
Abstract
This review is focused at exploring the strengths of modern technology driven data compiled in the areas of virus gene sequencing, virus protein structures and their implication to viral diagnosis and therapy. The information for virome analysis (viromics) is generated by the study of viral genomes (entire nucleotide sequence) and viral genes (coding for protein). Presently, the study of viral infectious diseases in terms of etiopathogenesis and development of newer therapeutics is undergoing rapid changes. Currently, viromics relies on deep sequencing, next generation sequencing (NGS) data and public domain databases like GenBank and unique virus specific databases. Two commonly used NGS platforms: Illumina and Ion Torrent, recommend maximum fragment lengths of about 300 and 400 nucleotides for analysis respectively. Direct detection of viruses in clinical samples is now evolving using these methods. Presently, there are a considerable number of good treatment options for HBV/HIV/HCV. These viruses however show development of drug resistance. The drug susceptibility regions of the genomes are sequenced and the prediction of drug resistance is now possible from 3 public domains available on the web. This has been made possible through advances in the technology with the advent of high throughput sequencing and meta-analysis through sophisticated and easy to use software and the use of high speed computers for bioinformatics. More recently NGS technology has been improved with single-molecule real-time sequencing. Here complete long reads can be obtained with less error overcoming a limitation of the NGS which is inherently prone to software anomalies that arise in the hands of personnel without adequate training. The development in understanding the viruses in terms of their genome, pathobiology, transcriptomics and molecular epidemiology constitutes viromics. It could be stated that these developments will bring about radical changes and advancement especially in the field of antiviral therapy and diagnostic virology.
Similar content being viewed by others


Introduction
In the context of this review we have taken an operative definition of “viromics” to include the study of the full genome nucleotide sequences of viruses, functional genes as well as “non coding” regions of the viral genome. We have chosen illustrative examples to delineate the understanding of human viruses and disease associations. Where necessary, we have also described host genomics, viral and host proteomics to complete the picture. The essential focus of this review is on viral pathogenesis and drug therapy from a molecular, especially genomic, perspective. The review also includes some information on the technological approaches to viromics.
The term “viromics” was introduced in 2001. Here, we have used the working definition of viromics only in relation to human viral infections. Viromics refers to the characterization of the virome in environmental niches. These could be an infected host, wherein, interactions among the viruses that are present in the host could be characterized. The information for virome analysis is generated by study of viral genomes and viral genes using high throughput sequencing (HTS) or next generation sequencing (NGS) [34]. Presently, it is possible to generate very long sequences (30 kb) using high quality Taq polymerase and this may be part of the sequencing for analysis by NGS instrumentation. MiSeq is the only desktop sequencer that can produce 2 × 300 paired-end reads in a single run. This allows small genome sequencing and enables detection of target variants with unmatched accuracy and could generate full reads for many viruses whose genome length (RNA or DNA) is usually in the range of 3.5–20 kb. However, several groups of viruses such as Herpesviridae and Poxviridae have genomes in the range of 220–360 kb [25, 37, 46]. DNA viruses have identifiable Open Reading Frames (ORFs) which code for a complete protein. These could be referred to as viral genes. This is typically seen, for example, in the Hepatitis B Virus (HBV) [5]. All negative stranded RNA viruses possess ORFs which could be formed by transcription of overlapping nucleotide sequences. In the case of positive strand RNA viruses, like enteroviruses, the viral genome is entirely transcribed into one mRNA (polycistronic code) which is translated into a polyprotein. By autolytic cleavage this polyprotein generates the individual viral proteins in the cytosol of the infected host cell [20].
The study of infectious diseases in terms of etiopathogenesis and development of newer therapeutics is now undergoing rapid changes. Within the next decade the classical approach to pathogen recognition by isolation and culture may not be relevant in the day-to-day practice of infectious disease medicine. The laboratory technologies are rapidly evolving and becoming affordable especially those based on identification of “signature sequence” of nucleotides in the genome of the pathogens. Conventional PCR and sequencing is now 3 decades old and is being replaced by NGS, which could identify the pathogen, its susceptibility to antimicrobial agents and even pathological markers of disease in appropriate clinical sample directly.
Humans suffer infections by bacteria, viruses, fungi, parasites and even algae. At times the infections may be polymicrobial or co-infectious in nature. The infection may be acute, chronic or latent. Latent infection by certain microbes or viruses may lead to opportunistic infection when the host is congenitally immunocompromised, due to infection, immunosuppressive therapy or certain endocrine disorders. Certain infections have now been incriminated in the evolution of the autoimmune disease processes. DNA microarrays and proteomics are now augmented by advanced sequencing methods to help in identifying disease processes. Multiple agents are now recognized to cause indistinguishable illnesses; the term ‘syndrome’ applies to such situations. Early and rapid diagnosis of the infecting agent would enable prompt and appropriate therapy. Many molecular techniques have been evaluated for rapid diagnosis of infectious syndromes, and include real-time multiplex PCR, DNA microarray, loop-mediated isothermal amplification, and other similar assays [48].
Diagnostic virology was previously based on cell culture methods, microscopy, serology and conventional molecular techniques. This is now dramatically changed by newer technologies. Portability for field studies has necessitated development of nanotechnology based diagnostics which could cut down costs, including the need for expensive instrumentation [40]. The performance and speed of sequencing of NGS platforms has added significantly to genomic studies, including viromics [1].
