
Abstract
ADAMTS13, a metalloproteinase, specifically cleaves unusually large multimers of von Willebrand factor (VWF), newly released from vascular endothelial cells. The ratio of ADAMTS13 activity to VWF antigen (ADAMTS13/VWF) and indicators of the alternative complement pathway (C3a and sC5b-9) are both related to the severity of COVID-19. The ADAMTS13/VWF ratio is generally moderately decreased (0.18–0.35) in patients with severe COVID-19. When these patients experience cytokine storms, both interleukin-8 and TNFα stimulate VWF release from vascular endothelial cells, while interleukin-6 inhibits both production of ADAMTS13 and its interaction with VWF, resulting in localized severe deficiency of ADAMTS13 activity. Platelet factor 4 and thrombospondin-1, both released upon platelet activation, bind to the VWF-A2 domain and enhance the blockade of ADAMTS13 function. Thus, the released unusually-large VWF multimers remain associated with the vascular endothelial cell surface, via anchoring with syndecan-1 in the glycocalyx. Unfolding of the VWF-A2 domain, which has high sequence homology with complement factor B, allows the domain to bind to activated complement C3b, providing a platform for complement activation of the alternative pathway. The resultant C3a and C5a generate tissue factor-rich neutrophil extracellular traps (NETs), which induce the mixed immunothrombosis, fibrin clots and platelet aggregates typically seen in patients with severe COVID-19.
Similar content being viewed by others


Introduction
COVID-19 caused by SARS-CoV-2 infection is often associated with the acute respiratory distress syndrome (ARDS), and complicated with thrombosis of many organs, meeting the diagnostic criteria for disseminated intravascular coagulation (DIC) [1]. During the clinical course, patients typically show a macrophage activation syndrome or cytokine storm, in which an excess production of inflammatory cytokines such as interferons, interleukins (IL), chemokines, tumor necrosis factors (TNFs) or colony-stimulating factors recruit more immune cells to the site of injury, thus leading to organ damage. Such thrombosis usually localizes to the lungs, termed ‘pulmonary intravascular coagulopathy’ [2], characterized by fibrin thrombus in pulmonary small arteries, and high plasma levels of inflammatory cytokines, ferritin, D-dimer, tissue-plasminogen activator (t-PA), and fibrinogen, together with slightly decreased platelet counts. Autopsies reveal pulmonary small vessels containing mixed microthrombi consisting of fibrin clots and platelet aggregates with or without megakaryocytes [3]. Some of these microthrombi react with an antibody against von Willebrand factor (VWF). These findings, in part, meet pathological criteria for thrombotic microangiopathy (TMA), manifesting thrombocytopenia, hemolytic anemia, and multi-organ failures, typically seen in such life-threatening diseases as thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) [4, 5], but differing from common TMA, because circulating platelet counts are usually not severely decreased and hemolytic anemia is rare.
Thromboembolic complications associated with COVID-19 are, therefore, assumed to be induced by several factors acting together, including coagulopathy, complementopathy and endotheliopathy, often under cytokine storm [6,7,8]. In fact, in COVID-19 patients, complement activation generates tissue factor (TF)-enriched neutrophil extracellular traps (NETs), which mediate both thrombosis and endotheliopathy [9, 10]. Important indicators for disease progression and thrombosis include elevated plasma levels of activated complement components [11] and of VWF with moderately decreased activity of ADAMTS13 (VWF-specific cleaving protease) [12,13,14].
As yet, a relationship between complement activation and VWF in COVID-19 thrombosis has received little attention; however, complement is often activated in concert with elevated VWF levels, as typically shown in congenital deficiency of ADAMTS13 activity [15,16,17]. Further, the A-domains of VWF share primary sequence homology with a 225 amino acid segment of complement factor B [18,19,20], indicating that both proteins are descended from a common ancestor, while factor B binds to complement C3b, a key protein of the complement activation cascade. These observations suggest that VWF participates in complement activation during COVID-19. The present review focuses on the role of VWF in COVID-19 associated ‘TMA’, in relation to the structure–function of VWF with the complement activation mechanism.
VWF, Weibel-Palade body and ADAMTS13
VWF is exclusively produced by vascular endothelial cells (ECs) and stored in Weibel-Palade bodies (WPBs), which are small intracellular organelles of vascular ECs, not only for storage of unusually large VWF multimers (UL-VWFMs) but also of various proteins involved in hemostasis, inflammation, and angiogenesis, including factor VIII, VWF-propeptide, P-selectin, angiopoietin-2 (Ang-2), IL-8, t-PA, etc. [21, 22]. It is well established that in vitro, several substances such as thrombin and histamine can release UL-VWFMs from cultured ECs [21, 22], while in vivo UL-VWFMs are released from WPBs upon their stimulation with epinephrine and 1-desamino-8-D-arginine vasopressin (DDAVP) [23], plus some inflammatory cytokines.
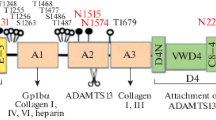
In addition to acting as a carrier protein for coagulation factor VIII, plasma VWF has an essential role in primary hemostasis by anchoring platelets onto denuded vascular ECs [24]. The VWF-cDNA codes for 2813 amino acids including 3 structural domains: (1) a signal peptide [22 amino acid (aa) residues], (2) the propeptide including the D1 and D2 assemblies (741 aa), and (3) the mature VWF subunit (2050 aa) including D’-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CK [24, 25], where each of the A1-A2-A3 domains has a loop structure, formed by intra-molecular disulfide bonds [26] (Fig. 1). Of note, it is well-known that a variety of ligands involved in thrombosis-hemostasis, complement activation-regulation and various toxins can bind to specific domain on the monomeric VWF subunit as shown in Fig. 1: factor VIII [27], platelet GPIb [28], heparin [29], heparan sulfate [30], P-selectin glycoprotein ligand-1 [31], collagen type I [32], DNA of NETs [33], platelet factor 4 (PF4) [34], thrombospondin-1(TSP-1) [35], β2-integrins [36], collagen type III [37], αIIbβ3 [38], αvβ3 [39], factor H [40], C3b [16], C3 [41], snake venoms: botrocetin [42], bitiscetin-1 [43], bitiscetin-2 [44], Staphylococcus aureus -Protein A [45] and -VWF binding protein [46], and pathologic E.coli-producing Shiga toxin [47]. Notably, the VWF-A1 domain is cryptic under normal physiological conditions, but once exposed under high shear stress, it turns to active conformation, to which a variety of the ligands can bind. In spite of VWF’s ability to bind many proteins, the physiological role(s) of this binding capacity is largely unexplored.

