
Abstract
Ultrametric spaces are widely used to depict evolutionary times in phylogenetic trees since they assume that every population/species is located at the tips of bifurcating branches of the same length. The discrete branching of ultrametric trees permits the measurement of distances between pairs of individuals that are proportional to their divergence time. Here the traditional ultrametric concept of bifurcating and divergent phylogenetic tree is overturned and a new type of non-ultrametric diagram is introduced. The objective of this study is the description of gene flows in branching species/populations in terms of converging trees instead of bifurcating trees. To provide an operational example, the paleoanthropological issue of the date of Neanderthal genome’s introgression in non-African humans is examined. Neanderthals and ancient humans are not anymore two species that exchange chunks of DNA, rather become a single, novel cluster of extant hominins that must be considered by itself. The novel converging, non-ultrametric phylogenetic trees permit the calibration of molecular clocks with a twofold benefit. When the date of the branching of two population/species from a common ancestor is known, the novel approach allows to calculate the time of subsequent introgressions. On the contrary, when the date of the introgression between two population/species is known, the novel approach allows to detect the time of their previous branching from a common ancestor.
Similar content being viewed by others

Introduction
Phylogenetic analysis protocols entail various steps, from the identification of sets of homologous sequences to the download of the achieved sequences, ending up with the production of a phylogenetic tree (PT) inferred from the aligned sequences (Soares et al., 2017; Tamura et al., 2011). PTs depict morphological/genetic evolutionary relationships among populations or species (Woese, 2000). Unlike other evolutionary diagrams such as additive trees and cladograms, the PT abscissa describes the amount of change of a given character that occurred over time, while the ordinate describes the evolutionary time. PT makes use of bifurcating branches in which the tips stand for groups of descendants’ populations/species and the nodes for their common ancestors. The branch lengths are proportional to the inferred evolutionary distance among the specimens under investigation (Hall, 2013). Rooted PT of clustering sequences is generally set in ultrametric spaces, i.e. non-Euclidean spaces in which all the tips are equidistant from the root. To provide an example, the widely used UPGMA and WPGMA methods (i.e. unweighted and weighted pair group methods with arithmetic mean) generate ultrametric trees characterized by the constant rate assumption that the distances from the root to every branch tip are equal (Sokal & Michener, 1958). Therefore, ultrametric trees are very useful to describe a peculiar aspect of the evolutionary history, i.e. the divergence time among populations/species (Page & Holmes, 1998).
Here an alternative approach to ultrametric trees, i.e. a novel non-ultrametric phylogenetic tree (NUPT) is proposed which allows the description of gene flows in terms of convergent instead of divergent branches. The model is illustrated and its theoretical advantages and operational implications are discussed, providing an example drawn from the paleoanthropological issue of ancient hominins’ DNA introgression in Late Pleistocene Homo sapiens. Putative gene flows from archaic hominins to Homo sapiens suggest a very intricate evolutionary history characterized by a single of multiple episodes of gene flow between populations/species (Prüfer et al., 2017). For example, genome sequencing of various hominins’ specimens points towards gene flows among Neanderthals, Denisovans, unknown archaic groups and early modern humans (Hajdinjak et al., 2021; Meyer et al., 2012; Prüfer et al., 2014; Reich et al., 2010).
The focus here is on the genetic relationships between Neanderthals and modern humans. The inferred timing of their last common ancestor is a hotly debated matter since small variations in the weighed parameters lead to very different estimates of their divergence time (Meyer et al., 2016). Mixed results from studies of cranial variation, ancient DNA, human mutation rate, mitochondrial sequence comparisons and average value of linkage disequilibrium suggest that the lineages leading to modern humans and Neanderthals diverged between 300,000 and 600,000 years ago, with little subsequent admixture (Krings et al., 1997; Noonan et al., 2006; Serre et al., 2004). More radical suggestions date back the last common ancestor to pre-800,000 years ago (Gómez-Robles A, 2019). It has been suggested that an introgression from Neanderthals to non-African human ancestors occurred 37,000–86,000 years ago, most likely between 47,000 and 65,000 years ago (Posth et al., 2017; Sankararaman et al., 2012). In the sequel, it is made clear how and why the novel NUPT approach sheds new light on the times of divergence and introgression of Neanderthals and ancient non-African human populations.