Technological approaches and metadata analysis
Advances in nucleotide sequencing
Conventionally virus isolation, viral antigen detection and virus particle demonstration have been relied upon for diagnosis from 1940s to 1980s. Since then viral genomic material detection has become established as a means of diagnosis. Subsequently, using nucleotide sequencing the genomes of viruses have been characterized. More recently, it has become possible for multiple virus detection and genetic analysis in host cells, tissues and body fluids. Table 1 gives information on these approaches.
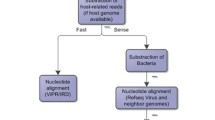
We describe here two widely used NGS platforms namely Illumina and Ion Torrent. These two approaches can give information on sequence sizes of 300–400 nucleotides. Library construction of sequences uses an adaptor which is incorporated at the 5′ end and in a two step PCR with M13-tailed primers (universal barcode). Ligation is achieved with a three prime adapter. Appropriate software is used to eliminate low quality sequences [47]. The steps involved in NGS is shown in a simplified manner in Fig. 1. The steps involved in genetic analysis, with a special reference to diagnosis of infectious viral syndrome, are shown in Fig. 2 which describes virus genome detection through amplification following specific capture and metadata analysis.

Schematic representation of steps involved in next generation sequencing

Schematic representation of the steps involved in viromics approach for diagnosis of infectious viral syndromes and genetic analysis
In experiments to study splice variants, epigenetic modifications (methylation) and Single Nucleotide Polymorphism (SNP) identification could be studied in paired-end runs. But suitable this may be costly and laborious, so single read ends are considered in RNA-Seq or ChIP-Seq methodologies [23].
Longer read runs are preferred to give accurate information and read lengths > 100 are required for transcriptomic studies. Each sequencing platform and instrument gives different number of reads. The length of the read is affected by efficiency of reverse transcription and amplification. Optimization is essential in any type of instrument. Viral and host mRNA analysis (transcriptomes) could provide crucial information on virus pathogenesis and possibly help towards antiviral drug development [https://www.illumina.com/techniques/popular-applications/gene-expression-transcriptome-analysis.html] [32].
The PCR method uses “fusion primers” to attach the Ion A and truncated P1 (trP1) adapters to the amplicons as they are generated in the PCR. The fusion primers contain the Ion A and trP1 sequences at their 5′-ends adjacent to the target-specific portions of the primers. The target region is the portion of the genome that will be sequenced in the sample(s) of interest. Two pairs of forward and reverse primers per target region are used to enable bidirectional sequencing [8]. NGS technology has improved with single-molecule real-time sequencing (SMRT) wherein, complete long reads can be obtained with less errors that could arise due to editing carried out by personnel without sufficient training both in instrumentation and the computer software (MiSeq reporter) [15].
Interesting insights have been derived from the study of the virus genome and proteome. Several virus genes together with their function and protein characterization are now available in the public domain and selected information from such information-banks is shown in Table 2. Information on virus anti-receptor and host cell receptors is similarly available as shown in Table 3. These information avenues are now being exploited for development of chemotherapy and vaccines, along with the study of virus transmission and global spread.
High Throughput Sequencing (HTS) technologies are capable of sequencing multiple DNA molecules in parallel, enabling hundreds of millions of DNA molecules to be sequenced at a time. This technology is emerging as a useful tool for study of viruses in clinical samples. This is especially so when the target sequence is unknown. The Roche-454 genome sequencer or Illumina genome analyzer platforms have been established for routine analysis in research facilities [9]. HTS methodology has not yet shown to be useful for routine viral diagnostics. A bioinformatics pipeline (ezVIR) has been designed to process HTS data from standard platforms for known human viruses. The pipeline works by identifying the most likely viruses present in the specimen given the sequencing data. ezVIR can also give information on typing, it can also can create and identify cross-contamination genome coverage histograms for analysis of specimens prepared in series using linux box (linux operating system) [41].
Presently, one of the new generation sequencing technologies developed is by Pacific BioSciences which is able to give information on long reads, which could help in Viromics and transcriptomics research [18]. Likewise, Oxford Nanopore technologies have a similar approach. Here, bionanopores are created on a stable matrix. The analyte (ssDNA) passes from, through nanopores by the effect of an electrical gradient applied to the matrix. An unique electrical signal (signature) generated by each nucleotide allows its recognition by its charged properties and orientation of the molecule [44].
Application of advanced nucleotide sequencing techniques
The earliest successful application of genomic information in the classification of viruses was established successfully for enteroviruses in comparison to standard cell culture techniques. Viral RNA radio labeling (32P) in cell culture followed by two dimentional electrophoresis of digested genome allowed for distinct variant recognition. This was validated by sequencing of cDNA by Sanger method. A member of enterovirus serotype/genotype was defined as a virus strain which had > 75% homology at nucleotide level and > 88% amino acid level [33, 38].
Metagenomics studies have revealed a significant role of gastrointestinal viruses in microbial dysbiosis or dysbacteriosis (a microbial flora alteration in the gut). Molecular interactions in the microbiome by studies on metagenomics, metatranscriptomics and viromics will unravel the importance of the human microbiome (including virome) in health and disease [7].
NGS has facilitated studies in the monitoring of influenza virus strain evolution, the study of quasispecies in the host infected with Human Immunodeficiency Virus (HIV) and Hepatitis C virus (HCV) viruses. More recently information on drug resistance directly from viral genome sequencing has been made available through NGS technology [4].