Structural domains of human von Willebrand factor (VWF) and its binding ligands. The cDNA of mature human VWF subunit includes the following structural domains: D’-D3-A1-A2-A3-D4-C1-C2-C3-C4-C5-C6-CK. The monomeric mature VWF subunit begins with amino acid residue number 763 and ends with 2813 (2050 amino acid residues). As shown in the upper panel, each A domain has a loop structure linked by an intramolecular disulfide bond. ADAMTS13 cleaves at the peptide bond of Y1605-M1606. LLG = Leucine-Leucine-Glycine motif, RGD = Arginine-Glycine-Aspartate motif. The lower panel lists the natural ligands that bind to each VWF domain involved in thrombosis/hemostasis, complement activation/inhibition, and others. TSP-1 = thrombospondin-1, PF4 = platelet factor 4, PSGL-1 = P-selectin glycoprotein ligand-1, NETs = neutrophil extracellular traps, SA-Protein A = Staphylococcus aureus Protein A. SA-VWFbp = Staphylococcus aureus VWF binding protein. Note that the ligands shown by the red bars bind to VWF domains in a shear-dependent manner. The binding of platelet GPIb to the A1-domain initiates platelet activation, alongside the enhanced proteolysis by ADAMTS13 [48], as does factor H [40]. Notably, both TSP-1 and PF4, released from α-granules of platelets upon activation, bind to the A2-domain, preventing cleavage by ADAMTS13. (See text in detail.)
Mature VWF subunits are linked by disulfide bonds in a head-to-head and tail-to-tail configuration to form large multimers ranging from 500 × 103 to 15 × 106 daltons [49]. Before being released into the circulation, UL-VWFMs undergo proteolytic cleavage at the peptide bond of Y1605-M1606 [50, 51] by ADAMTS13 under the high shear stress generated in microvasculatures. In the absence of ADAMTS13, UL-VWFMs are not cleaved, and stay anchored or are released without proteolytic processing [52]. In early studies, the anchor protein was postulated to be P-selectin or αvβ3-integrins [39, 53], although a subsequent report excluded this possibility [54]. Later, the finding that VWF can bind to negatively-charged heparin [29], demonstrated that VWF likely binds to heparan sulfate linked to syndecan-1 on the glycocalyx of vascular ECs [30].
Although several organs, including liver and vascular ECs, express ADAMTS13, the highest level of gene expression occurs in liver [55]. These studies revealed that ADAMTS13 is localized to liver stellate cells [56], which help maintain plasma levels of ADAMTS13 activity. Although the function of ADAMTS13 secreted by vascular ECs has not been well characterized [57], it may co-operate with plasma ADAMTS13 for the cleavage of newly-released UL-VWFMs on the surface of vascular ECs. Thus, a local inhibition of ADAMTS13 on the surface of the vascular ECs may foster the formation of thrombosis in COVID-19 patients. In vitro experiments in both rat primary hepatic stellate cells and human umbilical cells, suggest that a local reduction of ADAMTS13 activity may be caused by inflammatory cytokines such as IFN-γ, IL-4, and TNF-α which inhibit its production without inhibiting the release of UL-VWFMs [58]. In addition, TSP-1 and PF4, both released from platelet α-granules upon activation can bind to the unfolded VWF-A2 domain, causing steric hindrance for ADAMTS13 binding, and thus blocking cleavage of UL-VWFMs by ADAMTS13 [34, 35].
Pathological features of COVID-19 thrombosis
In COVID-19, the spike (S) protein of SARS-CoV-2 is cleaved by the host's serine protease (TMPRSS2) to form S1 and S2 moieties. The two moieties remain together as the S1 moiety binds to the host’s angiotensin-converting enzyme-2 (ACE-2) and the S2 moiety fuses with the host cell [59]. Many organs have high levels of ACE-2 mRNA: lung, heart, aorta, urinary tract, lymph-nodes, testis, ovary, salivary gland, mammary gland, gastrointestinal tract, brain (amygdala, cerebral cortex, brain stem) [60], platelets, and megakaryocytes [61].
Autopsies of COVID-19 patients who died of ARDS showed extensive necrosis of alveolar cells, type 2 alveolar cell hyperplasia, and fibrin deposition in the alveolar cells as well as considerable infiltration of CD4-T cells, but little infiltration of CD8-T cells [62, 63]. Pulmonary arterioles contained an incompletely occluded hyaline thrombus about 1–2 mm in diameter; vascular remodeling in alveolar capillaries was abnormal with the normal vessel hierarchy of the alveolar plexus substituted by bizarre blood vessel formation, termed ‘intussusceptive angiogenesis’. This was significantly increased in patients with long hospital stays [62]. Moreover, autopsies of patients who died of ARDS revealed numerous intrapulmonary arteriole thrombi including fibrin, CD61-positive platelets and megakaryocytes, with positive immunostaining of VWF [63]. These patients also had hypercellular bone marrow [64, 65]. The pathogenesis of mixed thrombi of fibrin and platelets in the lungs with or without megakaryocytes, and without a significant reduction of circulating platelets, is unknown. However, megakaryocytes in human lungs have long been known [66], while the ratio of platelets to red blood cells is higher in the cubital artery in the arm than in the vein, suggesting that the lung may produce platelets [67]. Proof that the human lung is a source of platelets is lacking, although in mice, half of the platelets in the circulating blood are produced in the lungs, and the other half in the bone marrow [68]. Notably, the following studies also identified ‘megakaryocytopathy’ occurring in hepatic, cardiac and renal microvasculatures [64, 65], as well as in cortical capillaries in the brain of a deceased COVID-19 patient who had experienced a brain fog [69].
From the early era of COVID-19, it has been noted that in severe cases, the plasma level of VWF antigen is markedly increased (278–772% of normal), and ADAMTS13 activity is moderately decreased (40–89% of normal), resulting in an average ADAMTS13/VWF ratio of 0.18–0.35 [70, 71], usually without severe thrombocytopenia. A mild-to-moderate reduction in plasma ADAMTS13 activity in COVID-19 patients has been thought simply to reflect a severe inflammation reaction because VWF is a marker of the acute phase, and is assumed to be a consequence of UL-VWFMs released from vascular endothelial cells under cytokine storms. Subsequent investigations have confirmed that a low ADAMTS13/VWF ratio in patient plasma is associated with a high prothrombotic risk [11,12,13]. Indeed, these patients sometimes have a variety of autoimmune complications, such as immune thrombocytopenia (ITP) [72], Guillain–Barré syndrome [73], antiphospholipid syndrome (APS) [74], immune TTP [75], and more recently anti-factor H associated aHUS [76]. However, anti-PF4 antibody [77, 78] rarely appears after SARS-CoV-2 vaccination. Root-Bernstein [79] recently proposed as an explanation that SARS-CoV-2-associated autoimmunity may be enhanced by coexisting bacterial or viral infections, as these pathogens have primary amino acid sequence homology with many human serum proteins. However, a direct link between the autoantibodies and the mild-to-moderate reduction of ADAMTS13 activity in COVID-19 patients is not known. A large increase in VWF released from vascular ECs, metaphorically a ‘VWF flood’, leaves UL-VWFMs not only on ECs, but also in circulation without efficient cleavage by ADAMTS13. These UL-VWFMs induce platelet activation and aggregation, but curiously without severe reduction of platelet counts. The elevated VWF levels of patient plasma often lack UL–VWFMs or high molecular weight (HMW)–VWFMs [80]. The proportion of UL–HMW–VWFMs tended to decline in long-term hospitalization patients in the ICU, possibly due to selective consumption of UL–HMW–VWFMs involved in platelet aggregates in TMA or to heightened cleavage by ADAMTS13 under high shear stress generated during extracorporeal membrane oxygenation (ECMO) applied to severely ill patients [80, 81].
In severely ill patients, additional markers for vascular EC damage, such as soluble (s) P-selectin, s-thrombomodulin (TM), and sCD40L were elevated, along with cytokines including TNFα, IL-6 and IL-10, but not IL-1b [82]. These cytokines are likely released both directly and indirectly from type 1 alveolar cells and alveolar resident cells, such as neutrophils, lymphocytes and macrophages.
