
Abstract
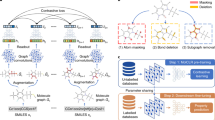
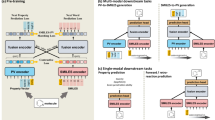
Molecular representation learning can preserve meaningful molecular structures as embedding vectors, which is a necessary prerequisite for molecular property prediction. Yet, learning how to accurately represent molecules remains challenging. Previous approaches to learning molecular representations in an end-to-end manner potentially suffered information loss while neglecting the utilization of molecular generative representations. To obtain rich molecular feature information, the pre-training molecular representation model utilized different molecular representations to reduce information loss caused by a single molecular representation. Therefore, we provide the MVGC, a unique multi-view generative contrastive learning pre-training model. Our pre-training framework specifically acquires knowledge of three fundamental feature representations of molecules and effectively integrates them to predict molecular properties on benchmark datasets. Comprehensive experiments on seven classification tasks and three regression tasks demonstrate that our proposed MVGC model surpasses the majority of state-of-the-art approaches. Moreover, we explore the potential of the MVGC model to learn the representation of molecules with chemical significance.
Graphical Abstract







Similar content being viewed by others

Data Availability
The benchmark datasets for molecular property prediction: https://moleculenet.org/.
References
Scalia G, Grambow CA, Pernici B et al (2020) Evaluating scalable uncertainty estimation methods for deep learning-based molecular property prediction. J Chem Inf Model 60(6):2697–2717. https://doi.org/10.1021/acs.jcim.9b00975
Walters WP, Barzilay R (2020) Applications of deep learning in molecule generation and molecular property prediction. Acc Chem Res 54(2):263–270. https://doi.org/10.1021/acs.accounts.0c00699
Xiong Z, Wang D, Liu X et al (2019) Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J Med Chem 63(16):8749–8760. https://doi.org/10.1021/acs.jmedchem.9b00959
Gilmer J, Schoenholz SS, Riley PF et al (2017) Neural message passing for quantum chemistry. arXiv. https://doi.org/10.48550/arXiv.1704.01212
Velickovic P, Cucurull G, Casanova A et al (2018) Graph attention networks. arXiv. https://doi.org/10.48550/arXiv.1710.10903
Kipf TN, Welling M (2016) Variational graph auto-encoders. arXiv. https://doi.org/10.48550/arXiv.1611.07308
Guo Z, Yu W, Zhang C et al (2020) GraSeq: graph and sequence fusion learning for molecular property prediction. In: Proceedings of the 29th ACM international conference on information & knowledge management, pp 435–443. https://doi.org/10.1145/3340531.3411981
Jin W, Coley C, Barzilay R et al (2017) Predicting organic reaction outcomes with Weisfeiler–Lehman network. arXiv. https://doi.org/10.48550/arXiv.1709.04555
Do K, Tran T, Venkatesh S (2019) Graph transformation policy network for chemical reaction prediction. In: Proceedings of the 25th ACM SIGKDD international conference on knowledge discovery & data mining, pp 750–760. https://doi.org/10.1145/3292500.3330958
Jin W, Barzilay R, Jaakkola T (2018) Junction tree variational autoencoder for molecular graph generation. arXiv. https://doi.org/10.48550/arXiv.1802.04364
Jin W, Barzilay R, Jaakkola T (2020) Hierarchical generation of molecular graphs using structural motifs. arXiv. https://doi.org/10.48550/arXiv.2002.03230
Du Y, Fu T, Sun J et al (2022) Molgensurvey: a systematic survey in machine learning models for molecule design. arXiv. https://doi.org/10.48550/arXiv.2203.14500
Zhu X, Vondrick C, Fowlkes CC et al (2016) Do we need more training data? Int J Comput Vis 119:76–92. https://doi.org/10.1007/s11263-015-0812-2
Hestness J, Narang S, Ardalani N et al (2017) Deep learning scaling is predictable, empirically. arXiv. https://doi.org/10.48550/arXiv.1712.00409
Brown N, Fiscato M, Segler MH et al (2019) GuacaMol: benchmarking models for de novo molecular design. J Chem Inf Model 59(3):1096–1108. https://doi.org/10.1021/acs.jcim.8b00839
Sagawa S, Raghunathan A, Koh PW et al (2020) An investigation of why overparameterization exacerbates spurious correlations. arXiv. https://doi.org/10.48550/arXiv.2005.04345
Gaulton A, Bellis LJ, Bento AP et al (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic acids Res 40(D1):D1100–D1107. https://doi.org/10.1093/nar/gkr777