Several viral pathogens, such as hemorrhagic fever agents and newly emerging zoonotic viral infections must only be investigated in Biosafety level laboratories 3 or 4 (BSL). The availability of HTS and NGS in laboratories within such facilities has significantly enhanced the information available on these agents, for example such studies have shed light on virus transmission to humans [58].
Study of viruses as single or multiple pathogens in infected host
Quantitative Insights into Microbial Ecology (QIIME) provides information through construction of a taxonomic tree in the Newick format. This approach may be applicable in viromics when one looks at information on multiple populations of virus in a given host environment, such as blood, in the case of blood borne viruses (HIV, HBV and HCV) [49]. The presence of individual or co-infection with multiple respiratory viruses could now be studied.
A study by Thorburn et al. [59], has provided some interesting information on respiratory infections. They documented viral contigs in 53/89 respiratory samples with reproducibility in 86.8% of samples. They did not detect mixed infections by NGS. The phylogenetic analysis using NGS is represented in Fig. 3, wherein, you see multiple respiratory viruses detected in the samples. An additional application of NGS could be analysis of wild strains of human influenza virus to search for evidence of evolutionary trends especially becauseof the implication for vaccine design. Recently, it has been shown that mutations accumulate in the H3N2 strain of Innfluenza virus due to propogation in eggs for vaccine preparation. Mutations affect antigenicity of the vaccine strains [67].

Neighbor-joining (NJ) phylogenetic tree of full-genome reference sequences and a subset of the NGS consensus sequences with full or near-full reference genome coverage. The branch annotations represent the bootstrap values (percentage of 1000 samples trees). Sequences generated in the study are shown as circles and squares. Courtesy: Thorburn et al. 2015
NGS output could be used with the FigTree application to generate a file in Biological Observation Matrix (Biom) format. This is referred to as a representing operational taxonomic unit (OUT) table; this format is compatible with the MEGAN software which requires matrix-type data. FigTree graphically represents phylogenetic relationships with nodes and branches as trees. It is suitable for use with Bayesian Evolutionary Analysis Sampling Tree (BEAST) output files thus, making a powerful analytical tool. Phylogenetic Diversity is called alpha-diversity analysis (diversity within a sample) and beta-diversity analysis (diversity across samples). This is supported through QIIME and other software packages which make possible metagenomic data handling. [16, 39].
It is opined by experience that Sanger sequencing has as compared to NGS has poor sensitivity and only detects consensus populations of > 20%. However, Ultradeep pyrosequencing can overcome certain limitations with improved sensitivity to detect mutations. It also has the ability to quantitate and indicate the presence of minor viral subpopulations. Furthermore, it offers the capacity of identification novel resistance mutations. NGS thus occurs in a massively parallel fashion the sequencing of hundreds of thousands to millions of DNA molecules in one reaction.
NGS has been shown to pick up several new mutations associated with drug resistance and has shown good concordance with Sanger sequencing method for the previously recognized mutations [35]. The concept of temporal viromics has evolved based on the availability of analytical data on virus infected host cell protein expression. This investigative approach gives information on virus gene expression, virus protein translation and host gene modulation (increased or decreased expression) in relation to time from infection. These studies are best carried out in cell culture host. For example, human cytomegalovirus (HCMV) infected cells have been investigated for transcriptomics analysis and host proteomics. Early and late genes expression has been studied [53, 61].
Identification of new viruses in clinical syndromes and understanding virus latency
The study of human endogenous retroviruses (HERVs) is important from several points of view. 1. The quantitation of the viral genome in clinical samples serves to ascertain virus loads of exogenous viruses of humans. 2. They are integrated in the chromosome and latent. The viruses are passed from one generation to another through the germ-line as a ‘provirus’. 3. HERVs constitute about 8% of the human genome and some play an important role in the maturation of the T cell limb of the immune system in the thymus. In fact it is now postulated that their antigens are expressed on thymic Hassall’s corpuscles cells and serve as ‘superantigens’. Immature thymocytes with high-affinity T cell receptors (TCR) for self antigens seen with context of MHC class 1 antigens are pushed to programmed cell death (apoptosis) because of the superantigens of HERVs. This prevents emergence of auto reactive T cells. The high affinity T-cells that leave the thymus are thus not directed against self-antigens [31]. More recently, HERV (HML-2) named K111 provirus has been detected in HIV infected patient which is activated by HIV-1 Tat protein [14]. Also, HERVs play a regulatory role in human genes through certain motifs in their long terminal repeats [51]. Some evidence suggests that HERVs could have a pathogenic role in certain neurological diseases [12].
Studies on HIV strains using four different NGS platforms (454/Roche pyrosequencing, Illumina, Ion torrent and PacBio) were able to identify major virus variants with similar efficiency. These techniques were useful in the determination of co-receptor usage by HIV strains (HIV co-receptor tropism i.e. CCR5 receptor usage) [2]. The HIV –SMART assay was improved to detect even low copy numbers of the viral RNA by using a concentrator method. This method is used post RNA extraction using a commercial kit [45]. Molecular surveillance is essential to monitor HIV diversity. An universal library preparation method (HIV-SMART [i.e.,switching mechanism at 5′ end of RNA transcript]) for NGS has been developed. Broad application of the HIV-SMART approach has been demonstrated. Multiplexing 8 or more libraries per MiSeq run results in full genome coverage at a median ∼ 2000 × depth. The method consistently identified viral sequence heterogeneity at viral loads of ≤ 4.5 log copies/ml has been shown. HIV-SMART provides an opportunity to identify diverse HIV strains. This approach could be adapted to sequence any RNA virus for viral characterization and surveillance [6].
