAW, Gaggiotti OE, Morrissey MB, Ritchie MG (2017) Identifying consistent allele frequency differences in studies of stratified populations. Methods Ecol Evol 8:1899–1909. https://doi.org/10.1111/2041-210X.12810
Willer H, Yussefi-Menzler M, Sorensen N (2008) The world of organic agriculture: Statistics and emerging trends 2008. FiBL, IFOAM - Organics International. https://orgprints.org/id/eprint/13123/
Wright S (1990) Evolution in Mendelian populations. Bull Math Biol 52:241–295. https://doi.org/10.1007/BF02459575
Xu Y, Jia Q, Zhou G et al (2017) Characterization of the sdw1 semi-dwarf gene in barley. BMC Plant Biol 17:1–10. https://doi.org/10.1186/s12870-016-0964-4
Yan L, Loukoianov A, Blechl A, Tranquilli G, Ramakrishna W, SanMiguel P, Bennetzen JL, Echenique V, Dubcovsky J (2004) The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303(5664):1640–1644. https://doi.org/10.1126/science.1094305
Yao Z, You FM, N’Diaye A et al (2020) Evaluation of variant calling tools for large plant genome re-sequencing. BMC Bioinformatics 21:360. https://doi.org/10.1186/s12859-020-03704-1
Zahn S, Koblenz B, Christen O et al (2020) Evaluation of wild barley introgression lines for agronomic traits related to nitrogen fertilization. Euphytica 216:1–14. https://doi.org/10.1007/s10681-020-2571-6
Zeileis A, Kleiber C, Jackman S (2008) Political Science Computational Laboratory, Stanford University: Package ‘ pscl.’ J Stat Softw 27:1–25. https://doi.org/10.18637/jss.v027.i08
Zohary D, Hopf M, Weiss E (2012) Domestication of plants in the old world: the origin and spread of domesticated plants in Southwest Asia. Europe, and the Mediterranean Basin. https://doi.org/10.1093/acprof:osobl/9780199549061.001.0001
Zotarelli L, Dukes MD, Romero CC, Migliaccio KW, Kelly T, Morgan KT (2010) Step by step calculation of the Penman-Monteith Evapotranspiration (FAO-56 Method). IFAS Extension, Univ. Florida. https://www.agraria.unirc.it/documentazione/materiale_didattico/1462_2016_412_24509.pdf. Accessed 3 Feb 2020
Acknowledgements
We are very grateful to the late Prof. G. Fischbeck for providing Hordeum vulgare spontaneum L. accession ISR 42-8 as well as D. Böltes and B. Defant for the crossing work. We owe special thanks to K. Müller and W. Bungert for taking care of the fieldwork over all these years and handling the populations. We thank L. Hördemann and A. Shrestha for helping to collect sample tissues and DNA extraction.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was partly funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under grants of LE813 and under Germany’s Excellence Strategy, EXC-2070 - 390732324 (PhenoRob).
Author information
Authors and Affiliations
Contributions
Conceptualization, J.L. and A.B.; methodology, J.L. and A.B.; formal analysis, M.S.; software M.S.; visualization, M.S.; supervision, J.L. and A.B.; writing—original draft, M.S.; writing—review and editing, M.S, A.B., and J.L.; funding acquisition, J.L.; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Schneider, M., Ballvora, A. & Léon, J. Deep genotyping reveals specific adaptation footprints of conventional and organic farming in barley populations—an evolutionary plant breeding approach. Agron. Sustain. Dev. 44, 33 (2024). https://doi.org/10.1007/s13593-024-00962-8
Accepted:
Published:
DOI: https://doi.org/10.1007/s13593-024-00962-8