Complement activation
The complement system is part of the innate immune system. It protects against pathogens in several ways including opsonization, which facilitates the phagocytosis of pathogens, activation of leukocytes, and the production of anaphylatoxins (C3a and C5a). The three complement pathways are the classical, lectin and alternative pathways (AP) [7]. The classical pathway is initiated by antibody production and subsequent binding of antibodies to microorganisms, in turn triggering a cascade of several complement proteins, whereas in the lectin pathway, a lectin such as mannose binding lectin (MBL) binds to mannose moieties on the surface of pathogens and initiates the complement cascade through mannose associated serine protease (MASP1, MASP2). The final product of the complement activation cascade, termed a membrane attack complex (MAC/C5b-9), results in phagocytosis of the microorganisms. In both the classic and lectin pathways, the central C3 convertase consists of two complement proteins, C4bC2a. However, the activation of the AP is unique and does not require either a pathogen recognition mechanism or C4b or C2a [7].
Figure 2 diagrams complement activation in the AP, according to Pangburn and Müller–Eberhard [83]. In this pathway, plasma C3, which has a thioester domain, is spontaneously hydrolyzed by H20 to form C3(H20), to which binds factor B, leading to proteolysis by factor D releasing a 32 kDa-Ba protein and thereby generating C3(H20)Bb, which is the initiation C3-convertase. This convertase then cleaves C3 into 2 fragments–C3a and metastable C3b. The latter further changes either to the fluid-phase C3b by hydrolysis or to the surface-bound C3b through covalent binding of a thioester bond to the cell surface [84,85,86].

Activation and amplification of the complement alternative pathway (AP), according to Pangburn and Müller-Eberhard [83]. In this pathway, plasma C3, which has a thioester domain, is spontaneously hydrolyzed by H20 to form C3(H20), to which binds factor B, leading to proteolysis by factor D releasing a 32 kDa-Ba protein and thereby generating C3(H20)Bb, which is the initiation C3-convertase. This convertase then cleaves C3 into 2 fragments–C3a and metastable C3b. The latter further changes either to the fluid-phase C3b by hydrolysis or to the surface-bound C3b through covalent binding of a thioester bond to the cell surface. The surface-bound C3b has been well characterized, but the fluid-phase ‘soluble’ C3b, particularly its function, has been little studied in vitro, presumably due to its instability in vivo (See text in detail.)
C3b when bound to microorganisms (activator surface) forms C3bBb, due to the activity of C3-convertase; C3bBb in turn binds to properdin (P or factor P) for stabilization. Together they form an amplification cycle of C3 activation, followed by the generation of C5-convertase in the alternative pathway (AP) that generates C3bBbC3b, resulting in formation of C5b-9 (MAC), and leading to the death of the microorganism [7]. The surface-bound C3b has been well characterized, but the fluid-phase ‘soluble’ C3b, particularly its function, has been little studied in vitro, presumably due to its instability in vivo (Table 1).
In contrast, under normal physiological conditions (non-activator surface), the surface-bound C3b is inactivated by complement factor I, a serine protease, in cooperation with two additional proteins: (1) complement factor H bound to sulfated glycosaminoglycan (GAG) expressed on the vascular EC surface, and (2) membrane cofactor protein (MCP) or CD46 (figure not shown) [7, 87]. The gene mutations responsible for such complement and its regulatory factors often induce the uncontrolled complement activation, termed atypical HUS [87].
NETs in COVID-19 thrombosis
The spike (S) protein of SARS-CoV-2 binds not only to ACE-2, but also in vitro to heparan sulfate on the glycocalyx of cultured vascular ECs, to which factor H, a complement regulatory protein, also binds under physiological conditions [88]. These two ligands may compete with each other for binding, resulting in uncontrolled activation of the AP. This finding is important, but its clinical significance needs to be evaluated, since most patients infected with SARS-CoV-2 have only low levels of the virus in their circulation and do not develop thromboses. A study updated by a French group indicated that severe COVID-19 patients with underlying TMA had mutations in complement and its regulatory factors that were indistinguishable from those in aHUS [89]. This finding is also relevant, because severe infections including influenza etc. are often strong inducers for TMA bouts in aHUS patients.
On the other hand, another recent study showed that human vascular ECs express little or almost no ACE-2 [90]. This finding may in part address why SARS-CoV-2 infection has not been confirmed by blood transfusion [91], and further strengthens the idea that COVID-19 endotheliopathy is associated with an indirect cause rather than with direct SARS-CoV-2 infection, as discussed above. For example, an anti-SARS-CoV-2 spike (S) IgG with an aberrant glycosylation site (low fucose and high galactose) on the Fc domain was found in severely ill COVID-19 patients [92, 93], and a similar antibody complexed with recombinant SARS-CoV-2 spike (S) protein induced platelet activation via binding of the complex to the platelet IgG-Fc receptor (FcγRIIA) [94]. Platelet activation was assumed to generate intracellular signal transduction to activate the platelet GPIb/IX complex, to which VWF binds and forms platelet thrombi in vitro. This suggests that the immune complex itself may activate the classic complement pathway. In contrast, the role of the lectin pathway in complement activation during SARS-CoV-2 infection seems clear, because it is initiated by the binding of mannose binding lectin (MBL) to the spike (S) protein of SARS-CoV-2, leading to the activation of MBL-associated serine protease 2 (MASP-2) [7].
In patients with COVID-19 thrombosis, plasma levels of NETs, TF, the thrombin–antithrombin (TAT) complex, and soluble (s) C5b-9 (MAC) are elevated [9, 10]. Also, neutrophils isolated from healthy individuals and stimulated with platelet-rich plasma from patients with COVID-19, but not with plasma devoid of platelets, efficiently release TF-bearing NETs, indicating a critical role of platelets. Importantly, the release of NETs from neutrophils was totally abolished by inhibition either of complement activation with a C5aR1 antagonist or with a thrombin inhibitor (dabigatran) or a protease-activated receptor-1 (PAR-1) inhibitor. These results indicate that TF-bearing NETs generated by a double-hit phenomenon function as a driver of COVID-19 thrombosis, where C5a binding to C5aR1 on the neutrophil surface generates intracellular TF, and thrombin is released from platelets activated by C3a through its binding to the surface receptor PAR-1 [9].
Activation and amplification of the alternative complement pathway in congenital deficiency of ADAMTS13 activity
Congenital TTP (cTTP), termed Upshaw-Schulman syndrome, is a hereditary deficiency of ADAMTS13 activity caused by biallelic ADAMTS13 gene mutations [95,96,97]. A hallmark of cTTP is severe neonatal hemolytic jaundice necessitating exchange blood transfusions, that is generated under high shear stress induced by shrinkage of the patent ductus arteriosus, usually within 2–3 days after birth [98]. Beyond this period, these patients exhibit either the early- or late-onset phenotype of TTP bouts [99, 100]. In both, however, there is often severe renal dysfunction during the acute phase [14,15,16]. Kidney biopsies revealed deposits of complement C3 and C5b-9 in the renal cortex [14]. Moreover, in these patients, the plasma level of sC5b-9 was remarkably increased during the acute phase [15], even without the accompanying gene mutations of complement and its regulatory factors responsible for atypical HUS [16, 17], while ex vivo assays showed that patient serum induced C3 and C5b-9 deposits on cultured vascular ECs. These deposits were eliminated by spiking cTTP serum with recombinant ADAMTS13, indicating that in these patients, complement is activated via the AP on the surface of vascular ECs [16].