Materials and methods
Ultrametric diverging trees: what, when and why
Ultrametric trees rely on the mathematical concept of ultrametricity, which is closely related to the concept of triangle inequality. In plane geometry, triangle inequality states that the length of every side of the triangle is lower than or equal to the sum of the other two, such that:d (x,z) ≤ d (x,y) + d (y,z)where x, y and z are the three vertices of a triangle and d is the distance between every pair of vertices (Fig. 1A). In this case, the triangle lies in the classical Euclidean space.

Composition and arrangement of ultrametric spaces. A According to the standard Euclidean geometry, triangle inequality means that every side of a triangle cannot be longer than the sum of the other two. B In the non-Euclidean case, strong triangle inequality comes into play so that just triangles with two or three sides of the same length are allowed. C An ultrametric tree is shaped as a triangle grid generated by the steps among x, y and z. Note that the distances between the branches are accurately preserved. The points x, y and z may stand not just for the vertices of equilateral or isosceles triangles, but also for features such as biological taxa, populations and species. D Regular branching tree of a translational invariant ultrametric space. The population/species are represented by the yellow balls at the tips of the distal branches, while the distances among the population/species are provided by the tree itself, according to the hierarchy of the balls’ sets and subsets. E Phylogenetic ultrametric tree of genetic flows between Neanderthals and ancient non-African humans (termed “humans” for sake of clarity). The split and introgression dates (500,000 and 60,000 years ago, respectively) are tentative and based on the current literature. If the two available sets are deemed to be separated, they can be located inside a diverging branching tree. F New NUPT of genetic flows between Neanderthals and non-African humans. In this case, the two sets partially superimpose, giving rise to phylogenetic trees that are convergent instead of bifurcating. Therefore, a large set of modern extant hominins is achieved which encompasses as subsets both introgressed Neanderthals and ancient humans
Yet, there exist a strongest version of triangle inequality, such that:d (x,z) ≤ max [d (x,y), (y,z)]
In plain words, the rule of strong triangle inequality suggests the sole occurrence of peculiar triangles, i.e. equilateral or isosceles with the basis smaller than the other two sides (Fig. 1B). Those triangles do not lie in Euclidean space, rather in a space termed ultrametric. This non-Euclidean space can be depicted either as a triangular grid where the distances are always preserved (Fig. 1C), or as a tree-like hierarchy of triangles/balls (Fig. 1D).
The ultrametric assumption requires that a tree must display the following unusual properties:
-
1)
The ultrametric distances are described by steps that are unavoidably discrete: continuum is not allowed.
-
2)
The ultrametric-based clocks rely on the assumption that the temporal distance always corresponds to the total time multiplied by a constant.
-
3)
Different ultrametric balls do not have points in common. This leads to counterintuitive outcomes:
-
4)
In an ultrametric space, two balls cannot overlap.
-
5)
In an ultrametric space, two balls always keep the same spacing and fixed distance.
