
Abstract
Major histocompatibility complex (MHC) genes have been widely studied to assess the immunological fitness and evolutionary adaptation of animal populations. Among the Canidae, the raccoon dog’s adventurous nature, omnivorous behavior, and high variability of intracellular pathogens make it ideal to study selection on MHC class I in a non-model canid species. Here, we examined allelic diversity and evolutionary patterns of MHC class I genes in the raccoon dog (Nyctereutes procyonoides). We identified 48 novel MHC class I alleles from 31 raccoon dogs from Japan and Russia. Some alleles were geographically restricted, whereas others were widely distributed across the species’ range. The rate of non-synonymous substitutions was greater than that of synonymous substitutions for both exon 2 and exon 3 encoding α1 and α2 domains, respectively, in the α chain of the MHC class I protein. Positively selected sites at the amino acid level were evident in both the α1 and α2 domains, and a recombination breakpoint was found in exon 3. Bayesian phylogenetic trees showed no evidence of trans-species polymorphism (TSP) with alleles from carnivoran species in other families but did detect TSP between raccoon dogs and the domestic dog, Canis familiaris, indicative of long-term balancing selection in canids. Our results indicate that the extensive allelic diversity of MHC class I in Japanese and Russian raccoon dogs has been influenced and maintained by pathogen-driven positive selection, recombination, and long-term balancing selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The major histocompatibility complex (MHC) has been widely studied because of its importance in evolutionary ecology as well as its role in the immune response to pathogens (Hedrick 1994; Sommer 2005; Piertney and Oliver 2006). The MHC is subdivided into three major subfamilies, classes I, II, and III. MHC class II proteins are expressed in antigen-presenting cells, which bind peptides from extracellular pathogens and present them to helper T-cells (CD4+) (Hughes and Yeager 1998). MHC class I proteins are expressed in all nucleated cells and are essential in the defense against intracellular pathogens such as viruses; they function by binding and presenting degraded pathogen peptides to cytotoxic T-cells (CD8+) (Sommer 2005). The class I protein consists of a single amino acid chain (α chain) encoded by seven exons. Among these exons, the most polymorphic are exons 2 and 3, which encode amino acids involved in forming peptide-binding grooves in the extracellular α1 and α2 domains, respectively (Bjorkman et al. 1987). The genetic diversity among MHC genes is influenced by various evolutionary factors, including variation in copy number (Piertney and Oliver 2006), balancing selection (Sommer 2005), and birth-and-death evolution (Nei et al. 1997). Studies of MHC class I evolution in mammals, including the giant panda (Ailuropoda melanoleuca) (Zhu et al. 2013), four badger species (Meles meles, M. canescens, M. leucurus, and M. anakuma) (Sin et al. 2012; Abduriyim et al. 2019), and the brown bear (Ursus arctos) (Kuduk et al. 2012), indicate that their allelic diversity has been greatly influenced by recombination and positive selection. Furthermore, in the four Meles species, the α1 and α2 domains appear to have undergone different evolutionary histories (Sin et al. 2012; Abduriyim et al. 2019). In the family Canidae, only the genus Canis has been well studied (Wagner et al. 2002; Ross et al. 2012; Liu et al. 2017; Miyamae et al. 2017; Venkataraman et al. 2017), and studies of MHC class I evolution are lacking in other non-model genera. At present, there have been four complete canine MHC, dog leukocyte antigen (DLA), and class I genes that have been cloned and sequenced, which are DLA-12, DLA-79, DLA-88, and DLA-64 (Burnett et al. 1997), and among them, DLA-88 is the most polymorphic (Graumann et al. 1998). The raccoon dog, Nyctereutes procyonoides, is a native species in East Asia, spanning from Northern Vietnam, through Eastern China, to Far Eastern Russia, Mongolia, and Japan. In the 1950s, this canid species was introduced to the European territories for fur production, and since then, it has become widespread in those regions (Kauhala and Saeki 2016). Currently, Japanese raccoon dogs are classified into two subspecies, N. p. viverrinus and N. p. albus. N. p. viverrinus is widely distributed in Honshu, Shikoku, Kyushu, and many small islands in Japan. On the other hand, N. p. albus can only be found on Hokkaido island. Recently, the population in Hokkaido is speculated to be a different species from the continental populations based on morphological and genetic data (Ward et al. 1987; Hong et. al 2018; Kim et al. 2013, 2015).