Additional evidence of a relationship between VWF and complement is that the A domain of VWF shares the primary sequence homology with complement factor B [18,19,20]. When factor B binds to C3b followed by cleavage by factor D, the complex becomes C3-convertase (C3bBb) with release of Ba. The Bb moiety consists of the N-terminal VWF-A (homologous) domain and the C-terminal serine protease domain. Competition assays and mass spectrometry showed that the recombinant (factor B) VWF-A domains are responsible for binding to C3b [19]. However, in spite of a high sequence homology between both A-domains, it had been uncertain whether C3b can bind to the VWF-A domain, but that was shown by Bettoni et al. [16] (below).
Under the flow of medium lacking ADAMTS13, confluent cell cultures of vascular ECs from human umbilical cord (HUVECs) released UL-VWFMs that formed strings anchored on the cell surface [52]. Washed platelets, subsequently added, adhered to these EC-anchored UL-VWFMs like a bead necklace. However, added ADAMTS13 immediately cleaved these UL-VWFMs. In addition, TNFα, IL-8, and a complex of IL-6 and sIL-6R, like histamine, induced UL-VWFM release from HUVECs. Curiously, IL-6 alone at a high concentration did not induce UL-VWFM release and rather significantly inhibited ADAMTS13 activity under flow conditions [101]. The inhibition mechanism is still unclear, but none occurred under non-flow static conditions, suggesting that inhibition by IL-6 occurs in a conformation-dependent manner through interaction either with VWF or ADAMTS13. This finding is extremely important and further indicates that under a cytokine storm with high concentrations of several cytokines, the hyperactive UL-VWFMs are not efficiently cleaved by ADAMTS13 and accumulate on the vascular EC surface via anchors, providing a platform for both platelet thrombosis and activation of the AP.
In 2013, long before the emergence of COVID-19, Turner and Moake [102] reported an extremely fascinating result, in which under ex vivo flow and in the absence of exogenous ADAMTS13, HUVECs upon stimulation with histamine released several members of the AP and their regulatory factors, which then bound to UL-VWFMs simultaneously released from and anchored onto HUVECs. These proteins included C3, factor B, factor D, properdin, C5, factor H, factor I, but notably not C4 [102]. This observation also raised a question on the role on EC-derived ADAMTS13 in regulating VWF multimeric size. In addition, in 2017 Bettoni et al. [16] demonstrated a consistent result, in which ‘soluble’ C3b can bind to the A2 domain of monomeric VWF coated onto microtiter wells under static conditions, and after normal human serum addition to the wells, form C3-/C5-convertase, in a Ca2+-dependent manner, that differs from a Ca2+-independent activation process of the AP on C3b covalently bound to cell surfaces [85, 86] (comparison shown in Table 1). More importantly, they also showed that the ‘soluble’ C3b bound neither to plasma-derived nor to recombinant multimeric VWF under static conditions, indicating that C3b binding to VWF depends on the VWF conformation [16]. Since both the classical and the lectin pathways require C4b to form C3- or C5-convertase, but the AP does not, complement activation in a VWF-dependent manner must be via the AP.
UL-VWFMs are involved in activation of the alternative complement pathway on the micro-vasculatures during COVID-19 thrombosis
Under physiological conditions, C3b bound covalently to the vascular ECs is inactivated by complement factor I, a serine protease, in cooperation with complement factor H bound to sulfated glycosaminoglycan (GAG) on the vascular ECs [7]. Factor H can also bind to the A1-A2 domain of VWF under high shear stress, promoting the enhanced proteolysis of VWF by ADAMTS13 [40]. In the absence of ADAMTS13, however, factor H bound to VWF fails to dysregulate AP activation presumably due to the physiologically different positional relationship of C3b and FH. Taking these scenarios together with the experimental data discussed above, a model of VWF-dependent complement activation of the AP during COVID-19 can be proposed as shown in Fig. 3. First, SARS-CoV-2 invades the lung alveoli from the respiratory tract and infects type 2 alveolar epithelial cells and macrophages. This causes release of cytokines from resident cells, such as macrophages, CD4-T lymphocytes and neutrophils. Inflammatory cytokines further stimulate release of cytokines from blood cells and vascular ECs, generating a cytokine storm [103]. Consequently, IL-8, TNFα, and a complex of IL-6 and its soluble receptor (sIL-6R) stimulate vascular ECs, and induce exocytosis of UL-VWFMs from WPBs. Under high shear flow, UL-VWFMs undergo a conformational change, allowing ADAMTS13 more accessibility; however, IL-6 interferes with this interaction, resulting in inhibition of ADAMTS13 activity [101]. This inhibitory effect could also be heightened by the binding of TSP-1 and/or PF4 to VWF-A2 domain [34, 35]. In such microenvironments, the 3-loop domains (A1-A2-A3) of VWF are exposed on the molecular surface. The A1-loop domain binds to the heparan sulfate of syndecan-1 on the vascular EC surface [30], while the A2 domain binds to C3b generated by AP activation, without interference of factor H, as mentioned.