Scientists use DNA sequence data gathered from hominin samples to test models of archaic admixture such as, in our case, the Neanderthal gene flow in modern non-African humans. To evaluate recombination events and build PT, scientists infer the date of the last genetic exchange between the ancestral populations/species by measuring the extent of admixture linkage disequilibrium (Sankararaman et al., 2016). A widely used procedure for dating gene flow in target populations/species evaluates a set of single nucleotide polymorphisms at which a single randomly chosen allele is derived relative to an ancestor (Sankararaman et al., 2016; Wall, 2000). Traditionally, scientists have preferred ultrametric trees for ancestral state reconstruction and phylogenetic inference because of their unvaluable practical advantages. Discrete branchings of ultrametric trees are characterized by a distance between any pair of (modern) sequences that is plainly proportional to their divergence time (Gavryushkin et al., 2016). In ultrametric trees, the amount of change in living populations/species is related to the amount of time (Cusimano & Renner, 2014). This allows the building of molecular clocks able to correlate phenotypic evolution and genomic/molecular/protein sequence data with branch lengths. Indeed, the molecular clock assumption suggests that the mutation rates are always constant on all the branches. This means that the mutation distances are proportional to the divergence time and equal between any pair of modern sequences and their most recent common ancestor (Page & Holmes, 1998).
Ultrametric trees have been used to build up PTs of gene flows among disparate hominins. Several tasks have been accomplished such as the genome sequence of Neanderthals from the Altai Mountains and Vindija Cave (Prüfer et al., 2017), the putative date of interbreeding between Neanderthals and modern humans (Sankararaman et al., 2012), the Neanderthal ancestry in Initial Upper Palaeolithic European humans (Hajdinjak et al., 2021), the Denisovan and Neanderthal gene flow in Icelandic genomes (Skov et al., 2020), the ancient gene flow from early modern humans into Eastern Neanderthals (Kuhlwilm et al., 2016) and the genetic evidence for archaic admixture in Africa (Hammer et al., 2011). It is noteworthy that the statistic across pairs of introgressed alleles is expected to have an exponential decay with genetic distance since linkage disequilibrium decays at a constant rate per generation. This means that recombination events are expected to break down the genetic segments shared by different populations/species such as modern humans and Neanderthals (Sankararaman et al., 2012).
Even though ultrametric trees are favoured by scientists to ascertain gene flows between populations/species, they have some limitations. Distances between populations/species have been successfully used to shed light into their evolutionary past (Zhivotovsky, 2001). The resulting scenarios are generally understood to be a snapshot of the data, in the sense that they can be used as a starting point to guide exploration of a dataset. However, only in rare cases are the distances assumed to have arisen under a molecular clock (i.e. to satisfy the ultrametric property). The molecular clock assumption is generally expected to hold below the species level, which is the reason why tools like UPGMA have been applied successfully in these types of studies. In general, trees that do not have the same amount of time between the tips and the roots and cannot be regarded as being properly ultrametric, unless the cases in which evolution is strictly proportional to time. Indeed, the rates of neutral evolution on large time scales are neither constant nor are they a function of mutation alone. Since human evolution, including that of the fossil populations from which genomes may be recovered, is far from mutation drift equilibrium, variation in population size through time and across populations can be expected. Moreover, modifications in the population size change the degree to which mildly deleterious and mildly fitness-enhancing mutations may behave as though they are neutral.
Towards a novel converging tree
Given these premises, we suggest a non-ultrametric alternative to the widespread ultrametric trees, i.e. a novel PT able to assess recombination events in evolutionary times. Ultrametric trees entail the philosophical and methodological implication that PTs are bifurcating. Every branch leads to a single population/species that is kept conceptually distinct from the others, even in the event of introgressive episodes (Fig. 1E). Therefore, in ultrametric trees, the tips (i.e. the population or the species under assessment) shall be regarded as separated sets. A variant of PT is proposed here in which the ultrametric assumption is partially lessened. In the novel framework, the two population/species undergoing introgression give rise to a SINGLE population, so that the branching tree tends to converge in a single cluster, rather than bifurcating in two smaller clusters (Fig. 1F). In mathematical terms, the non-overlapping sets located at the numerous tips of bifurcating ultrametric trees become overlapping subsets located at the unique tip of convergent NUPT.