Raccoon dogs carry infectious diseases, such as Asian tick-borne meningoencephalitis, canine distemper, paratyphoid fever, anthrax, tuberculosis, and rabies. This species is among the most invasive mammals in European countries, where it is the main vector for rabies, alveolar echinococcosis, and sarcoptic mange, which are dangerous to human health (Kauhala and Kowalczyk 2011). In Japan, raccoon dogs infected with canine distemper virus have been reported from Wakayama Prefecture (Suzuki et al. 2015). The raccoon dog’s omnivorous mode of feeding and high prevalence of intracellular pathogens make it ideal for studying selection on MHC class I in a non-model canid species. The aim of the present study was to characterize the allelic diversity of the MHC class I α1 and α2 domains and their patterns of evolution in raccoon dogs from Japan and Russia.
Materials and methods
Samples and DNA extraction
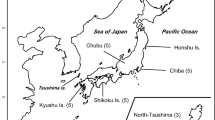
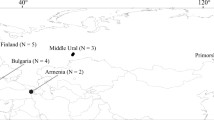
In total, 31 blood or tissue samples from raccoon dogs were used (Fig. 1: Table 1; Supplementary Table 1), 23 from the Japanese Islands, four from Far Eastern Russia, and four from Western Russia. Some samples were common to those in the MHC class II study by Bartocillo et al. (2020). DNA from all samples was extracted by using a DNeasy Blood and Tissue Kit (Qiagen) and then stored at 4°C until use.

Sampling locations for native raccoon dogs examined in this study (except the introduced population of Western Russia). Numbers (N) in parentheses indicate sample sizes
Polymerase chain reaction
Exon 2 forward primer 5′-TCTCACCCGTCGGCTCCGCAG -3′ and exon 3 reverse primer 5′-AGGCGAGATCGGGGAGGC-3′ targeting the dog MHC class I DLA-88 alleles (Wagner et al. 2000) were used for polymerase chain reaction (PCR) amplification. Reactions were performed in 25-μL reaction volumes, each containing 0.625 U PrimeSTAR GXL (Takara Bio), 5 μL of 5× PrimeSTAR GXL buffer, 2 μL of 2.5mM dNTP, 0.3 μL (25 pmol/μL) of each phosphorylated forward and reverse primers, and 1 μL of DNA extract (60–100 ng of genomic DNA). Reaction conditions in a Takara Dice Touch Thermal Cycler were 98°C for 2 min, 30–35 cycles of 98°C for 10 s and 68°C for 45 s, and 68°C for 10 min, with a final hold at 4°C. PCR products were electrophoresed on a 2% agarose gel and visualized with ethidium bromide florescence under UV illumination and then purified by using a QIAquick PCR Purification Kit (Qiagen).
Cloning and sequencing
Cloning and sequencing were done as described by Bartocillo et al. (2020). On average, 27 positive clones per individual were sequenced by using M13 forward and reverse primers. Sequences were aligned and checked by using MUSCLE in MEGA 7 (Kumar et al. 2016). DnaSP v.5 (Librado and Rozas 2009) was used to determine identical sequences among all sequences obtained. Sequences were then identified as bona-fide variants based on criteria associated with the nomenclature rules for dog DLA (Kennedy et al. 2000). An accepted bona-fide sequence in this study was defined as having identical sequences derived from at least two raccoon dog individuals or from two separate PCR reactions, while unique single sequences were rejected as chimeras. BLAST-N searches (Altschul et al. 1990) of the NCBI GenBank database were conducted to identify identical or homologous sequences. Bona-fide true alleles were named according to the rules for MHC nomenclature in non-human species (Klein et al. 1990) in descending order of frequency of occurrence. Nucleotide sequences obtained in this study were deposited in the DNA Databank of Japan (DDBJ) under accession numbers LC554227-LC554274.
Data analysis
Putative functional Nypr-MHC1 alleles were analyzed by calculating Tajima’s D (Tajima 1989) to determine whether the DNA sequences evolved by directional or balancing selection. To determine whether the antigen-binding sites (ABSs), non-ABSs, and the overall amino acid sequences of the α1 and α2 domains were under positive selection, ratios between non-synonymous (dN) and synonymous (dS) substitution rates per nucleotide site were calculated by using the Nei-Gojobori method (Zhang et al. 1998) with the Jukes-Cantor correction (Jukes and Cantor 1969). ABS positions were inferred from the ABSs in human MHC proteins (Bjorkman et al. 1987). The mixed effect model of evolution (MEME) and genetic algorithm for recombination detection (GARD) in DATAMONKEY (Murrell et al. 2012) were used to identify amino acid sites that have undergone positive selection or represent recombination breakpoints, respectively. MrBayes v.3.2.6 (Ronquist and Huelsenbeck 2003) was used to obtain a Bayesian phylogenetic tree for allelic sequences of the raccoon dog MHC class I (Nypr-MHC1) with the orthologs from representative species in Felidae, Canidae, Mustelidae, Ursidae, and human. KAKUSAN 4 (Tanabe 2007) was used to select optimal models of nucleotide substitution. Phylogenetic analyses were conducted separately for four data partitions—the partial open reading frame (ORF; exons 2 and 3 concatenated), exon 2, exon 3, and intron 2. All phylogenetic trees were reconstructed from two Markov Chain Monte Carlo (MCMC) runs of 10 million generations for exon 3 and intron 2, 15 million generations for exon 2, and 20 million generations for the concatenated exon 2 and 3. All the chains were sampled every 1000 generations, with the first 25% samples discarded as burn-in.