The role of UL-VWFMs in activation of the alternative complement pathway (AP) in the microvasculatures during COVID-19 thrombosis. SARS-CoV-2 invades lung alveoli from the respiratory tract and infects type 2 alveolar pneumocytes and macrophages. This causes release of cytokines from resident cells, such as macrophages, CD4-T lymphocytes and neutrophils. Inflammatory cytokines further stimulate release of cytokines from blood cells and vascular ECs, generating a cytokine storm. Consequently, IL-8, TNFα, and a complex of IL-6 and its soluble receptor (sIL-6R) stimulate vascular ECs, and induce exocytosis of UL-VWFMs from Weibel-Palade Bodies (WPBs). Under a high shear flow, UL-VWFMs undergo a conformational change, allowing ADAMTS13 more accessibility; however, IL-6 interferes with this interaction, resulting in inhibition of ADAMTS13 activity. In such microenvironments, the A1-loop domain of VWF binds platelets, forming platelet aggregates with or without involving resident megakaryocytes. The activated platelets release PF4 and TSP-1 from the α-granules, both of which bind to the A2-domain of VWF and block cleavage by ADAMTS13. The A1-loop domain of VWF binds to the heparan sulfate of syndecan-1 on the vascular EC surface, while the A2 domain binds to C3b generated by the AP activation. C3b bound to UL-VWFM anchored on the EC surface binds factor B, which is proteolyzed by factor D. Then binding of properdin to the C3b moiety as a stabilizer results in the formation of C3-convertase in the AP. Subsequent activation through the AP (C5-convertase) (C3bBbC3b) produces C5b, to which C6 ~ C9 bind, finally forming C5b-9 (MAC), which in turn activates endothelial cells (EC) together with endotheliopathy, UL-VWFM is a major constituent of WPBs, which also contain IL-8, Ang-2, t-PA, etc. The secretion of IL-8 into the circulation enhances UL-VWFM release and accelerates C3b binding to UL-VWFM on the vascular EC surface, promoting platelet thrombi formation. The released t-PA generates plasmin under microenvironments in which thrombomodulin (TM) on the vascular EC surface undergoes shedding. TM binds to thrombin to form a complex that inhibits thrombin activity, but activates protein C and thrombin-activatable fibrinolysis inhibitor (TAFI) to TAFIa (carboxypeptidase), which can proteolytically inactivate both C3a and C5a. Shedding of TM loses the antithrombotic function of vascular ECs, turning them into the thrombogenic surface
In summary, C3b bound to UL-VWFM anchored on the EC surface binds factor B, which is proteolyzed by factor D, and then binding of properdin, as a stabilizer, to the C3b moiety results in the formation of C3-convertase in the AP. Subsequent activation through the AP (C5-convertase) (C3bBbC3b) produces C5b, to which C6 ~ C9 bind, finally forming C5b-9 (MAC), which in turn causes EC activation together with endotheliopathy,
UL-VWFM is a major constituent of WPBs, which also contain IL-8, Ang-2, t-PA, etc. [21, 22], as described above. The secretion of IL-8 into the circulation enhances UL-VWFM release and also accelerates binding of C3b to UL-VWFM on the vascular EC surface, promoting platelet thrombi formation. The released t-PA generates plasmin under microenvironments in which TM on the vascular EC surface undergoes shedding. Physiologically, TM binds to thrombin to form a complex, inhibiting thrombin activity, but activating protein C and thrombin-activatable fibrinolysis inhibitor (TAFI) to TAFIa (carboxypeptidase), which can proteolytically inactivate both C3a and C5a. The shedding of TM loses the antithrombotic function of vascular ECs, turning them into the thrombogenic surface. Further, both VWF and Ang-2 are constituents of WPBs and co-operate in vascular angiogenesis, where VWF signals via αIIbβ3-integrins to promote smooth muscle proliferation, and Ang-2 via VEGFR2 signaling for endothelial cell migration/proliferation [104, 105]. Thus, the imbalance between these two signals may in part address why fragile blood vessels, as in ‘intussusceptive angiogenesis’ in the lung, are formed during longer hospitalization of COVID-19 patients [62].
Conclusions
NETs are an important driver for COVID-19 immunothrombosis [10], more profoundly in concert with complement activation; thus the plasma levels of activated complement components are excellent indicators for the disease severity [11]. In this setting, both classical and lectin pathways of complement activation are well characterized [7], but the AP has been poorly understood. Binding of the spike (S) protein of SARS-CoV-2 to heparan sulfate on vascular ECs [88] indicates competition for binding with factor H, thus mediating activation of the AP. However, a majority of infected patients do not develop thrombosis, emphasizing the requirement of the amplification mechanism for AP activation. Such absence of blood-born infection of SARS-CoV-2 might be because human vascular ECs apparently express little or almost no ACE-2 [91].
A low ADAMTS13/VWF ratio has been an independent indicator of COVID-19 severity apart from complement activation [12,13,14]. Whereas, amplification of AP activation is a phenomenon lately recognized in congenital deficiency of ADAMTS13 activity [15, 16]. Although COVID-19 patients do not show severe deficiency of plasma ADAMTS13 activity, during a cytokine storm, large amounts of VWF are released and ADAMTS13 activity is suppressed specifically by IL-6 under the high shear stress generated in microenvironments in concert with binding of TSP-1 and PF4 which bind to the VWF-A2 domain, where VWF functions as a platform for AP activation and amplification. This hypothesis is suggested by historical reviews concerning the common decent of VWF and complement factor B [18,19,20], and appears to fit with the direction of current therapeutic options. Further, recent studies on SARS-CoV-2 vaccine-induced thrombotic thrombocytopenia have focused on its association with PF4, ADAMTS13 and complement activation. In particular, autoantibodies against PF4 resulting in increased stability of UL-VWFMs have been implicated in vaccine-induced thrombosis (VITT), although a possible role of the alternative complement pathway in VITT has not been characterized [77, 78].
In sum, the clinical and experimental data on blood clotting disorders together with the clinical data on COVID-19 point to the complex scheme outlined in Fig. 3 whereby infection with SARS-CoV-2 in the lungs induces a cytokine storm that in turn acts on the vascular endothelium to release VWF and other proteins such as t-PA from vascular ECs, at the same time inhibiting secretion and function of the protease ADAMTS13, that normally cleaves UL-VWFMs. The alternative complement pathway is activated, further promoting release of VWF, t-PA and thrombomodulin from vascular ECs. The end result of the combination of excess UL-VWFMs tethered to the vascular endothelial surface plus elevated concentrations of several clotting factors is wide-spread microthrombosis.
References
Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. Thromb Haemost. 2020;4:844–7.
McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020;2(7):e437–45.
Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020;2:198–209.
George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654–66. https://doi.org/10.1056/NEJMra1312353 (PMID: 25119611).
Scully M. How to evaluate and treat the spectrum of TMA syndromes in pregnancy. Hematology Am Soc Hematol Educ Program. 2021;2021(1):545–51. https://doi.org/10.1182/hematology.2021000290 (PMID: 34889427).
Gu SX, Tyagi T, Jain K, Gu VW, Lee SH, Hwa JM, et al. Thrombocytopathy and endotheliopathy: crucial contributors to COVID-19 thromboinflammation. Nature Rev Cardiol. 2021;18(3):194–209. https://doi.org/10.1038/s41569-020-00469-1 (Epub 2020 Nov 19. 2020. s41569-020-00469-1).
Noris M, Benigni A, Remmuzzi G. The case of complement activation in COVID-19 multiorgan impact. Kidney Int. 2020;98(2):314–22.
Chen LYC, Hoiland RL, Stukas S, Wellington CL, Sekhon MS. Confronting the controversy: interleukin-6 and the COVID-19 cytokine storm syndrome. Eur Respir J. 2020;56(4):2003006. https://doi.org/10.1183/13993003.03006-2020.
Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos D, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. 2020;130(11):6151–7. https://doi.org/10.1172/JCI141374.
Yang J, Wu Z, Long Q, Huang J, Hong T, Liu W, et al. Insights into immunothrombosis: the interplay among neutrophil extracellular trap, von Willebrand Factor, and ADAMTS13. Front Immunol. 2020;2(11): 610696. https://doi.org/10.3389/fimmu.2020.610696.
Henry BM, Szergyuk I, de Oliveira MHS, Lippi G, Benoit J, Vikse J, et al. Complement levels at admission as a reflection of coronavirus disease 2019 (COVID-19) severity state. J Med Virol. 2021;93(9):5515–22. https://doi.org/10.1002/jmv.27077 (Epub 2021 May 19).
Favaloro EJ, Henry BM, Lippi G. Increased VWF and decreased ADAMTS-13 in COVID-19: creating a milieu for (micro)thrombosis. Semin Thromb Hemost. 2021;47(4):400–18. https://doi.org/10.1055/s-0041-1727282 (Epub 2021 Apr 23. PMID: 33893632 Review).
Joly BS, Darmon M, Dekimpe C, Dupont T, Dumas G, Yvin E, et al. Imbalance of von Willebrand factor and ADAMTS13 axis is rather a biomarker of strong inflammation and endothelial damage than a cause of thrombotic process in critically ill COVID-19 patients. J Thromb Haemost. 2021;19(9):2193–8. https://doi.org/10.1111/jth.15445 (Epub 2021 Jul 27. PMID: 34219357).