Sterling T, Irwin JJ (2015) ZINC 15-ligand discovery for everyone. J Chem Inf Model 55(11):2324–2337. https://doi.org/10.1021/acs.jcim.5b00559
Nakata M, Shimazaki T (2017) PubChemQC project: a large-scale first-principles electronic structure database for data-driven chemistry. J Chem Inf Model 57(6):1300–1308. https://doi.org/10.1021/acs.jcim.7b00083
Hu W, Liu B, Gomes J et al (2019) Strategies for pre-training graph neural networks. arXiv. https://doi.org/10.48550/arXiv.1905.12265
You Y, Chen T, Sui Y et al (2020) Graph contrastive learning with augmentations. arXiv. https://doi.org/10.48550/arXiv.2010.13902
Feng S, Ni Y, Lan Y et al (2023) Fractional denoising for 3d molecular pre-training. arXiv. https://doi.org/10.48550/arXiv.2307.10683
Liu S, Wang H, Liu W et al (2021) Pre-training molecular graph representation with 3d geometry. arXiv. https://doi.org/10.48550/arXiv.2110.07728
Jing L, Tian Y (2020) Self-supervised visual feature learning with deep neural networks: a survey. IEEE Trans Pattern Anal Mach Intell 43(11):4037–4058. https://doi.org/10.1109/TPAMI.2020.2992393
Stärk H, Beaini D, Corso G et al (2022) 3d infomax improves GNNS for molecular property prediction. arXiv. https://doi.org/10.48550/arXiv.2110.04126
Weininger D (1988) SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 28(1):31–36. https://doi.org/10.1021/ci00057a005
Bengio Y, Ducharme R, Vincent P (2000) A neural probabilistic language model. NeurIPS. https://doi.org/10.5555/944919.944966
Oliveira AF, Da Silva JL, Quiles MG (2022) Molecular property prediction and molecular design using a supervised grammar variational autoencoder. J Chem Inf Model 62(4):817–828. https://doi.org/10.1021/acs.jcim.1c01573
Wang S, Guo Y, Wang Y (2019) Smiles-bert: large scale unsupervised pre-training for molecular property prediction. In: Proceedings of the 10th ACM international conference on bioinformatics, computational biology and health informatics, pp 429–436. https://doi.org/10.1145/3307339.3342186
Chithrananda S, Grand G, Ramsundar B (2020) ChemBERTa: large-scale self-supervised pretraining for molecular property prediction. arXiv. https://doi.org/10.48550/arXiv.2010.09885
Kusner MJ, Paige B, Hernández-Lobato JM et al (2017) Grammar variational autoencoder. In: International conference on machine learning. arXiv. https://doi.org/10.48550/arXiv.1703.01925
Liu S, Demirel MF, Liang Y (2019) N-gram graph: Simple unsupervised representation for graphs, with applications to molecules.arXiv. https://doi.org/10.48550/arXiv.1806.09206
You Y, Chen T, Shen Y et al (2021) Graph contrastive learning automated. In: International conference on machine learning. arXiv. https://doi.org/10.48550/arXiv.2106.07594
Ying C, Cai T, Luo S et al (2021) Do transformers really perform badly for graph representation? arXiv. https://doi.org/10.48550/arXiv.2106.05234
Wu Z, Ramsundar B, Feinberg EN et al (2018) MoleculeNet: a benchmark for molecular machine learning. Chem Sci 9(2):513–530. https://doi.org/10.1039/C7SC02664A
Zhu Y, Chen D, Du Y et al (2022) Featurizations matter: a multiview contrastive learning approach to molecular pretraining. In: ICML 2022 2nd AI for Science Workshop. https://openreview.net/forum?id=Pm1Q1X3avx1
Carhart RE, Smith DH, Venkataraghavan R et al (1985) Atom pairs as molecular features in structure-activity studies: definition and applications. J Chem Inf Comput Sci 25(2):64–73. https://doi.org/10.1021/ci00046a002
Bento AP, Hersey A, Félix E et al (2020) An open source chemical structure curation pipeline using RDKit. J Cheminf 12:1–16. https://doi.org/10.1186/s13321-020-00456-1
Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50(5):742–754. https://doi.org/10.1021/ci100050t
Church KW (2017) Word2Vec. Nat Lang Eng 23(1):155–162. https://doi.org/10.1017/S1351324916000334
Jaeger S, Fulle S, Turk S (2018) Mol2vec: unsupervised machine learning approach with chemical intuition. J Chem Inf Model 58(1):27–35. https://doi.org/10.1021/acs.jcim.7b00616
Devlin J, Chang MW, Lee K et al (2018) Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv. https://doi.org/10.48550/arXiv.1810.04805
Liu Y, Zhang R, Li T et al (2023) MolRoPE-BERT: an enhanced molecular representation with Rotary Position Embedding for molecular property prediction. J Mol Graph Model 118:108344. https://doi.org/10.1016/j.jmgm.2022.108344