Results
Allelic diversity of raccoon dog MHC class I genes
MHC class I fragments of 708–810 bp long were sequenced from 31 raccoon dog individuals from Japan and Russia, with an average of 27 plasmid clones sequenced per individual. Each sequence consisted of exon 2 (267 bp; 89 amino acids), exon 3 (273 bp; 92 amino acids), and the intervening intron 2 (168–270 bp). A total of 48 alleles were identified in this study, and all of them were novel (Table 1). One to six putative functional alleles were detected per individual, indicating that there are at least three MHC class I loci in the raccoon dog. In comparison with exon 2 (encoding α1 domain), exon 3 (α2 domain) was somewhat higher in nucleotide diversity, and α2 domain included more polymorphic amino acid sites as a result (Tables 2 and 3). Intervening intron 2 sequences (Supplementary Fig. S1) showed remarkably low allelic variation compared to their exon sequences. The distribution of the MHC class I alleles among the geographical populations (Table 1) shows that although no alleles were shared among all regions, Nypr-MHC1*01 and *02 were the most broadly distributed in six of the eight sampling locations, excluding Hokkaido and Far Eastern Russia. Nypr-MHC1*02 was the allele most frequently detected among the individuals analyzed. Nypr-MHC1*08 was shared by raccoon dogs from Hokkaido, Honshu, and Shikoku Islands, and Nypr-MHC1*20 was shared only by raccoon dogs from Northern, Central, and Southern Honshu. The native population in Far Eastern Russia shared three alleles (Nypr-MHC1*04, *06, and *09) with the introduced population in Western Russia. The degree of geographical restriction varied considerably, ranging from no alleles specific to a region (central Honshu) to 10 region-specific alleles (Hokkaido).
Selection mechanism of raccoon dog MHC class I genes
Selection pressures on functional domains in the raccoon dog MHC class I protein were analyzed to determine possible evolutionary differences between exon 2 and exon 3. High positive Tajima’s D values indicate balancing selection, while the values lower than zero indicate purifying selection. Tajima’s D values for both exons and the intervening intron 2 were significantly greater than zero (Table 3), indicating that balancing selection has influenced the allelic diversity in both exons and an intron. The ω (dN/dS) value was markedly greater than 1 for the ABS codons in each exon (Table 4), indicating that positive selection on codons encoding ABSs in both exons 2 and 3 has influenced allelic diversity. The MEME analysis provided evidence of positive selection at the amino acid level for both domains (asterisks in Fig. 2 (α1 domain) and Fig. 3 (α2 domain)), but the α2 domain displayed more sites (three) under positive selection than the α1 domain (one). Three out of the four sites under positive selection coincided with ABS codons inferred from the human HLA-A2 locus (Bjorkman et al. 1987). The GARD analysis detected one recombination breakpoint in exon 3, but none for exon 2. Our results indicate that in the α1 domain, positive selection was the main factor leading to allelic diversity in the raccoon dog, whereas in the α2 domain, both positive selection and recombination could have influenced the allelic diversity.

Alignment of amino acid sequences deduced from partial sequences of MHC class I (Nypr-MHC1) exon 2 from raccoon dogs. Dots indicate the identity with the Nypr-MHC1*01 sequence. Number at the top indicates amino acid positions in the α1 domain. Gray shading indicates antigen binding sites (ABSs) predicted from human MHC class I (Bjorkman et al. 1987). The asterisk indicates an amino acid site under positive selection, as calculated by a MEME (mixed effect model of evolution) analysis

Alignment of amino acid sequences deduced from partial sequences of MHC class I (Nypr-MHC1) exon 3 from raccoon dogs. A triangle indicates a recombination break point, determined by a GARD (genetic algorithm for recombination detection) analysis. For other information, see the caption of Fig. 2
Phylogenetic analysis of raccoon dog MHC class I genes
Analysis of the partial ORF consisting of exons 2 and 3 (Supplementary Fig. S2) showed only weak or no evidence of trans-species polymorphism (TSP). A separate analysis of exon 2 (Fig. 4) showed a more strongly supported pattern of TSP, in which two clades including both raccoon dog and domestic dog sequences emerged; one of these clades showed relatively high nodal support (posterior probability = 0.98). In other words, in the two clades, raccoon dog sequences were more closely related to a group of dog sequences than other raccoon dog sequences. The same situation was evident from exon 3 (Fig. 5), although the groupings of dog and raccoon dog sequences were different than for exon 2. However, phylogenetic analysis of concatenated exon 2 and 3 sequences (partial ORF) showed ambiguous evidence for TSP; one clade of dog DLA alleles formed the sister group to a monophyletic subclade containing all the Nypr-MHC1 alleles, with other dog DLA alleles outside these two subclades, although the node linking the two clades was weakly supported (posterior probability = 0.89). In addition, intron 2 sequences mostly formed clades by species in the phylogenetic analysis (Supplementary Fig. S3), with the exception that one group of raccoon dog sequences was more closely related to the dog sequences (DLA) than to other raccoon dog sequences. No specific patterns in the geographical distribution of alleles were detected in any phylogenetic trees.