The use of HTS and NGS is expanding viral metagenomics (viromics) giving information on virus circulation, quasispecies in infected patients, drug susceptibility data. Researchers have used Illumina MiSeq to generating complete genome information on different genotypes of Dengue virus, enterovirus and RSV [3]. Using NGS, Herpes simplex virus 1 (HSV-1), HSV-2 and VZV were detected in the CSF samples of patients with meningoencephalitis [22]. In this study, the number of ‘unique reads’ of the identified viral genes ranged from 144 to 44,205 (93.51–99.57%). The coverage of identified viral genes ranged from 12 to 98% with a ‘depth value’ of 1.1–35, respectively. NGS could therefore be used for “pan-viral” or even “pan-microbial” screening of CSF for diagnosis of CNS infectious diseases. NGS has now been established in a few public health laboratories of high stature and may take a while to actually enter good hospital laboratories. The CDC in the US is using this for molecular epidemiology of epidemiological studies of influenza virus [https://blogs.cdc.gov/publichealthmatters/2017/03/using-amd-to-fight-flu/]. The Public Health England, Genomic Services and Development Unit, London offers on-demand services [https://www.gov.uk/guidance/genomic-services-and-development-unit-gsdu] which could be utilized by diagnostic laboratories to identify novel pathogens.
The association of several human cancers with viruses has been studied over the last 5 decades, and the application of conventional PCR techniques has produced strong lines of evidence to support this. Several DNA viruses (HPV 16 and 18) and some RNA viruses, especially retroviruses (HTLV-1 and HTLV-II) and Hepatitis C virus are linked to human cancers. Certain molecular mechanisms have been identified, such as the role of G protein-coupled receptors (GPCRs) in activation of several signaling pathways that affect cellular proliferation in the causation of cancer [65]. Presently, with the availability of NGS, it is now possible to study viral genomics, transcriptomics, immunomics, host genomics (metagenomics) and their interactions to understand the complete pathobiology of viral induced cancers [62].
Using the National Center for Biotechnology Information (NCBI) taxonomy database a study using NGS in high grade gliomas and glioblastomas has revealed an association with EBV [13, 19]. In the case of HTLV-1 associated T-cell leukemia, NGS has identified the presence of multiple mutations in SUZ12, DNMT1, DNMT3A, DNMT3B, TET1, TET2, IDH1, IDH2, MLL, MLL2, MLL3 and MLL4. The mutations in the TET gene was dominant in adult T-cell leukemia [64]. Also several neurological tumors are being linked to viruses using metagenomcis with a combination of NCBI and Cancer Genome Atlas [54]. The virus-tumor map will facilitate the study of tumor associated viruses using transcriptome data analysis through fusion genes and the FusionMap, especially in the study of provirus integration site like in HPV induced neoplasia [57].
Application of viromics in development of antiviral therapeutic agents and vaccines
In this section we have focused on the developments that would have a bearing on the new approaches to antiviral therapy and development of vaccines based on information obtained from functional genomics. Presently, this is a significant approach for microbial vaccine research. It is now possible to identify target gene sequences and their expressed proteins as potential protective antigens/epitope. T-and B-cell responses to these may be robust and give protective immunity [50].
Antiviral therapeutic agents
Advances in HIV genome characterization, especially the proteomics of HIV-1, has similarly revealed mutations associated with HIV drug resistance. Several mutations have a critical effect on the geometry of active site of the protease gene complex. Numerous single nucleotide polymorphism (SNPs) were described and the 3D structure of the protein has been shown in in silico analysis to have lowered affinity for anti-HIV drugs such as reverse transcriptase-inhibitors [30]. Studies of HIV-1 protease inhibitors with different molecular structures have been made available describing the affinity of the inhibitor to the viral protease [26, 43].
NGS is now becoming an important tool in identifying quasispecies population and drug resistant mutants in infected individuals [10]. The influenza virus A is associated with seasonal influenza and variants were associated with human pandemic influenza (pdH1N1). In patients infected with the influenza virus, it is possible to show variants simultaneously in the virus population showing synonymous and non-synonymous mutations in the heamagglutinin (HA) and neuraminidase (NA) genes. Furthermore, resistance to NA inhibitors have been documented both in individuals before and during the course of oseltamivir therapy. The Ion Torrent PGM has been successfully used in clinical studies [60, 66].
Presently, the understanding of virus replication in the context of viromics and proteomics is facilitating the development of antiviral molecules against enteroviruses towards future development of drugs. Transgenic murine model expressing the poliovirus receptor (TgPVR21) has been used to evaluate the developmental drug (a kinase inhibitor, A4) which blocks poliovirus replication. This evaluation is in a preclinical stage [20].
Experimental Dengue virus infection of Huh 7 cells was used to document decreased mRNA levels of host RNAi factors, like Dicer, Drosha, Ago1, and Ago2. This could play a role in suppression of genes in virus-infected cells enabling increased dengue virus replication. Downregulation of human microRNAs (miRNAs) in response to viral infection was also documented. NS4B of all four dengue virus serotypes was seen to be a potent RNAi suppressor. The NS4B N-terminal region, and the signal sequence 2 K, has been shown to have interferon (IFN)-antagonistic properties [27].
