
Abstract
Background
Dropping cost and increasing clinical application of whole genome sequencing (WGS) lead a necessity of efficient (accurate and rapid) variant calling procedures from a personal WGS data (n = 1). A number of variant calling pipelines have been introduced utilizing the human genome reference GRCh38 as a reference and a benchmark dataset called ‘NA12878’, which are both ‘standard’ but limited ethnic origin. Considering the nature of variant calling algorithms and recent updates in sequencing protocol, however, it is necessary to revisit the efficiency of the current best pipelines for a personal WGS data from diverse ethnicity.
Objective
We discuss the most efficient practices for variant calling of a personal WGS reads, with a particular emphasis on whether (1) ethnic match or mismatch between the reference genome and a WGS data produces a distinct result and more importantly (2) there is an ethnic-specific optimal workflow.
Methods
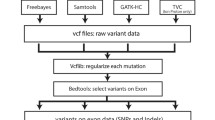
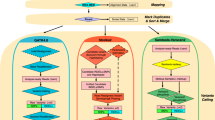
Here, we generate an appropriate WGS data, DNA array, and sufficient number of Sanger validated variants from a single Korean subject to perform such a comprehensive comparison. We applied this WGS reads and the ‘NA12878’ reads to 8 different variant calling pipelines with 2 different reference genomes (GRCh38 and KOREF, a Korean reference genome) to which the WGS reads from different ethnic origins are aligned.
Results
We evaluated the performance of the pipelines with the matched array genotype data and Sanger sequencing validation and demonstrated that: regardless to the ethnic match/mismatch (1) Novoalign-GATK4 showed the most efficient performance with the exceptional calls in MHC region; (2) the overall performance was better with GRCh38, while a significant difference in recall was observed. In addition, we found it is largely reduced computing cost maintaining performance to remove ‘markduplication’ step with PCR-free WGS data.
Conclusion
For variant calling of a personal PCR-free WGS data, regardless of ethnicity consideration, we recommend the use of the Novoalign + GATK4 with GRCh38 and without ‘markduplication’.






Similar content being viewed by others

Data availability
FASTQ file of a Korean subject and the genotype true set list from Sanger validation are available (SRR22944222).
References
Ballouz S, Dobin A, Gillis JA (2019) Is it time to change the reference genome? Genome Biol 20(1):159
Beck TF, Mullikin JC, Program NCS, Biesecker LG (2016) Systematic evaluation of sanger validation of next-generation sequencing variants. Clin Chem 62(4):647–654
Chen J, Li X, Zhong H, Meng Y, Du H (2019) Systematic comparison of germline variant calling pipelines cross multiple next-generation sequencers. Sci Rep 9(1):9345
Cho YS, Kim H, Kim HM, Jho S, Jun J, Lee YJ, Chae KS, Kim CG, Kim S, Eriksson A, Edwards JS, Lee S, Kim BC, Manica A, Oh TK, Church GM, Bhak J (2016) An ethnically relevant consensus Korean reference genome is a step towards personal reference genomes. Nat Commun 7:13637
Cornish A, Guda C (2015) A comparison of variant calling pipelines using genome in a bottle as a reference. Biomed Res Int 2015:456479
Ebbert MT, Wadsworth ME, Staley LA, Hoyt KL, Pickett B, Miller J, Duce J, I Alzheimer’s Disease Neuroimaging, Kauwe JS, Ridge PG (2016) Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinf 17(7):239
Heldenbrand JR, Baheti S, Bockol MA, Drucker TM, Hart SN, Hudson ME, Iyer RK, Kalmbach MT, Kendig KI, Klee EW, Mattson NR, Wieben ED, Wiepert M, Wildman DE, Mainzer LS (2019) Recommendations for performance optimizations when using GATK3.8 and GATK4. BMC Bioinf 20(1):557
Hwang KB, Lee IH, Li H, Won DG, Hernandez-Ferrer C, Negron JA, Kong SW (2019) Comparative analysis of whole-genome sequencing pipelines to minimize false negative findings. Sci Rep 9(1):3219
Hwang S, Kim E, Lee I, Marcotte EM (2015) Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci Rep 5:17875