Bayesian phylogenetic tree for exon 2 of MHC class I alleles from raccoon dogs (indicated by red characters); representative species of canids (blue), felids (purple), mustelids (green), ursids (brown), and humans (light blue). Numbers near nodes are posterior probability values. DDBJ/EMBL/GenBank accession numbers for nucleotide sequences are included after allele names for sequences retrieved from GenBank. Abbreviations within allele names indicate different species: Nypr, raccoon dog; DLA-88, domestic dog; FLA, domestic cat; Meme, European badger; Aime, giant panda; HLA, human

Bayesian phylogenetic tree for exon 3 of MHC class I alleles from raccoon dogs; representative species of canids, felids, mustelids, ursids, and humans. Alleles of different species are shown in different colors. For other information, see the caption of Fig. 4
Discussion
Allelic diversity of raccoon dog MHC class I genes
This is the first study to characterize MHC class I genes in the raccoon dog. Overall, our study identified 48 novel Nypr-MHC1 alleles, one to six alleles per individual (one to three loci). The level of allelic variation was higher than in most other mammalian species: 16 alleles from 26 individuals of the giant panda (Zhu et al. 2013); 111 alleles (30 alleles of Patr-A, 41 alleles of Patr-B, 29 alleles of Patr-C, and 11 alleles of Patr-AL) from 30 individuals of the chimpanzee (including 20 Pan troglodytes schweinfurthii, four P. t. verus, and six P. t. troglodytes) (Maibach et al. 2017); 17 transcribed MHCI alleles (six DLA-12, two DLA64, two DLA-79, and seven DLA-88) from the transcriptome of the four wolf, Canis lupus blood samples (Liu et al. 2017); 33 alleles from 12 individuals of the domestic cat, Felis catus (Holmes et al. 2013); 37 alleles (13 alleles of Papa-A, 13 alleles of Papa-B, and 11 alleles of Papa-C) from 21 individuals of the bonobo, Pan paniscus (Maibach et al. 2017); and 37 alleles from 234 individuals of the brown bear (Kuduk et al. 2012). The domestic dog is an apparent exception among canids, showing higher allelic variation, with 81 known alleles (73 DLA-88, one DLA-12, one DLA-64, and six DLA-79) (Wagner et al. 2002; Ross et al. 2012; Miyamae et al. 2017; Venkataraman et al. 2007, 2017). This might be the result of higher sampling effort in this well-studied model species. By the same token, our study was limited to the Japanese and Russian raccoon dog populations, and we might have detected additional alleles if raccoon dogs from other areas had been included and other primer sets were used.
Exposure over a long period of time to a wide variety of intracellular pathogens, such as the canine distemper virus (Suzuki et al. 2015), SARS (IASR 2005), rabies (Kurosawa et al. 2017), Trichinella sp. (Pozio 2000), and six bacterial and five viral pathogens (Sutor et al. 2014), may have been the driving force for the high variation in Nypr-MHC1 in raccoon dogs. The high allelic MHC class I variation in raccoon dogs, along with their high dispersal capability, adaptation to a wide range of climatic and environmental conditions (Caut et al. 2008), omnivorousness, and a high reproductive rate (Kauhala and Kowalczyk 2011) could have contributed to the raccoon dog’s successful expansion in Europe after being introduced there from their native range in East Asia.
Bartocillo et al. (2020) reported similar high variation of MHC class II DRB alleles in raccoon dogs, with both locally restricted and widespread alleles. Locally restricted alleles could have resulted from balancing selection with the presence of some native pathogens (Hughes and Yeager 1998). Interestingly, the MHC class I allelic diversity of raccoon dogs observed in this study showed that the highest number of locally restricted alleles (10 alleles) was found in Hokkaido. However, the seven most frequent alleles identified in this study were not found in this island. The geographical isolation of Hokkaido from other parts of Japan by the Tsugaru straits during the last glacial maximum (Ohshima 1991) could have prevented gene flow and resulted in increased genetic drift in Hokkaido, which would have promoted genetic divergence of MHC I between the raccoon dog populations in Hokkaido and the other islands of Japan. On the other hand, widespread pathogens might explain alleles shared among regions. Kurosawa et al. (2017) reported that rabies originated in Japan locally and then spread throughout the country. Not only the distribution but also the pattern of spread of infectious disease might have had an effect on the allelic variation of MHC I genes in raccoon dogs.