Flaviviridae display intricate mechanisms to engage the host cell machinery for their purpose. In experimental animals and infected cells, proteomics study shows changes in protein expression in the host cell which has indicated increases in certain protein expressions and decrease in others. This alteration of the proteins and homeostasis (proteostasis) of cellular proteins results in abnormality at a cellular level like upregulation of Hsp70 in Dengue virus infection in cultured cells [56]. The virus proteins engage host proteins during virus entry and replication. In the whole animal (infected host), there are several immunological effects because of expression of pro-inflammatory cytokines [11, 21]. Studies with enterovirus in infected cells have shown the virus is entirely dependent for its intracytoplasmic replication on host protein homeostasis machinery. The virus capsids and viral RNA accumulate rapidly for the formation of mature virus particles. It is postulated that cellular chaperones are involved in supporting the replicating cycle [63]. Studies on virus gene expression in host cells and changes in host cell protein homeostasis i.e. expression of proteins in normal vs infected cells provides insights on viral pathogenesis at a cellular level. Presently, evidence is accumulated of autophagy in the cytosol of virus infected cells as part of cell repair mechanism and conservation of building blocks like amino acids [17].
Messenger RNA (mRNA) has emerged as a useful and highly effective platform to deliver vaccine antigens and therapeutic proteins. The immune modulatory therapies may best work at low viral levels. This includes RNA interference and clustered regularly interspaced short palindromic repeats (CRISPR/Cas9). Further invitro studies and clinical trials are warranted [24, 42].
Presently, there are a considerable number of good treatment options for HBV/HIV/HCV, but these viruses are showing the development of drug resistance [55].
The drug susceptibility regions of the genomes are sequenced and the prediction of drug resistance is now possible from 3 public domains available on the web. They are:
HBV Drug resistance database: Stanford University
HBVseq accepts user-submitted HBV RT sequences. HBVrt DB was constructed by annotating the body of publicly available HBV RT sequences.
HIV Drug resistance database: Stanford University
This database provides extensive comments and a highly transparent scoring system that is ‘hyperlinked’. This HIVdb has listed the individual mutations and combination of mutations to a given drug or group of drugs with a scoring system. The scoring system relates to drug resistance mutations (DRMs). The primary information here is the combination of genotyping, phenotyping and clinical experience results.
HCV Drug resistance database: Stanford University
This database has been created for information on mutations in the HCV genome which relate to drug resistence as evidenced by clinical failure of the drug. The direct-acting antivirals (DAAs) could inhibit both structural and non structural proteins and hence interfere with HCV replication. This has been possible because of advances in virus gene analysis and transcriptomics studies.
The caveat at this stage is: One has to carefully evaluate the utility of NGS in drug resistance mutations detection especially, in the case of HIV/HBV/HCV. Clinically drug resistance data generated in databases is population sequence based wherein; the sequence of the dominant circulating virus genome (80%) is reflected. NGS approach may result in identifying mutations even in less than 1% of the viral genomic copies. This could have implication in choosing drug combinations for therapy [52].
Viral vaccines
The recognition of poliovirus vaccine derived polioviruses (VDPV) circulating in the community where OPV is used has also revealed the presence of Sabin virus 2 neurovirulent mutants in cases of vaccine-associated paralytic polio. The NGS method has been successfully implemented to generate PV genomes for molecular epidemiology of the most recent PV isolates. Researchers have used the Nextera XT DNA library preparation successfully with Illumina MiSeq technique. Successful viral reads were obtained in 85–95% of the samples covering over 90% of PV genome [36].
Hantaviruses are viral pathogens that causes hantavirus cardiopulmonary syndrome (HCPS) in the Americas. Linear B-cell epitopes for hantaviruses that are specific to genotypes causing HCPS in humans have been studies using in silico prediction servers. An epitope IMASKSVGS/TAEEKLKKKSAF was identified as the best candidate B-cell epitope specific for hantaviruses causing HCPS andpromiscuous epitopes were identified in the C-terminal of the protein [28]. Zika virus, though first identified in late 1950s, emerged in 2014 as a significant Aedesaegypti mosquito borne encephalitic illness with high risk of congenital disease of the newborns to mothers infected during early trimester of pregnancy. As of mid-March 2017, 151 Zika virus genome sequences are available in the GenBank database. Though it is a current major public health problem globally, there is no ZIKV-specific treatment or vaccine at present. Development of a safe and effective vaccine is hence a high priority. It is now eminently possible to use metagenomic and proteomics approach to make “Designer vaccines” suitable for geographical regions which are virus B-cell or T-cell epitope based as in the case of hantaviruses [29].
Concluding remarks
A study of the virus genome especially its genes and their expression in host cells and the host genetic changes through upregulation of certain constitutional genes has revealed important facets of viral infections and the ensuing pathology. The virus produces effects of physical damage locally in the infected cells and systemically in the host through upregulation of proinflammatory cytokines in the course of infection. However, we are on the threshold of breakthrough especially in the field of viral detection and therapy. This has been made possible through advances in the technology with the advent of NGS, meta-analysis through sophisticated and easy to use software and high speed computers for bioinformatics. Information emanating from viromics and proteomics of the host has started revealing new facets in the diagnosis of viral infections and development of antiviral agents including vaccines. We believe this will have a huge positive impact on prevention and control of viral infections in childhood and adults and those infections with epidemic to pandemic potential.