Rostami M, Mansouritorghabeh H, Parsa-Kondelaji M. High levels of Von Willebrand factor markers in COVID-19: a systematic review and meta-analysis. Clin Exp Med. 2021;6:1–11. https://doi.org/10.1007/s10238-021-00769-x (Online ahead of print. PMID: 34741678).
Tati R, Kristoffersson A-C, Ståhl A-L, Rebetz J, Wang L, Licht C, et al. Complement activation associated with ADAMTS13 deficiency in human and murine thrombotic microangiopathy. J Immunol. 2013;191(5):2184–93. https://doi.org/10.4049/jimmunol.1301221.
Bettoni S, Galbusera M, Gastoldi S, Donadelli R, Tentori C, Sparta G, et al. Interaction between multimeric von Willebrand factor and complement: a fresh look to pathophysiology of microvascular thrombosis. J Immunol. 2017;199(3):1021–40. https://doi.org/10.4049/jimmunol.1601121 (Epub 2017 Jun 26).
Fan X, Kremer-Hovinga JA, Shirotani-Ikejima H, Eura Y, Hirai H, Honda S, et al. Genetic variations in complement factors in patients with congenital thrombotic thrombocytopenic purpura with renal insufficiency. Int J Hematol. 2016;103(3):283–91. https://doi.org/10.1007/s12185-015-1933-7 (Epub 2016 Feb 1 PMID: 26830967).
Sadler JE, Shelton-Inloes BB, Sorace JM, Harlan JM, Titani K, Davie EW. Cloning and characterization of two cDNAs coding for human von Willebrand factor. Proc Natl Acad Sci USA. 1985;82(19):6394–8. https://doi.org/10.1073/pnas.82.19.6394.
Shelton-Inloes BB, Titani K, Sadler JE. cDNA sequences for human von Willebrand factor reveal five types of repeated domains and five possible protein sequence polymorphisms. Biochemistry. 1986;25(11):3164–71. https://doi.org/10.1021/bi00359a014.
Hinshelwood J, Spencer DIR, Edwards YJK, Perkins SJ. Identification of the C3b binding site in a recombinant vWF-A domain of complement factor B by surface-enhanced laser desorption-ionisation affinity mass spectrometry and homology modelling: implications for the activity of factor B. J Mol Biol. 1999;294(2):587–99. https://doi.org/10.1006/jmbi.1999.3223.
Ewenstein BM, Warhol MJ, Handin RI, Pober JS. Composition of the von Willebrand factor storage organelle (Weibel-Palade body) isolated from cultured human umbilical vein endothelial cells. J Cell Biol. 1987;104(5):1423–33. https://doi.org/10.1083/jcb.104.5.1423.
van Mourik JA, de Wit TR, Voorberg J. Biogenesis and exocytosis of Weibel-Palade bodies. Histochem Cell Biol. 2002;117(2):113–22. https://doi.org/10.1007/s00418-001-0368-9.
Reiter RA, Knöbl P, Varadi K, Turecek PL. Changes in von Willebrand factor-cleaving protease (ADAMTS13) activity after infusion of desmopressin. Blood. 2003;101(3):946–8. https://doi.org/10.1182/blood-2002-03-0814 (Epub 2002 Sep 19).
Ruggeri ZM. Von Willebrand factor. Curr Opin Hematology. 2003;10(2):142–9. https://doi.org/10.1097/00062752-200303000-00008.
Zhou Y-Z, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood. 2012;120(2):449–58. https://doi.org/10.1182/blood-2012-01-405134 (Epub 2012 Apr 6).
Marti T, Rösselet SJ, Titani K, Walsh KA. Identification of disulfide-bridged substructures within human von Willebrand factor. Biochemistry. 1987;26(25):8099–109. https://doi.org/10.1021/bi00399a013.
Foster PA, Fulcher CA, Marti T, Titani K, Zimmerman TS. A major factor VIII binding domain resides within the amino-terminal 272 amino acid residues of von Willebrand factor. J Biol Chem. 1987;262(18):8443–6.
Fujimura Y, Titani K, Holland LZ, Russell SR, Roberts JR, Elder JH, et al. von Willebrand factor A reduced and alkylated 52/48-kDa fragment beginning at amino acid residue 449 contains the domain interacting with platelet glycoprotein Ib. J Biol Chem. 1986;261(1):381–5.
Fujimura Y, Titani K, Holland LZ, Roberts JR, Kostel P, Ruggeri ZM, et al. A heparin-binding domain of human von Willebrand factor. Characterization and localization to a tryptic fragment extending from amino acid residue Val-449 to Lys-728. J Biol Chem. 1987;262:1734–9.
Kalagara T, Moutsis T, Yang Y, Pappelbaum KI, Farken A, Cladder-Micus L, et al. The endothelial glycocalyx anchors von Willebrand factor fibers to the vascular endothelium. Blood Adv. 2018;18:2347–57.
Pendu R, Terraube V, Christophe OD, Gahmberg CG, de Groot PG, Lenting PJ, et al. P-selectin glycoprotein ligand 1 and β2-integrins cooperate in the adhesion of leukocytes to von Willebrand factor. Blood. 2006;108(12):3746–52. https://doi.org/10.1182/blood-2006-03-010322 (Epub 2006 Aug 22).
Pareti FI, Fujimura Y, Dent JA, Holland LZ, Zimmerman TS, Ruggeri ZM. Isolation and characterization of a collagen binding domain in human von Willebrand factor. J Biol Chem. 1986;261(32):15310–5.
Grässle S, Huck V, Pappelbaum KI, Gorzelanny C, Aponte-Santamaria C, Baldauf C, et al. von Willebrand factor directly interacts with DNA from neutrophil extracellular traps. Arterioscler Thromb Vasc Biol. 2014;34(7):1382–9. https://doi.org/10.1161/ATVBAHA.113.303016 (Epub 2014 May 1).
Nazy I, Elliott TD, Arnold DM. Platelet factor 4 inhibits ADAMTS13 activity and regulates the multimeric distribution of von Willebrand factor. Br J Haematol. 2020;190(4):594–8. https://doi.org/10.1111/bjh.16553 (Epub 2020 Mar 4).
Wang A, Liu F, Dong N, Ma Z, Zhang J, Su J, et al. Thrombospondin-1 and ADAMTS13 competitively bind to VWF A2 and A3 domains in vitro. Thromb Res. 2010;126(4):e260–5. https://doi.org/10.1016/j.thromres.2010.07.009 (Epub 2010 Aug 11).
Koivunen E, Ranta T-M, Annila A, Taube S, Uppala A, Jokinen M, et al. Inhibition of β2 integrin-mediated leukocyte cell adhesion by Leucine-Leucine-Glycine motif-containing peptides. J Cell Biol. 2002;153:905–15.
Kalafatis K, Takahashi Y, Girma JP, Meyer D. Localization of a collagen-interactive domain of human von Willebrand factor between amino acid residues Gly 911 and Glu 1,365. Blood. 1987;70(5):1577–83.
Girma JP, Chopek MK, Titani K, Davie EW. Limited proteolysis of human von Willebrand factor by Staphylococcus aureus V-8 protease: isolation and partial characterization of a platelet-binding domain. Biochemistry. 1986;25(11):3156–63. https://doi.org/10.1021/bi00359a013 (LPMID: 3015200).