Evolution of raccoon dog MHC class I genes
We found genetic evidence of historical positive selection on both the α1 and α2 domains of MHC class I proteins in Japanese and Russian raccoon dogs, based on the ratio of the non-synonymous and synonymous substitution rates (Table 4). Our results were congruent with a study on wolf MHC class I genes (Liu et al. 2017), which showed strong positive selection. Similarly, Bartocillo et al. (2020) detected strong positive selection in MHC class II DRB exon 2 in raccoon dogs. Separate analysis of the two exons indicated stronger positive selection on exon 2 (encoding the α1 domain) than on exon 3 (encoding α2 domain) (Table 4). Specifically, for exon 2, only ABS codons showed evidence of strong positive selection, which was in agreement with MHC class I studies in badgers (Sin et al. 2012; Abduriyim et al. 2019). In contrast, for exon 3, not only ABS codons but also non-ABS codons (i.e., all codons) showed evidence of strong positive selection. In addition, there were fewer polymorphic amino acid sites in both domains in the raccoon dog (Table 2) compared to other carnivoran species reported by Sin et al. (2012) and Abduriyim et al. (2019). Nevertheless, even with a lower number of polymorphic amino acid sites and ABS-restricted polymorphic sites, we found high allelic variation in raccoon dogs. This indicates that positive selection could have contributed to maintaining the high allelic variation of MHC class I in raccoon dogs. Positive selection on ABSs is favorable to a population, as it gives a wider range of antigen peptides for recognition of a wider variety of pathogens and increases the chance of survival from pathogenic infections (Hughes and Nei 1992; Hughes and Yeager 1998; Jeffery and Bangham 2000).
TSP is a phylogenetic pattern that results from the retention of ancestral allelic lineages across divergence events, including speciation (Penn and Potts 1999), and a typical phenomenon showing the contribution of balancing selection on a gene (Klein et al. 1998). A phylogenetic analysis of MHC class II DRB (Bartocillo et al. 2020) found no pattern of TSP; instead, all raccoon dog sequences formed a monophyletic clade within the Canidae clade. Our analyses provided some evidence for TSP in MHC class I between the raccoon dog and domestic dog (Figs. 4 and 5), but not between the raccoon dog and representatives of other carnivoran families (Felidae, Mustelidae, Ursidae).
Indications of TSP for exon 2 (Fig. 4) or exon 3 (Fig. 5) (especially for exon 2) were congruent with the Tajima’s D values (D = 3.1924 for exon 2; D = 3.8264 for exon 3), which indicate that these sequences have evolved under balancing selection. However, no clear evidence of TSP was observed based on the partial ORF which consists of exons 2 and 3, even though the Tajima’s D value is sufficiently high, perhaps because the raccoon dogs diverged long time ago from other canids and that no ancestral alleles have been retained. In addition, data on MHC class I genes in canids are scarce, and studies on other species should be conducted to further understand the extent of TSP in canids.
Intron 2 sequences (Supplementary Fig. S1) in the raccoon dog showed remarkably low allelic variation compared to their exon sequences. The phylogenetic analysis of intron sequences was quite interesting (Supplementary Fig. S3), as it showed two distinct clades of raccoon dog sequences, one of which was highly supported (posterior probability = 1), and the other of which was the sister group to domestic dog intron sequences. The results were similar to the study of introns in HLA-A, HLA-B, and HLA-C by Cereb et al. (1996), whereby introns 1, 2, and 3 were relatively conserved compared with their neighboring exons 2 and 3. Introns of HLA-A, HLA-B, and HLA-C genes in humans have been reported to be conserved and locus-specific, i.e., a byproduct of interallelic recombination and subsequent genetic drift which then leads to the homogenization of introns over evolutionary time (Cereb et al. 1997). It has been established before that introns have no apparent purpose, but studies provide evidence that introns may play a vital role in the long-term survival of genes by preventing degenerative interlocus recombination (Kricker et al. 1992). Overall, the presence of two highly conserved clades of introns in the raccoon dog is still a mystery, and studies on introns of MHC classs I genes in the Canidae should be conducted.