References
Alekseeva AE, Brusnigina NF. Metagenomic studies and infectious diseases diagnostics. Zh Mikrobiol Epidemiol Immunobiol. 2015;2:81–9.
Archer J, Weber J, Henry K, Winner D, Gibson R, Lee L, Paxinos E, Arts EJ, Robertson DL, Mimms L, Quiñones-Mateu ME. Use of four next-generation sequencing platforms to determine HIV-1 coreceptor tropism. PLoS ONE. 2012;7(11):e49602.
Baronti C, Piorkowski G, Leparc-Goffart I, de Lamballerie X, Dubot-Pérès A. Rapid next-generation sequencing of dengue, EV-A71 and RSV-A viruses. J Virol Methods. 2015;226:7–14.
Barzon L, Lavezzo E, Militello V, Toppo S, Palù G. Applications of next-generation sequencing technologies to diagnostic virology. Int J Mol Sci. 2011;12:7861–84.
Beck J, Nassal M. Hepatitis B virus replication. World J Gastroenterol. 2007;7(13):48–64.
Berg MG, Yamaguchi J, Alessandri-Gradt E, Tell RW, Plantier JC, Brennan CA. A Pan-HIV strategy for complete genome sequencing. J Clin Microbiol. 2016;54:868–82.
Bilkel S, Valdez-Lara A, Cornejo-Granados F, Rico K, Canizales-Quinteros S, Soberón X, Del Pozo-Yauner L, Ochoa-Leyva A. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput Struct Biotechnol J. 2015;13:390–401.
Blomquist TM, Crawford EL, Lovett JL, Yeo J, Stanoszek LM, Levin A, Li J, Lu M, Shi L, Muldrew K, Willey JC. Targeted RNA-sequencing with competitive multiplex-PCR amplicon libraries. PLoS ONE. 2013;8:e79120.
Cheval J, Sauvage V, Frangeul L, Dacheux L, Guigon G, Dumey N, Pariente K, Rousseaux C, Dorange F, Berthet N, Brisse S, Moszer I, Bourhy H, Manuguerra CJ, Lecuit M, Burguiere A, Caro V, Eloit M. Evaluation of high-throughput sequencing for identifying known and unknown viruses in biological samples. J Clin Microbiol. 2011;49:3268–75.
Chimukangara B, Samuel R, Naidoo K, de Oliveira T. Primary HIV-1 drug resistant minority variants. AIDS Rev. 2017; 19(2).:89–96.
Chiu HC, Hannemann H, Heesom KJ, Matthews DA. Davidson AD1. High-throughput quantitative proteomic analysis of dengue virus type 2 infected A549 cells. PLoS ONE. 2014;9(3):e93305.
Christensen T. Human endogenous retroviruses in neurologic disease. APMIS. 2016;1:116–26.
Cimino PJ, Zhao G, Wang D, Sehn JK, Lewis JS Jr, Duncavage EJ. Detection of viral pathogens in high grade gliomas from unmapped next-generation sequencing data. Exp Mol Pathol. 2014;96:310–5.
Contreras-Galindo R, Kaplan MH, He S, Contreras-Galindo AC, Gonzalez-Hernandez MJ, Kappes F, Dube D, Chan SM, Robinson D, Meng F, Dai M, Gitlin SD, Chinnaiyan AM, Omenn GS. Markovitz DM. HIV infection reveals widespread expansion of novel centromeric human endogenous retroviruses. Genome Res. 2013;23:1505–13.
Dilernia DA, Chien JT, Monaco DC, Brown MP, Ende Z, Deymier MJ, Yue L, Paxinos EE, Allen S, Tirado-Ramos A, Hunter E. Multiplexed highly-accurate DNA sequencing of closely-related HIV-1 variants using continuous long reads from single molecule, real-time sequencing. Nucleic Acids Res. 2015;43:e129.
Drummond AJ, Rambaut A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;8(7):214.
Fang S, Su J, Liang B, Li X, Li Y, Jiang J, Huang J, Zhou B, Ning C, Li J, Ho W, Li Y, Chen H, Liang H, Ye L. Suppression of autophagy by mycophenolic acid contributes to inhibition of HCV replication in human hepatoma cells. Sci Rep. 2017;9(7):44039.
Feng Y, Zhang Y, Ying C, Wang D, Du C. Nanopore-based fourth-generation DNA sequencing technology. Genom Proteom Bioinform. 2015;13(1):4–16.
Flippot R, Malouf GG, Su X, Khayat D, Spano JP. Oncogenic viruses: lessons learned using next-generation sequencing technologies. Eur J Cancer. 2016;61:61–8.
Ford Siltz LA, Viktorova EG, Zhang B, Kouiavskaia D, Dragunsky E, Chumakov K, Isaacs L, Belov GA. New small-molecule inhibitors effectively blocking picornavirus replication. J Virol. 2014;88:11091–107.
Gerold G, Bruening J, Weigel B, Pietschmann T. Protein interactions during the flavivirus and hepacivirus life cycle. Mol Cell Proteom. 2017;4:S75–91.