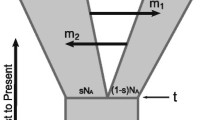
To describe NUPT characterized by converging instead of bifurcating trees, a novel type of diagram is operationally required for gene flows calculation. The diagram for the assessment the novel NUPT approach consists of a square. A square has been chosen since all the sides and the angles must be identical both to preserve translational symmetries and to avoid non-replicable distortions due to confounding factors such as geometric transformations, projections, displacement, shift operators, affine connexions and parallel transport. The diagram, illustrated in Fig. 2, describes the relationships between two branching population/species, termed Ancient Sample I and Ancient Sample II. The grid on the left portrays a temporal clock with time running from the past (top) to the present-day (bottom). The left and the right sides of the square represent, respectively, the branching of the Ancient Sample I and of the Ancient Sample II after the split from a common ancestor. The upper side of the square is calibrated on the supposed dating of the Ancient Samples I and II’s bifurcation from a common ancestor. The lower side of the square describes the merging of the Ancient Samples I and II’s DNAs in the extant population/species. The lower side of the square encompasses a percent scale from 0 to 100% which measures both the amount of Ancient Sample I’s DNA still preserved in the current population/species and the amount of Ancient Sample II’s DNA still preserved in the current population/species.

Novel non-ultrametric approach for detection of genetic flows timing. After the split (upper side of the square) at a given time in the past, two branches are generated: the branch of the Ancient Sample I (left side) and the branch of the Ancient Sample II (right side). When the Ancient Sample II’s DNA is introgressed by the Ancient Sample I’s DNA (or vice versa), the final result is a single modern population/species containing percentages of both the genetic materials. To provide an example, the figure suggests that in the lower side of the square the 65% of the single modern population’s DNA comes from the Ancient Sample II, while the remaining 35% from the Ancient Sample I. The arrow from the upper right vertex to the lower side of the square gives rise to the angle β. The two numbered yellow circles illustrate the two steps of the procedure described in the main text
The procedure requires two further steps, illustrated by the two numbered yellow circles in Fig. 2.
-
1)
The first step consists of drawing a line from the right upper vertex of the Ancient Sample II’s right side of the square to a percent value of the lower side. This percent value stands for the estimated percentage of the Ancient Sample’s II DNA still preserved in the modern population/species. The operation of drawing the line leads to the formation of the angle β.
-
2)
The second step consists of translating the angle β to the lower part of the diagram, with one of the vertices located at the chosen percent value. A triangle is formed such that one of its sides meets the Ancient Sample I’s left side of the square at one point. This point corresponds to the date of the Ancient Sample I’s DNA introgression in the Ancient Sample II’s DNA.
In the next section, we will examine the specific procedure related with Neanderthals and ancient non-African humans relationships.
Results
Two divergent population/species of Neanderthals and non-African archaic humans branching from a common ancestor are located on the left and the right sides of the square, respectively. The calibration of the temporal clock portrayed on the left grid may vary according to the available estimates. Two possible dates for the split between Neanderthals and ancient non-African humans, namely 400,000 and 600,000 years ago, are provided by Fig. 3A and B, respectively. After the split (upper side of the square) at a given time in the past, two branches were generated: the branch of the Neanderthals (left side) and the branch of the ancient non-African humans (right side).

Neanderthals (red square termed N) and ancient non-African humans (blue square termed H) branching from a common ancestor. Two scenarios are illustrated, each of them relying on a square with distinct molecular clock calibration. Our method suggests what follows: if the split took place 400,000 years ago, the introgression occurred about 40,000 years ago (A); if the split took place 600,000 years ago, the introgression occurred about 75,000 years ago (B)
The percent scale in the lower side of the square quantifies the percentage amount of ancient human’s DNA and Neanderthal’s DNA preserved in the current non-African human populations. Therefore, the first step consists of drawing a line from the right upper vertex of the ancient humans to the 96% value on the lower side. Indeed, estimates suggest that the modern population of non-Africans preserves about 96% of archaic human ancestors’ DNA, while the 4% is provided by the Neanderthal’s DNA introgression. The arrow from the upper right vertex to the lower side of the square gives rise to the angle β, which stands for the current non-African humans’ genomic divergence after the Neanderthals/ancient non-African humans split. The second step consists of translating the angle β to the lower side of the square, with one of the vertices located at the value of 96%. One of the sides of the triangle formed by the angle β meets the Neanderthal’s left side of the square in a point corresponding to the date of the Neanderthal’s DNA introgression in the archaic humans.