Our results from positive selection and recombination analyses revealed one codon (amino acid site) in exon 2 (Fig. 2) and three codons in exon 3 (Fig. 3) that are under positive selection and a recombination breakpoint only in exon 3 (Fig. 3). Positive selection at the amino acid level could have contributed to maintaining the high allelic diversity of exon 2, whereas for exon 3, both positive selection and recombination could have contributed to allelic diversity. Overall, differences in the intensity of positive selection between the two exons, different phylogenetic relationships evident in their trees, and the presence of recombination in only exon 3 suggest that the exons 2 and 3 encoding α1 and α2 domains, respectively, of MHC class I genes in the raccoon dog have different evolutionary histories. This agrees with the conclusion of Sin et al. (2012) and Abduriyim et al. (2019) that separate evolutionary analyses are necessary for the two MHC class I domains.
In conclusion, our findings confirmed that historical positive selection and recombination were the main forces that have acted in the evolution of MHC class I genes in raccoon dogs. Taken together, this work sheds light on the molecular evolutionary adaptation of non-model species and free-ranging animal populations in mammals, especially in the Canidae. These findings add to the growing body of literature that may guide policy makers in assessing conservation actions among raccoon dogs in Japan and Russia and/or help in the policy making of introducing raccoon dogs to non-native geographical areas for fur production. We believe that this work could be a framework for more exploration in the evolutionary and population genetics and conservation actions for raccoon dogs.
References
Abduriyim S, Nishita Y, Kosintsev PA, Raichev E, Väinölä R, Kryukov AP, Abramov AV, Masuda R (2019) Evolution of MHC class I genes in Eurasian badgers, genus Meles (Carnivora, Mustelidae). Heredity 122:205–218. https://doi.org/10.1038/s41437-0818-0100-3
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S00222836(05)80360-2
Bartocillo AMF, Nishita Y, Abramov AV, Masuda R (2020) Molecular evolution of MHC class II DRB exon 2 in Japanese and Russian raccoon dogs, Nyctereutes procyonoides (Carnivora: Canidae). Biol J Linn Soc 129:61–73. https://doi.org/10.1093/biolinnean/blz153
Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC (1987) Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329:506–512. https://doi.org/10.1038/329506a0
Burnett RC, DeRose SA, Wagner JL, Storb R (1997) Molecular analysis of six dog leukocyte antigen class I sequences including three complete genes, two truncated genes, and one full-length processed gene. Tissue Antigens 49:484–495. https://doi.org/10.1111/j.1399-0039.1997.tb02783.x
Caut S, Angulo E, Courchamp F (2008) Dietary shift of an invasive predator: rats, seabirds and sea turtle. J Appl Ecol 45:428–437. https://doi.org/10.1111/j.13652664.2007.01438.x
Cereb N, Kong Y, Lee S, Maye P, Yang SY (1996) Nucleotide sequences of MHC class I introns 1, 2, and 3 in humans and intron 2 in nonhuman primates. Tissue Antigens 47:498–511. https://doi.org/10.1111/j.1399-0039.1996.tb02592.x
Cereb N, Hughes A, Yang SY (1997) Locus-specific conservation of the HLA class I introns by intra-locus homogenization. Immunogenetics 47:30–36. https://doi.org/10.1007/s002510050323
Graumann MB, DeRose SA, Ostrander EA, Storb R (1998) Polymorphism analysis of four canine MHC class I genes. Tissue Antigens 51:374–381. https://doi.org/10.1111/j.1399-0039.1998.tb02976.x
Hedrick PW (1994) Evolutionary genetics at the major histocompatibility complex. Am Nat 143:945–964 https://www.jstor.org/stable/2462890
Holmes JC, Holmer SG, Ross P, Buntzman AS, Frelinger JA, Hess PR (2013) Polymorphisms and tissue expression of the feline leukocyte antigen class I loci FLAI-E, FLAI-H, and FLAI-K. Immunogenetics 65:675–689. https://doi.org/10.1007/s00251-013-0711-z
Hong YJ, Kim KS, Kimura J, Kauhala K, Voloshina I, Goncharuk MS, Yu L, Zhang Y, Sashika M, Lee H, Min MS (2018) Genetic diversity and population structure of East Asian raccoon dog (Nyctereutes procyonoides): genetic features in central and marginal populations. Zool Sci 35:249–259. https://doi.org/10.2108/zs170140
Hughes AL, Nei M (1992) Maintenance of MHC polymorphism. Nature 355:402–403. https://doi.org/10.1023/A:1026494212540