Guan H, Shen A, Lv X, Yang X, Ren H, Zhao Y, Zhang Y, Gong Y, Ni P, Wu H, Zhu Y, Cui L. Detection of virus in CSF from the cases with meningoencephalitis by next-generation sequencing. J Neurovirol. 2016;22:240–5.
Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11:476–86.
Herrera-Carrillo E, Berkhout B. Attacking HIV-1 RNA versus DNA by sequence-specific approaches: RNAi versus CRISPR-Cas. Biochem Soc Trans. 2016;44:1355–65.
Jia H, Guo Y, Zhao W, Wang K. Long-range PCR in next-generation sequencing: comparison of six enzymes and evaluation on the MiSeq sequencer. Sci Rep. 2014;4:5737.
Junaid M, Lapins M, Eklund M, Spjuth O, Wikberg JE. Proteochemometric modeling of the susceptibility of mutated variants of the HIV-1 virus to reverse transcriptase inhibitors. PLoS ONE. 2010;5(12):e14353.
Kakumani PK, Ponia SS, Sood V, Chinnappan M, Banerjea AC, Medigeshi GR, Malhotra P, Mukherjee SK, Bhatnagar RK. Role of RNA interference (RNAi) in dengue virus replication and identification of NS4B as an RNAi suppressor. J Virol. 2013;87:8870–83.
Kalaiselvan S, Sankar S, Ramamurthy M, Ghosh AR, Nandagopal B, Sridharan G. Prediction of pan-specific B-cell epitopes from nucleocapsid protein of hantaviruses causing hantavirus cardiopulmonary syndrome. J Cell Biochem. 2017. https://doi.org/10.1002/jcb.25887.
Kalaiselvan S, Sankar S, Ramamurthy M, Ghosh AR, Nandagopal B, Sridharan G. Prediction of B cell epitopes among hantavirus strains causing hemorragic fever with renal syndrome. J Cell Biochem. 2017;118:1182–8.
Kandathil AJ, Joseph AP, Kannangai R, Srinivasan N, Abraham OC, Pulimood SA, Sridharan G. Structural basis of drug resistance by genetic variants of HIV type 1 clade c protease from India. AIDS Res Hum Retrovir. 2009;25:511–9.
Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol. 2014;14:377–91.
Law GL, Korth MJ, Benecke AG, Katze MG. Systems virology: hostdirected approaches to viral pathogenesis and drug targeting. Nat Rev Microbiol. 2013;11(7):455–66.
Lee YF, Kitamura N, Nomoto A, Wimmer E. Sequence studies of poliovirus RNA. IV. Nucleotide sequence complexities of poliovirus type 1, type 2 and two type 1 defective interfering particles RNAs, and fingerprint of the poliovirus type 3 genome. J Gen Virol. 1979;44(2):311–22.
Lotze MT, Kost TA. Viruses as gene delivery vectors: application to gene function, target validation, and assay development. Cancer Gene Ther. 2002;9:692–9.
Lowe CF, Merrick L, Harrigan PR, Mazzulli T, Sherlock CH, Ritchie G. Implementation of next-generation sequencing for hepatitis B virus resistance testing and genotyping in a clinical microbiology laboratory. J Clin Microbiol. 2016;54:127–33.
Montmayeur AM, Ng TF, Schmidt A, Zhao K, Magaña L, Iber J, Castro CJ, Chen Q, Henderson E, Ramos E, Shaw J, Tatusov RL, Dybdahl-Sissoko N, Endegue-Zanga MC, Adeniji JA, Oberste MS, Burns CC. High-throughput next-generation sequencing of polioviruses. J Clin Microbiol. 2017;55:606–15.
Moss B. Poxviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology. Philadelphia: Lippincott Williams & Wilkins; 2007. p. 2905–46.
Oberste MS, Maher K, Kilpatrick DR, Pallansch MA. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol. 1999;73(3):1941–8.
Oulas A, Pavloudi C, Polymenakou P, Pavlopoulos GA, Papanikolaou N, Kotoulas G, Arvanitidis C, Iliopoulos I. Metagenomics: tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform Biol Insights. 2015;9:75–88.
Perumal V, Sridharan G, Padma S. Emergent technologies In the detection of emerging and re-emerging viruses with reference to infections in humans. Sri Ramachandra J Med. 2010;3:22–8.
Petty TJ, Cordey S, Padioleau I, Docquier M, Turin L, Preynat-Seauve O, Zdobnov EM, Kaiser L. Comprehensive human virus screening using high-throughput sequencing with a user-friendly representation of bioinformatics analysis: a pilot study. J Clin Microbiol. 2014;52:3351–61.
Pham EA, Perumpail RB, Fram BJ, Glenn JS, Ahmed A, Gish RG. Future therapy for hepatitis B virus: role of immunomodulators. Curr Hepatol Rep. 2016;15:237–44.
Qu S, Huang S, Pan X, Yang L, Mei H. Constructing interconsistent, reasonable, and predictive models for both the kinetic and thermodynamic properties of HIV-1 protease inhibitors. J Chem Inf Model. 2016;56(10):2061–8.
Rhoads A, Au KF. PacBio sequencing and its applications. Genom Proteom Bioinform. 2015;13(5):278–89.
Rodgers MA, Wilkinson E, Vallari A, McArthur C, Sthreshley L, Brennan CA, Cloherty G, de Oliveira T. Sensitive next-generation sequencing method reveals deep genetic diversity of HIV-1 in the Democratic Republic of the Congo. J Virol. 2017;91(6):e01841-16.