Illumina (2018) Illumina sequencing platforms
Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, Saunders CT (2018) Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15(8):591–594
Kishikawa T, Momozawa Y, Ozeki T, Mushiroda T, Inohara H, Kamatani Y, Kubo M, Okada Y (2019) Empirical evaluation of variant calling accuracy using ultra-deep whole-genome sequencing data. Sci Rep 9(1):1784
Kumaran M, Subramanian U, Devarajan B (2019) Performance assessment of variant calling pipelines using human whole exome sequencing and simulated data. BMC Bioinf 20(1):342
Li H, Dawood M, Khayat MM, Farek JR, Jhangiani SN, Khan ZM, Mitani T, Coban-Akdemir Z, Lupski JR, Venner E, Posey JE, Sabo A, Gibbs RA (2021) Exome variant discrepancies due to reference-genome differences. Am J Hum Genet 108(7):1239–1250
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760
Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, Pfeifer JD (2013) ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res 41(14):e142
Miller JR, Koren S, Sutton G (2010) Assembly algorithms for next-generation sequencing data. Genomics 95(6):315–327
Momozawa Y, Mizukami K (2021) Unique roles of rare variants in the genetics of complex diseases in humans. J Hum Genet 66(1):11–23
Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, Axelrod N, Busam DA, Strausberg RL, Venter JC (2008) Genetic variation in an individual human exome. PLoS Genet 4(8):e1000160
Poplin R, Chang PC, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, Gross SS, Dorfman L, McLean CY, DePristo MA (2018) A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 36(10):983–987
Souilmi Y, Lancaster AK, Jung JY, Rizzo E, Hawkins JB, Powles R, Amzazi S, Ghazal H, Tonellato PJ, Wall DP (2015) Scalable and cost-effective NGS genotyping in the cloud. BMC Med Genomics 8:64
Supernat A, Vidarsson OV, Steen VM, Stokowy T (2018) Comparison of three variant callers for human whole genome sequencing. Sci Rep 8(1):17851
Suwinski P, Ong C, Ling MHT, Poh YM, Khan AM, Ong HS (2019) Advancing personalized medicine through the application of whole exome sequencing and big data analytics. Front Genet 10:49
Zhang J, Chiodini R, Badr A, Zhang G (2011) The impact of next-generation sequencing on genomics. J Genet Genom 38(3):95–109
Zhao S, Agafonov O, Azab A, Stokowy T, Hovig E (2020) Accuracy and efficiency of germline variant calling pipelines for human genome data. Sci Rep 10(1):20222
Zhao Y, Fang LT, Shen TW, Choudhari S, Talsania K, Chen X, Shetty J, Kriga Y, Tran B, Zhu B, Chen Z, Chen W, Wang C, Jaeger E, Meerzaman D, Lu C, Idler K, Ren L, Zheng Y, Shi L, Petitjean V, Sultan M, Hung T, Peters E, Drabek J, Vojta P, Maestro R, Gasparotto D, Koks S, Reimann E, Scherer A, Nordlund J, Liljedahl U, Foox J, Mason CE, Xiao C, Hong H, Xiao W (2021) Whole genome and exome sequencing reference datasets from a multi-center and cross-platform benchmark study. Sci Data 8(1):296
Acknowledgements
We would like to thank to gimLAB members for the insightful discussions and Editage (www.editage.co.kr) for English language editing. This research was supported by the Korea Brain Research Institute basic research program funded by the Ministry of Science and ICT (22-BR-03-05) and the Healthcare AI Convergence Research & Development Program through the National IT Industry Promotion Agency of Korea (NIPA) funded by the Ministry of Science and ICT (No.1711120216) from the Republic of Korea. This research was also supported by U01-AG062602, funded by NIA NIH HHS from the United States.
Author information
Authors and Affiliations
Contributions
HSP performed all the analyses and wrote the manuscript. JSG designed the work and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Park, H., Gim, J. A comparative investigation of single nucleotide variant calling for a personal non-Caucasian sequencing sample. Genes Genom 45, 1527–1536 (2023). https://doi.org/10.1007/s13258-023-01439-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-023-01439-w