Hughes AL, Yeager M (1998) Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet 32:415–435. https://doi.org/10.1146/annurev.genet.32.1.415
IASR (2005) Zoonoses in Japan. Infectious Agent Surveillance Report 26:193–194. https://idsc.nih.go.jp/iasr/26/306/tpc306.html
Jeffery KJM, Bangham CRM (2000) Do infectious diseases drive MHC diversity? Microbes Infect 2:1335–1341. https://doi.org/10.1016/S1286-4579(00)01287-9
Jukes TH, Cantor CR (1969) Evolution of protein molecules. In: Munro HM (ed) Mammalian protein metabolism. Academic Press, New York, pp 21–132. https://doi.org/10.1016/B978-1-4832-3211-9.50009-7
Kauhala K, Kowalczyk R (2011) Invasion of the raccoon dog Nyctereutes procyonoides in Europe: history of colonization, features behind its success, and threats to native fauna. Curr Zool 57:584–598. https://doi.org/10.1093/czoolo/57.5.584
Kauhala K, Saeki M (2016) Nyctereutes procyonoides. The IUCN red list of threatened species 2016. Available at http://dx.doi.org/10.2305/IUCN.UK.2016-1.RLTS.T14925A85658776.en (date last accessed, 1 March 2016)
Kennedy LJ, Altet L, Angeles JM, Barnes A, Carter SD, Fracino O, Gerlach JA, Happ GM, Ollier WE, Polvi A, Thomson W, Wagner JL (2000) Nomenclature for factors of the dog major histocompatibility system (DLA), 1998: first report of the ISAG DLA Nomenclature Committee. Anim Genet 31:52–61. https://doi.org/10.1046/j.1365-2052.2000.00492.x
Kim SI, Park SK, Lee H, Oshida T, Kimura J, Kim YJ, Min MS (2013) Phylogeography of Korean raccoon dogs: implications of peripheral isolation of a forest mammal in East Asia. J Zool 290:225–235. https://doi.org/10.1111/jzo.12031
Kim SI, Oshida T, Lee H, Min MS, Kimura J (2015) Evolutionary and biogeographical implications of variation in skull morphology of raccoon dogs (Nyctereutes procyonoides, Mammalia: Carnivora). Biol J Linn Soc 116:856–872. https://doi.org/10.1111/bij.12629
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllensten UB, Heise ER, Jones PP, Parham P, Wakeland EK, Watkins DI (1990) Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetic 31:217–219. https://doi.org/10.1007/BF00204890
Klein J, Sato A, Nagl S, Colm O (1998) Molecular trans-species polymorphism. Annu Rev Ecol Syst 29:1–21. https://doi.org/10.1146/annurev.ecolsys.29.1.1
Kricker MC, Drake JW, Radman M (1992) Duplication-targeted DNA methylation and mutagenesis in the evolution of eukaryotic chromosomes. Proc Natl Acad Sci USA 89:1075–1079. https://doi.org/10.1073/pnas.89.3.1075
Kuduk K, Babik W, Bojarska K, Śliwińska EB, Kindberg J, Taberlet P, Swenson JE, Radwan J (2012) Evolution of major histocompatibility complex class I and class II genes in the brown bear. BMC Evol Biol 12:197. https://doi.org/10.1186/1471-2148-12-197
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Kurosawa A, Tojinbara K, Kadowaki H, Hampson K, Yamada A, Makita K (2017) The rise and fall of rabies in Japan: a quantitative history of rabies epidemics in Osaka Prefecture, 1914–1933. PLoS Negl Trop Dis 11:1–19. https://doi.org/10.1371/journal.pntd.0005435
Librado P, Rozas J (2009) DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Liu G, Zhang H, Sun G, Zhao C, Shang S, Gao X, Xia T, Yang X (2017) Characterization of the peripheral blood transcriptome and adaptive evolution of the MHC I and TLR gene families in the wolf (Canis lupus). BMC Genomics 18:1–14. https://doi.org/10.1186/s12864-017-3983-0
Maibach V, Hans JB, Hvilsom C, Marques-Bonet T, Vigilant L (2017) MHC class I diversity in chimpanzees and bonobos. Immunogenetics 69:661–676. https://doi.org/10.1007/s00251-017-0990-x
Miyamae J, Suzuki S, Katakura F, Uno S, Tanaka M, Okano M, Matsumoto T, Kulski JK, Morimoto T, Shiina T (2017) Identification of novel polymorphisms and two distinct haplotype structure in dog leukocyte antigen class I genes: DLA-88, DLA-12 and DLA-64. Immunogenetics 70:237–255. https://doi.org/10.1007/s00251-017-1031-5
Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL (2012) Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8:e1002764. https://doi.org/10.1371/journal.pgen.1002764
Nei M, Gu X, Sitnikova T (1997) Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc Natl Acad Sci USA 94:7799–7806. https://doi.org/10.1073/pnas.94.15.7799
Ohshima K (1991) The late-quaternary sea-level change of the Japanese Islands. J Geogr 100:967–975. https://doi.org/10.5026/jgeography.100.6_967
Penn DJ, Potts WK (1999) The evolution of mating preferences and major histocompatibility complex genes. Am Nat 153:145–164. https://doi.org/10.1086/303166