If the split between Neanderthals and archaic humans is estimated at 400,000 years ago, the Neanderthal introgression took place about 40,000 years ago (Fig. 3A). As an alternative, if the split between Neanderthals and archaic humans is estimated at 600,000 years ago, the Neanderthal introgression took place about 75,000 years ago (Fig. 3A).
These findings lend themselves to manifold conclusions that will be tackled in the next section.
Conclusions
We propose a new operational approach to phylogenetic trees, termed NUPT, able to estimate the date of introgression between two branching populations or species, e.g. Neanderthals and ancient non-African humans. NUPT is an extension of traditional clustering methods in phylogenetics and provides a useful resource in a scientific stage where there is increasing amounts of evidence for introgression or reticulate evolution (Garrick et al., 2019). In particular, our primary purpose was to calculate the time of Neanderthal’s DNA introgression in modern non-African humans. Our assumption is straightforward: when the ancient human DNA was introgressed by Neanderthal DNA, the end result is a converging, single modern population containing both the genetic materials.
Our square allows the graphic depiction of many parameters related to the genetic dynamics occurring between two populations or species. For example, the estimates suggesting that about 96% of the single modern population’s DNA comes from ancient humans, while about 4% from Neanderthals, can be graphically depicted in the lower side of our square by drawing just a few lines. A low amount of genetic divergence between two species that subsequently tend to converge suggests that their merging occurred very later after their original split. Such amount of genetic divergence can be graphically depicted by the angle β, which is correlated with the current population/species’ genomic divergence occurred after the split between the two populations/species. The lower the angle β, the closer to the current-day the introgression took place.
The temporal grid can be calibrated in different ways according to the various estimated dates of the original split between Neanderthals and ancient non-African humans. Our method accomplishes a valuable result, i.e. the different calibration of the molecular clock leads to different temporal estimates of the introgressive event. The date of introgression modifies according to the choice of the temporal grid: if the split between Neanderthals and ancient non-African humans took place 400,000 years ago, the introgression occurred about 40,000 years ago; if the split took place 600,000 years ago, the introgression occurred about 75,000 years ago.
Our novel NUPT approach has another twofold virtue. If the alleged date of the bifurcation between two population/species from a common ancestor is known, the method allows to calculate the date of their subsequent introgression. In turn, if the date of the introgression between two population/species is known, the method allows to detect the date of their previous split from a common ancestor. Researchers need simply to change the order of the operations one and two on the square: the angle β is drawn at first of the lower side of the square, then it is translated to the left side of the square. In this case, the upper side of the square is not calibrated on the supposed dating of the Ancient Samples I and II’s bifurcation from a common ancestor. The vertex of the achieved angle will meet the left side of the square in a point corresponding to the dating of the split between the two populations/species.
Discussion
In this study, a new approach for estimating rooted ultrametric, non-fully bifurcating phylogenetic trees is anticipated. The premise here is that the widely used ultrametric trees do not describe exactly the state of matter (Balaban et al., 2019) since they favour the description of the differences between population/species, instead of their similarities. In turn, our novel NUPT approach considers the clustering of populations/species instead of their distinctions. Starting from two overlapping sets with a few features in common, a coalescent model of phylogenetic tree can be built that is characterized by converging branches. The paper is concerned with the problem of introgression between two species. As such, we aim to provide a novel method to overcome the issue that distances used for reconstructing the evolutionary past between species need not be ultrametric. In particular, a simple procedure to build non-ultrametric phylogenetic trees is proposed to calculate the hypothetical date of interbreeding among different populations/species.