Huang J, Roth R, Heuser JE, Sadler JE. Integrin αvβ3 on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood. 2009;113(7):1589–97. https://doi.org/10.1182/blood-2008-05-158584 (Epub 2008 Oct 16).
Feng S, Liang X, Cruz MA, Vu H, Zhou Z, Pemmaraju N, et al. The Interaction between Factor H and Von Willebrand Factor. PLoS ONE. 2013;8(8): e73715. https://doi.org/10.1371/journal.pone.0073715 (eCollection 2013).
Nolasco JG, Nolasco LH, Da Q, Cirlos S, Ruggeri ZM, Moake JL, et al. Complement component C3 binds to the A3 domain of von Willebrand Factor. TH Open. 2018;2(3):e338–45. https://doi.org/10.1055/s-0038-1672189.
Sugimoto M, Mohri H, McClintock RA, Ruggeri ZM. Identification of discontinuous von Willebrand factor sequences involved in complex formation with botrocetin. A model for the regulation of von Willebrand factor binding to platelet glycoprotein Ib. J Biol Chem. 1991;266(27):18172–8 (PMID: 1917949).
Matsui T, Hamako J, Matsushita T, Nakayama T, Fujimura Y, Titani K. Binding site on human von Willebrand factor of bitiscetin, a snake venom-derived platelet aggregation inducer. Biochemistry. 2002;41(25):7939–46. https://doi.org/10.1021/bi020004b (PMID: 12069583).
Obert B, Houllier A, Meyer D, Girma JP. Conformational changes in the A3 domain of von Willebrand factor modulate the interaction of the A1 domain with platelet glycoprotein Ib. Blood. 1999;93(6):1959–68 (PMID: 10068669).
O’Seaghdha M, van Schooten CJ, Kerrigan SW, Emsley J, Silverman GJ, Cox D, et al. Staphylococcus aureus protein A binding to von Willebrand factor A1 domain is mediated by conserved IgG binding regions. FEBS J. 2006;273(21):4831–41. https://doi.org/10.1111/j.1742-4658.2006.05482.x (Epub 2006 Sep 25).
Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vandenbriele C, Vanhoorelbeke K, et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von Willebrand factor-binding protein. Blood. 2014;124(10):1669–76. https://doi.org/10.1182/blood-2014-02-558890 (Epub 2014 Jun 20).
Lo NC, Turner NA, Cruz MA, Moake J. Interaction of Shiga toxin with the A-domains and multimers of von Willebrand factor. J Biol Chem. 2013;288(46):33118–23. https://doi.org/10.1074/jbc.M113.487413 (Epub2013 Oct 4).
Nishio K, Anderson PJ, Zheng XL, Sadler JE. Binding of platelet glycoprotein Iba to von Willebrand factor domain A1 stimulates the cleavage of the adjacent domain A2 by ADAMTS13. Proc Natl Acad Sci USA. 2004;101(29):10578–83. https://doi.org/10.1073/pnas.0402041101.
Titani K, Kumar S, Takio K, Ericsson LH, Wade RD, Ashida K, et al. Amino acid sequence of human von Willebrand factor. Biochemistry. 1986;25(11):3171–84. https://doi.org/10.1021/bi00359a015 (PMID: 3524673).
Dent JA, Berkowitz SD, Ware J, Kasper CK, Ruggeri ZM. Identification of a cleavage site directing the immunochemical detection of molecular abnormalities in type IIA von Willebrand factor. Proc Natl Acad Sci USA. 1990;87(16):6306–10. https://doi.org/10.1073/pnas.87.16.6306.
Furlan M, Robles R, Lämmle B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996;87(10):4223–34.
Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–9. https://doi.org/10.1182/blood-2002-05-1401 (Epub 2002 Jul 25 PMID: 12393397).
Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, et al. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood. 2004;103(6):2150–6. https://doi.org/10.1182/blood-2003-08-2956 (Epub 2003 Nov 20 PMID: 14630802).
Chauhan AK, Goerge T, Schneider SW, Wagner DD. Formation of platelet strings and microthrombi in the presence of ADAMTS-13 inhibitor does not require P-selectin or β3 integrin. J Thromb Haemost. 2007;5(3):583–9.
Plaimauer B, Zimmermann K, Völkel D, Antoine G, Keschbaumer R, Jenab P, et al. Cloning, expression, and functional characterization of the von Willebrand factor-cleaving protease (ADAMTS13). Blood. 2002;100(10):3626–32. https://doi.org/10.1182/blood-2002-05-1397 (Epub 2002 Jul 12).
Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Ishikawa M, Iwamoto T, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005;106(3):922–4. https://doi.org/10.1182/blood-2005-01-0152 (Epub 2005 Apr 26).
Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS-13. J Thromb Haemost. 2006;4:1396–404.
Cao WJ, Niiya M, Zheng XW, Shang DZ, Zheng XL. Inflammatory cytokines inhibit ADAMTS13 synthesis in hepatic stellate cells and endothelial cells. J Thromb Haemost. 2008;6(7):1233–5. https://doi.org/10.1111/j.1538-7836.2008.02989.x (Epub 2008 Jul 1).
Hoffmann M, Kleine-Weber H, Schroeder S, Klüger N, Herrier T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271-280.e8. https://doi.org/10.1016/j.cell.2020.02.052 (Epub 2020 Mar 5).
Lukiw WJ, Pogue A, Hill JM. SARS-CoV-2 infectivity and neurological targets in the brain. Cell Mol Neurobiol. 2020;25:1–8. https://doi.org/10.1007/s10571-020-00947-7.
Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. 2020;13(1):120. https://doi.org/10.1186/s13045-020-00954-7.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laeger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. New Engl J Med. 2020;383(2):120–8.
Fox SE, Akmatbekov A, Harbet JL, Guang L, Brown JQ, Van der Heide RS. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med. 2020;8(7):681–6.
Rapkiewiewicz AV, Mai X, Carson SE, Pittaluga S, Kleiner DE, Berger JS, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClin Med. 2020;24:100434. https://doi.org/10.1016/j.eclinm.2020 (100434.eCollection 2020 Jul).
Thachill J, Lisman T. Pulmonary megakaryocytes in coronavirus disease 2019 (COVID-19): roles in thrombi and fibrosis (review). Semin Thromb Hemost. 2020;46(7):831–4. https://doi.org/10.1055/s-0040-1714274 (Epub 2020 Sep 3).
Aschoff L. Üeber capilläre Embolie von riesenkernhaltigen Zellen. Virchows. Arch Pathol Anat Physiol Klin Med. 1893;134:11–26.
Howell WH, Donahue DD. The production of blood platelets in the lungs. J Exp Med. 1937;65:177–203.
Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544(7648):105–9. https://doi.org/10.1038/nature21706 (Epub 2017 Mar 22).
Nauen DW, Hooper JE, Stewart CM, Solomon IH. Assessing brain capillaries in coronavirus disease 2019. JAMA Neurol. 2021;78(6):760–2. https://doi.org/10.1001/jamaneurol.2021.0225.
Peyvandi F, Artoni A, Novembrino C, Aliberti S, Panigada M, Boscarino M, et al. Hemostatic alterations in COVID-19. Haematologica. 2021;106(5):1472–5. https://doi.org/10.3324/haematol.2020.262634.