Lin Z, Zhang Y, Duan L et al (2023) MoVAE: a variational AutoEncoder for molecular graph generation. In: Proceedings of the 2023 SIAM international conference on data mining (SDM). Society for Industrial and Applied Mathematics, pp 514–522. https://doi.org/10.1137/1.9781611977653.ch58
Kishimoto A, Kajino H, Hirose M et al (2023) MHG-GNN: combination of molecular hypergraph Grammar with graph neural network. arXiv. https://doi.org/10.48550/arXiv.2309.16374
Xie Y, Xu Z, Zhang J et al (2022) Self-supervised learning of graph neural networks: a unified review. IEEE Trans Pattern Anal Mach Intell 45(2):2412–2429. https://doi.org/10.1109/TPAMI.2022.3170559
Rong Y, Bian Y, Xu T et al (2020) Self-supervised graph transformer on large-scale molecular data. arXiv. https://doi.org/10.48550/arXiv.2007.02835
Zhang Z, Liu Q, Wang H et al (2021) Motif-based graph self-supervised learning for molecular property prediction. arXiv. https://doi.org/10.48550/arXiv.2110.00987
Wang Y, Wang J, Cao Z et al (2022) Molecular contrastive learning of representations via graph neural networks. Nat Mach Intell 4(3):279–287. https://doi.org/10.1038/s42256-022-00447-x
Li P, Wang J, Qiao Y et al (2020). Learn molecular representations from large-scale unlabeled molecules for drug discovery. arXiv. https://doi.org/10.48550/arXiv.2012.11175
Xu K, Hu W, Leskovec J et al (2018) How powerful are graph neural networks? arXiv. https://doi.org/10.48550/arXiv.1810.00826
Rarey M, Dixon JS (1998) Feature trees: a new molecular similarity measure based on tree matching. J Comput Aided Mol Des 12:471–490. https://doi.org/10.1023/A:1008068904628
Gasteiger J, Groß J, Günnemann S (2020) Directional message passing for molecular graphs. arXiv. https://doi.org/10.48550/arXiv.2003.03123
Loukas A (2019) What graph neural networks cannot learn: depth vs width. arXiv. https://doi.org/10.48550/arXiv.1907.03199
Hy TS, Trivedi S, Pan H et al (2018) Predicting molecular properties with covariant compositional networks. J Chem Phys 148(24):241745. https://doi.org/10.1063/1.5024797
Fey M, Yuen JG, Weichert F (2020) Hierarchical inter-message passing for learning on molecular graphs. arXiv. https://doi.org/10.48550/arXiv.2006.12179
Hopcroft JE, Motwani R, Ullman JD (2001) Introduction to automata theory, languages, and computation. ACM Sigact News 32(1):60–65. https://doi.org/10.1145/568438.568455
Vaswani A, Shazeer N, Parmar N et al (2017) Attention is all you need. arXiv. https://doi.org/10.48550/arXiv.1706.03762
Axelrod S, Gomez-Bombarelli R (2022) GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Sci Data 9(1):185. https://doi.org/10.1038/s41597-022-01288-4
Sun FY, Hoffmann J, Verma V et al (2019) Infograph: unsupervised and semi-supervised graph-level representation learning via mutual information maximization. arXiv. https://doi.org/10.48550/arXiv.1908.01000
Hu Z, Dong Y, Wang K et al (2020) Gpt-gnn: Generative pre-training of graph neural networks. In: Proceedings of the 26th ACM SIGKDD international conference on knowledge discovery & data mining, pp 1857–1867. https://doi.org/10.1145/3394486.3403237
Xu M, Wang H, Ni B et al (2021) Self-supervised graph-level representation learning with local and global structure. arXiv. https://doi.org/10.48550/arXiv.2106.04113
Van der Maaten L, Hinton G (2008) Visualizing data using t-SNE. J Mach Learn Res 9(11):2579–2605. https://www.jmlr.org/papers/volume9/vandermaaten08a/vandermaaten08a.pdf
Du W, Yang X, Wu D et al (2023) Fusing 2D and 3D molecular graphs as unambiguous molecular descriptors for conformational and chiral stereoisomers. Brief Bioinf 24(1):bbac560. https://doi.org/10.1093/bib/bbac560
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22373043). We would also like to thank Pro. Ruisheng Zhang for his help in this paper.
Author information
Authors and Affiliations
Contributions
Yunwu Liu: Writing, Data analysis, and Research design; Ruisheng Zhang: Supervision. All authors read the final manuscript and gave some suggestions for revision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, Y., Zhang, R., yuan, Y. et al. A Multi-view Molecular Pre-training with Generative Contrastive Learning. Interdiscip Sci Comput Life Sci (2024). https://doi.org/10.1007/s12539-024-00632-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12539-024-00632-z