In this paper, we focus on the instance of Neanderthal’s DNA introgression in modern non-African humans. Nevertheless, the novel procedure can be generalized to all the cases of genetic recombination between populations/species, ranging from introgression to hybridization, from reticulation to repeated lineage splitting and lineage fusion, i.e. the complete merging of two or more populations resulting in a single panmictic group (Pickrell and Pritchard, 2012; Garrick et al., 2014; Hunley et al., 2016; Garrick et al., 2020). NUPT allows the prediction of the branching date between two populations/species when the amount of genetic material’s introgression is known. This could be useful for the assessment of far-flung issues, such as interbreeding of wild and domestic animal populations, creation and fitness of new artificial varieties of hybrid plants and the study of the evolution of SARS-Cov-2 after the conjectured split of common coronavirus ancestors from bat and pangolin strains (Touati et al., 2020).
NUPT has limitations. The use of sequence divergence to date the nodes in non-ultrametric trees of different hominins groups (see Prüfer et al., 2017) is complicated by mixture among lineages. When the novel population is the product of merging between groups that have been evolving independently for a while, phylogenetic trees will sample sequences from both lineages and will make the population resemble, without corrections for admixture, the root state more than it would if it had continued evolving at a rate proportional to time.
Furthermore, the NUPT assumption cannot be fully consistent in two cases:
-
1)
When the conversion of ancestral variation among-group is achieved via random genetic drift occurring at different rates.
-
2)
When sequences that were polymorphic in the ancestral population subsequently segregate in the two branched lineages and in the admixed population.
Nevertheless, leaving apart the abovementioned limitations, NUPT leads to intriguing theoretical consequences. The hot disputes between the Out-Of-Africa and multiregional evolution models of human evolution in the Pleistocene (Wolpoff et al., 2000; Bräuer et al., 2004; Groucutt et al., 2015; Montinaro et al., 2021) can be tackled in terms of ancient populations, instead of species. These populations are able to diverge, converge and clustering in a worldwide network of genic exchanges that leads to “modern humans”, i.e. individuals characterized by local genetic differences.
Data availability
All data generated or analyzed during this study are included in the manuscript.
References
Balaban, M., Moshiri, N., Mai, U., Jia, X., & Mirarab, S. (2019). TreeCluster: Clustering biological sequences using phylogenetic trees. PLoS ONE, 14(8), e0221068. https://doi.org/10.1371/journal.pone.0221068
Bräuer, G., Collard, M., & Stringer, C. (2004). On the reliability of recent tests of the Out of Africa hypothesis for modern human origins. The Anatomical Record. Part A, Discoveries in Molecular, Cellular, and Evolutionary Biology, 279(2), 701–7. https://doi.org/10.1002/ar.a.20064
Cusimano, N., & Renner, S. S. (2014). Ultrametric trees or phylograms for ancestral state reconstruction: Does it matter? Taxon. 63(4), 721–726. https://www.jstor.org/stable/taxon.63.4.721
Garrick, R. C., Benavides, E., Russello, M. A., Hyseni, C., & Edwards, D. L. (2014). Lineage fusion in Galápagos giant tortoises. Molecular Ecology, 23(21), 5276–90. https://doi.org/10.1111/mec.12919
Garrick, R. C., Banusiewicz, J. D., Burgess, S., Hyseni, C., & Symula, R. E. (2019). Extending phylogeography to account for lineage fusion. Journal of Biogeography, 46(2), 268–278. https://doi.org/10.1111/jbi.13503
Garrick, R. C., Hyseni, C., & Arantes, I. C. (2020). Efficient summary statistics for detecting lineage fusion from phylogeographic datasets. Journal of Biogeography, 47(10), 2129–2140. https://doi.org/10.1111/jbi.13932
Gavryushkin, A., & Drummond, A. J. (2016). The space of ultrametric phylogenetic trees. Journal of Theoretical Biology, 403, 197–208. https://doi.org/10.1016/j.jtbi.2016.05.001
Gómez-Robles A. (2019). Dental evolutionary rates and its implications for the Neanderthal–modern human divergence. Science Advances, 5(5). https://doi.org/10.1126/sciadv.aaw1268
Groucutt, H. S., Petraglia, M. D., Bailey, G., Scerri, E. M. L., & Parton, A. (2015). Rethinking the dispersal of Homo sapiens out of Africa. Evolutionary Anthropology, 24(4), 149–164. https://doi.org/10.1002/evan.21455
Hajdinjak, M., Mafessoni, F., Skov, L., Vernot, B., & Hübner, A. (2021). Initial Upper Palaeolithic humans in Europe had recent Neanderthal ancestry. Nature, 592, 253–257.