Roizm B, Pellett EP. The family Herpesviridae: A brief introduction. In: Knipe MD, Howley MP, editors. The fields virology. Philadelphia:Lippincott Williams & Wilkins; 2001. pp. 1929–39.
Salipante SJ, Kawashima T, Rosenthal C, Hoogestraat DR, Cummings LA, Sengupta DJ, Harkins TT, Cookson BT, Hoffman NG. Performance comparison of illumina and ion torrent next-generation sequencing platforms for 16S rRNA-based bacterial community profiling. Appl Environ Microbiol. 2014;80:7583–91.
Sankar S, Ramamurthy M, Nandagopal B, Srikanth P, Venkatraman G, Sridharan G. Molecular and nanotechnologic approaches to etiologic diagnosis of infectious syndromes. Mol Diagn Ther. 2011;15:145–58.
Sauvage V, Eloit M. Viral metagenomics and blood safety. Transfus Clin Biol. 2016;23:28–38.
Seib KL, Dougan G, Rappuoli R. The key role of genomics in modern vaccine and drug design for emerging infectious diseases. PLoS Genet. 2009;5(10):e1000612.
Sokol M, Jessen KM, Pedersen FS. Utility of next-generation RNA-sequencing in identifying chimeric transcription involving human endogenous retroviruses. APMIS. 2016;124:127–39.
Spencer DH, Tyagi M, Vallania F, Bredemeyer AJ, Pfeifer JD, Mitra RD, Duncavage EJ. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J Mol Diagn. 2014;16:75–88.
Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, Mann M, Ingolia NT, Weissman JS. Decoding human cytomegalovirus. Science. 2012;338(6110):1088–93.
Strong MJ, Blanchard E, Lin Z, Morris CA, Baddoo M, Taylor CM, Ware ML, Flemington EK. A comprehensive next generation sequencing-based virome assessment in brain tissue suggests no major virus—tumor association. Acta Neuropathol Commun. 2016;4:71.
Sundaram V, Kowdley K. Management of chronic hepatitis B infection. BMJ. 2015;351:h4263.
Taguwa S, Maringer K, Li X, Bernal-Rubio D, Rauch JN, Gestwicki JE, Andino R, Fernandez-Sesma A, Frydman J. Defining Hsp70 subnetworks in dengue virus replication reveals key vulnerability in flavivirus infection. Cell. 2015;163(5):1108–23.
Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, Larsson E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun. 2013;4:2513.
Thomson E, Ip CL, Badhan A, Christiansen MT, Adamson W, Ansari MA, Bibby D, Breuer J, Brown A, Bowden R, Bryant J, Bonsall D, Da Silva Filipe A, Hinds C, Hudson E, Klenerman P, Lythgow K, Mbisa JL, McLauchlan J, Myers R, Piazza P, Roy S, Trebes A, Sreenu VB, Witteveldt J, STOP-HCV Consortium, Barnes E, Simmonds P. Comparison of next-generation sequencing technologies for comprehensive assessment of full-length hepatitis C viral genomes. J Clin Microbiol. 2016;54:2470–84.
Thorburn F, Bennett S, Modha S, Murdoch D, Gunson R, Murcia PR. The use of next generation sequencing in the diagnosis and typing of respiratory infections. J Clin Virol. 2015;69:96–100.
Van den Hoecke S, Verhelst J, Vuylsteke M, Saelens X. Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing. BMC Genom. 2015;14(16):79.
Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, Lehner PJ, Gygi SP. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell. 2014;157:1460–72.
White MK, Pagano JS, Khalili K. Viruses and human cancers: a long road of discovery of molecular paradigms. Clin Microbiol Rev. 2014;27:463–81.
Xia H, Wang P, Wang GC, Yang J, Sun X, Wu W, Qiu Y, Shu T, Zhao X, Yin L, Qin CF, Hu Y, Zhou X. Human enterovirus nonstructural protein 2CATPase functions as both an RNA helicase and ATP-independent RNA chaperone. PLoS Pathog. 2015;11(7):e1005067.
Yeh CH, Bellon M, Pancewicz-Wojtkiewicz J, Nicot C. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc Natl Acad Sci USA. 2016;113(24):6731–6.
Zhang J, Feng H, Xu S, Feng P. Hijacking GPCRs by viral pathogens and tumor. Biochem Pharmacol. 2016;15(114):69–81.
Zhao J, Liu J, Vemula SV, Lin C, Tan J, Ragupathy V, Wang X, Mbondji-Wonje C, Ye Z, Landry ML, Hewlett I. Sensitive detection and simultaneous discrimination of influenza A and B viruses in nasopharyngeal swabs in a single assay using next-generation sequencing-based diagnostics. PLoS ONE. 2016;11:e0163175.
Zost SJ, Parkhouse K, Gumina ME, Kim K, Diaz Perez S, Wilson PC, Treanor JJ, Sant AJ, Cobey S, Hensley SE. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc Natl Acad Sci USA. 2017; 114(47):12578–83.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ramamurthy, M., Sankar, S., Kannangai, R. et al. Application of viromics: a new approach to the understanding of viral infections in humans. VirusDis. 28, 349–359 (2017). https://doi.org/10.1007/s13337-017-0415-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-017-0415-3