Piertney SB, Oliver MK (2006) The evolutionary ecology of the major histocompatibility complex. Heredity 96:7–21. https://doi.org/10.1038/sj.hdy.6800724
Pozio E (2000) Factors affecting the flow among domestic synanthropic and sylvatic cycles of Trichinella. Vet Parasitol 93:241–262. https://doi.org/10.1016/s0304-4017(00)00344-7
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574. https://doi.org/10.1093/bioinformatics/btg180
Ross P, Buntzman AS, Vincent BG, Grover EN, Gojanovich GS, Collins EJ, Frelinger JA, Hess PR (2012) Allelic diversity at the DLA-88 locus in Golden Retriever and Boxer breeds is limited. Tissue Antigens 80:175–183. https://doi.org/10.1111/j.1399-0039.2012.01889.x
Sin YW, Dugdale HL, Newman C, Macdonald DW, Burke T (2012) Evolution of MHC class I genes in the European badger (Meles meles). Ecol Evol 2:1644–1662. https://doi.org/10.1002/ece3/285
Sommer S (2005) The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front Zool 2:1–18. https://doi.org/10.1186/1742-9994-2-16
Sutor A, Schwarz S, Conraths FJ (2014) The biological potential of the raccoon dog (Nyctereutes procyonoides, Gray 1834) as an invasive species in Europe-new risks for disease spread? Acta Theriol 59:49–59. https://doi.org/10.1007/s13364-013-0138-9
Suzuki J, Nishio Y, Kameo Y, Terada Y, Kuwata R, Shimoda H, Suzuki K, Maeda K (2015) Canine distemper virus infection among wildlife before and after the epidemic. J Vet Med Sci 77:1457–1463. https://doi.org/10.1292/jvms.15-0237
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tanabe AS (2007) KAKUSAN: a computer program to automate the selection of nucleotide substitution model and the configuration of a mixed model on multilocus data. Mol Ecol Notes 7:962–964. https://doi.org/10.1111/j.1471-8286.2007.01807.x
Venkataraman GM, Stroup P, Graves SS, Storb R (2007) An improved method for dog leukocyte antigen 88 typing and two new major histocompatibility complex class I alleles, DLA-88*01101 and DLA-88*01201. Tissue Antigens 70:53–57. https://doi.org/10.1111/j.1399-0039.2007.00839
Venkataraman GM, Kennedy LJ, Little MTE, Graves SS, Harkey MA, Torok-Storb BJ, Storb R (2017) Thirteen novel canine dog leukocyte antigen-88 alleles identified by sequence-based typing. HLA 90:165–170. https://doi.org/10.1111/tan.13077
Wagner JL, Creer SA, Storb R (2000) Dog class I gene DLA-88 histocompatibility typing by PCR-SSCP and sequencing. Tissue Antigens 55:564–567. https://doi.org/10.1034/j.1399-0039.2000.550607.x
Wagner JL, Sarmiento UM, Storb R (2002) Cellular, serological, and molecular polymorphism of the class I and class II loci of the canine major histocompatibility complex. Tissue Antigens 59:205–210. https://doi.org/10.1034/j.1399-0039.2002.590304.x
Ward OG, Wurster-Hill DH, Ratty FJ, Song Y (1987) Comparative cytogenetics of Chinese and Japanese raccoon dogs, Nyctereutes procyonoides. Cytogenet Cell Genet 45:177–186. https://doi.org/10.1159/000132451
Zhang J, Rosenberg HF, Nei M (1998) Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA 95:2708–3713. https://doi.org/10.1073/pnas.95.7.3708
Zhu Y, Sun DD, Ge YF, Yu B, Chen YY, Wan QH (2013) Isolation and characterization of class I MHC genes in the giant panda (Ailuropoda melanoleuca). Chin Sci Bull 58:2140–2147. https://doi.org/10.1007/s11434-012-5582-4
Acknowledgements
We thank Sergei Avdeyuk, Vladimir Ivanov, and Sergei Fokin for providing Russian specimens, Seiichiro Dakemoto and various zoos (listed in Supplementary Table 1) for providing specimens, and Dr. Matthew H. Dick for comments on an early draft and for editing the manuscript.
Funding
This study was supported by a Joint Research Project Grant from the Japan Society for the Promotion of Science (JSPS No. JPJSBP120194802) and the Russian Foundation for Basic Research (RFBR No. 19-54-50001), a JSPS KANKENHI Grant (No. 18H05508), and a Joint Research Program Grant from the Japan Arctic Research Network Center.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Astrid V. Stronen
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bartocillo, A.M.F., Nishita, Y., Abramov, A.V. et al. Evolution of MHC class I genes in Japanese and Russian raccoon dogs, Nyctereutes procyonoides (Carnivora: Canidae). Mamm Res 66, 371–383 (2021). https://doi.org/10.1007/s13364-021-00561-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-021-00561-y