Hall, B. G. (2013). Building phylogenetic trees from molecular data with MEGA. 2013. Molecular Biology and Evolution, 30(5), 1229–1235. https://doi.org/10.1093/molbev/mst012
Hammer, M. F., Woerner, A. E., Mendez, F. L., Watkins, J. C., & Wall, J. D. (2011). Genetic evidence for archaic admixture in Africa. PNAS, 108(37), 15123–15128. https://doi.org/10.1073/pnas.1109300108
Hunley, K. L., Cabana, G. S., & Long, J. C. (2016). The apportionment of human diversity revisited. American Journal of Physical Anthropology, 160(4), 561–569.
Krings, M., Stone, A., Schmitz, R. W., Krainitzki, H., & Stoneking, M. (1997). Neanderthal DNA sequences and the origin of modern humans. Cell, 90(1), 19–30. https://doi.org/10.1016/S0092-8674(00)80310-4
Kuhlwilm, M., Gronau, I., Hubisz, M. J., de Filippo, C., & Prado-Martinez, J. (2016). Ancient gene flow from early modern humans into Eastern Neanderthals. Nature, 530(7591), 429–433. https://doi.org/10.1038/nature16544
Meyer, M., Arsuaga, J.-L., Nagel, S., Aximu-Petri, A., & Martínez, I. (2016). Nuclear DNA sequences from the Middle Pleistocene Sima de los Huesos hominins. Nature, 531, 504–507.
Meyer, M., Kircher, M., Gansauge, M.-T., Li, H., & Racimo, F. (2012). A high-coverage genome sequence from an archaic Denisovan individual. Science, 338(6104), 222–226.
Montinaro, F., Pankratov, V., Yelmen, B., Pagani, L., & Mondal, M. (2021). Revisiting the out of Africa event with a deep-learning approach. American Journal of Human Genetics, 108(11), 2037–2051. https://doi.org/10.1016/j.ajhg.2021.09.006
Noonan, J. P., Coop, G., Kudaravalli, S., Smith, D., Krause, J., Alessi, J., Chen, F., Platt, D., Pääbo, S., & Pritchard, J. K. (2006). Sequencing and analysis of Neanderthal genomic DNA. Science, 314, 1113–1118.
Page, R., & Holmes, E. (1998). Molecular evolution: A phylogenetic approach. Blackwell Science ISBN 0–86542–889–1.
Pickrell, J., & Pritchard. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. Nature Precedings, 1–1
Posth, C., Wißing, C., Kitagawa, K., Pagani, L., & van Holstein, L. (2017). Deeply divergent archaic mitochondrial genome provides lower time boundary for African gene flow into Neanderthals. Nature Communications, 8, Article number: 16046.
Prüfer, K., de Filippo, C., Grote, S., Mafessoni, F., & Korlević, P. (2017). A high-coverage Neanderthal genome from Vindija Cave in Croatia. Science, 358(6363), 655–658. https://doi.org/10.1126/science.aao1887
Prüfer, K., Racimo, F., & Patterson, N. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature, 505, 43–49. https://doi.org/10.1038/nature12886
Reich, D., Green, R., Kircher, M., Krause, J., & Patterson, N. (2010). Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature, 468, 1053–1060. https://doi.org/10.1038/nature09710
Sankararaman S, Mallick, S., Patterson, N., & Reich, D. (2016). The combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Current Biology, 26(9), 1241–1247. Published online 2016 Mar 28. https://doi.org/10.1016/j.cub.2016.03.037
Sankararaman, S., Patterson, N., Li, H., Pääbo, S., & Reich, D. (2012). The date of interbreeding between Neanderthals and modern humans. PLOS Genetics. https://doi.org/10.1371/journal.pgen.1002947
Serre, D., Langaney, A., Chech, M., Teschler-Nicola, M., Paunovic, M., Mennecier, P., Hofreiter, M., Possnert, G., & Pääbo, S. (2004). No evidence of Neanderthal mtDNA contribution to early modern humans. PLoS Biology, 2, e57. https://doi.org/10.1371/journal.pbio.0020057
Skov, L., Coll Macià, M., Sveinbjörnsson, G., et al. (2020). The nature of Neanderthal introgression revealed by 27,566 Icelandic genomes. Nature, 582, 78–83.
Soares, A., Râbelo, R., & Delbem, A. (2017). Optimization based on phylogram analysis. Expert Systems with Applications, 78, 32–50. https://doi.org/10.1016/j.eswa.2017.02.012
Sokal, R., & Michener, C. (1958). A statistical method for evaluating systematic relationships. University of Kansas Science Bulletin, 38, 1409–1438.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Touati, R., Haddad-Boubaker, S., Ferchichi, I., Messaoudi, I., Elloumi, O., A., et al. (2020). Comparative genomic signature representations of the emerging COVID-19 coronavirus and other coronaviruses: High identity and possible recombination between Bat and Pangolin coronaviruses. Genomics, 112(6), 4189–4202. https://doi.org/10.1016/j.ygeno.2020.07.003
Wall, J. D. (2000). Detecting ancient admixture in humans using sequence polymorphism data. Genetics, 154(3), 1271–1279.
Woese, C. R. (2000). Interpreting the universal phylogenetic tree. PNAS, 97(15), 8392–8396. https://doi.org/10.1073/pnas.97.15.8392
Wolpoff, M. H., Hawks, J., & Caspari, R. (2000). Multiregional, not multiple origins. American Journal of Physical Anthropology, 112(1), 129–136. https://doi.org/10.1002/(SICI)1096-8644(200005)112:1%3c129::AID-AJPA11%3e3.0.CO;2-K
Zhivotovsky, L. A. (2001). Estimating divergence time with the use of microsatellite genetic distances: Impacts of population growth and gene flow. Molecular Biology and Evolution, 18(5), 700–709.
Acknowledgements
The author would like to thank his intellectual brother Norbert Jausovec, who gave precious suggestions and instructive insights. Norbert, sadly, left us a few months ago.
Author information
Authors and Affiliations
Contributions
The author performed: study concept and design, acquisition of data, analysis and interpretation of data, drafting of the manuscript, critical revision of the manuscript for important intellectual content, statistical analysis, obtained funding, administrative, technical, material support and study supervision.
Corresponding author
Ethics declarations
Ethics statement
This research does not contain any studies with human participants or animals performed by the Author.
Conflict of interest
The authors declare no competing interests.
Disclaimer
The author had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. The author warrants that the article is original, does not infringe on any copyright or other proprietary right of any third part, is not under consideration by another journal and has not been previously published.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tozzi, A. Non-ultrametric phylogenetic trees shed new light on Neanderthal introgression. Org Divers Evol 23, 1013–1021 (2023). https://doi.org/10.1007/s13127-023-00613-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-023-00613-y