Mancini I, Baronciani L, Artoni A, Colpani P, Biganzoli M, Cozzi G, et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J Thromb Haemost. 2021;19(2):513–21. https://doi.org/10.1111/jth.15191.
Bomhof G, Mutsaers PGNJ, Leebeek FWG, Te Boekhorst PAW, Hofland J, Croles FN, et al. COVID-19-associated immune thrombocytopenia. Br J Haematol. 2020;190(2):e61–4. https://doi.org/10.1111/bjh.16850.
Sedaghat Z, Karimi N. Guillain-Barre syndrome associated with COVID-19 infection: a case report. J Clin Neurosci. 2020;76:233–5.
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, et al. Coagulopathy and antiphospholipid antibodies in patients with Covid-19. New Engl J Med. 2020;382: e38.
Zulfiqar AA, Lorenzo-Villalba N, Hassler P, Andrès E. Immune thrombocytopenic purpura in a patient with Covid-19. New Engl J Med. 2020;382(18): e43.
Khandelwal P, Krishnasamy S, Govindarajan S, Kumar M, Marik B, Sinha A, et al. Anti-factor H antibody associated hemolytic uremic syndrome following SARS-CoV-2 infection. Pediatr Nephrol. 2022. https://doi.org/10.1007/s00467-021-05390-4.
Szóstek-Mioduchowska A, Kordowitzki P. Shedding Light on the Possible Link between ADAMTS13 and vaccine-induced thrombotic thrombocytopenia. Cells. 2021;10(10):2785. https://doi.org/10.3390/cells10102785 (PMID: 34685765).
Kelton JG, Arnold DM, Nazy I. Lessons from vaccine-induced immune thrombotic thrombocytopenia. Nat Rev Immunol. 2021;21(12):753–5. https://doi.org/10.1038/s41577-021-00642-8.
Root-Bernstein R. COVID-19 coagulopathies: Human blood proteins mimic SARS-CoV-2 virus, vaccine proteins and bacterial co-infections inducing autoimmunity: Combinations of bacteria and SARS-CoV-2 synergize to induce autoantibodies targeting cardiolipin, cardiolipin-binding proteins, platelet factor 4, prothrombin, and coagulation factors. BioEssays. 2021;43(12): e2100158. https://doi.org/10.1002/bies.202100158 (Epub 2021 Oct 22).
Hayakawa M, Takano K, Kayashima M, Kasahara K, Fukushima H, Matsumoto M. Management of a COVID-19 Patient during ECMO: Paying attention to acquired von Willebrand syndrome. J Atheroscler Thromb. 2020. https://doi.org/10.5551/jat.58362.
Tauber H, Ott H, Streif W, Weigel G, Loacker L, Fritz J, et al. Extracorporeal membrane oxygenation induces short-term loss of high-molecular-weight von Willebrand factor multimers. Anesthesia Analg. 2015;120(4):730–6.
Arulkumaran N, Thomas M, Brealey D, Alwan F, Singh D, Lunn M, et al. Plasma exchange for COVID-19 thrombo-inflammatory disease. EJHaem. 2020. https://doi.org/10.1002/jha2.140.
Pangburn MK, Müller-Eberhard HJ. Relation of putative thioester bond in C3 to activation of the alternative pathway and the binding of C3b to biological targets of complement. J Exp Med. 1980;152(4):1102–14. https://doi.org/10.1084/jem.152.4.1102.
Mathern DR, Peter S, Heeger PS. Molecules great and small: the complement system. Clin J Am Soc Nephrol. 2015;10(9):1636–50. https://doi.org/10.2215/CJN.06230614.
Law SK, Levine RP. Interaction between the third complement protein and cell surface macromolecules. Proc Natl Acad Sci USA. 1977;74(7):2701–5. https://doi.org/10.1073/pnas.74.7.2701.
Pangburn MK, Ferreira VP, Cortes C. Discrimination between host and pathogens by the complement system. Vaccine. 2008;26(Suppl 8):I15-21. https://doi.org/10.1016/j.vaccine.2008.11.023.
Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. New Engl J Med. 2009;361(17):1676–87.
Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, Brodsky RA. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. 2020;136(18):2080–9. https://doi.org/10.1182/blood.2020008248.
Sissy CE, Saldman A, Zanetta G, et al. COVID-19 as a potential trigger of complement-mediated atypical HUS. Blood. 2021;138(18):1777–82. https://doi.org/10.1182/blood.2021012752.
McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S, et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation. 2021;143(8):865–8. https://doi.org/10.1161/CIRCULATIONAHA.120.052824 (Epub 2021 Jan 6).
Cappy P, Candotti D, Sauvage V, Lucas Q, Boizeau L, Gomez J, et al. No evidence of SARS-CoV-2 transfusion transmission despite RNA detection in blood donors showing symptoms after donation. Blood. 2020;136(16):1888–91. https://doi.org/10.1182/blood.2020008230.
Larsen MD, de Graaf EL, Sonneveld ME, Plomp HR, Nouta J, Hoepel W, et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science. 2021;371(6532):eabc 8378. https://doi.org/10.1126/science.abc8378 (Epub 2020 Dec 23).
Chakraborty S, Gonzalez J, Edwards K, Mallajosyula V, Buzzanco AS, Sherwood R, et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat Immunol. 2021;22(1):67–73. https://doi.org/10.1038/s41590-020-00828-7 (Epub 2020 Nov 9).
Bye AP, Hoepel W, Mitchell JL, Jégouic S, Loureiro S, Sage T, et al. Aberrant glycosylation of anti-SARS-CoV-2 spike IgG is a pro-thrombotic stimulus for platelets. Blood. 2021;138(16):1481–9. https://doi.org/10.1182/blood.2021011871.
Levy GG, Nichols WC, Lian EC, Foroud F, McClintick JN, McGee BM, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488–94. https://doi.org/10.1038/35097008.
Fujimura Y, Matsumoto M, Isonishi A, Yagi H, Kokame K, Soejima K, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9(Suppl 1):283–301. https://doi.org/10.1111/j.1538-7836.2011.04341 (State-of-Art Review 2011).
Kremer-Hovinga JA, George J. Hereditary thrombotic thrombocytopenic purpura. N Engl J Med. 2019;381(17):1653–62. https://doi.org/10.1056/NEJMra1813013.
Fujimura Y, Lämmle B, Tanabe S, Sakai K, Kimura T, Kokame K, et al. Patent ductus arteriosus generates neonatal hemolytic jaundice with thrombocytopenia in Upshaw-Schulman syndrome. Blood Adv. 2019;3(21):3191–5. https://doi.org/10.1182/bloodadvances.2019000601.
Alwan F, Vendramin C, Liesner R, Clark A, Lester W, Dutt T, et al. Characterization and treatment of congenital thrombotic thrombocytopenic purpura. Blood. 2019;133(15):1644–51. https://doi.org/10.1182/blood-2018-11-884700.
Tarasco E, Bütikofer L, Friedman KD, George JN, Hrachovinova I, Knöbl PN, et al. Annual incidence and severity of acute episodes in hereditary thrombotic thrombocytopenic purpura. Blood. 2021;137(25):3563–75. https://doi.org/10.1182/blood.2020009801.
Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004;104(1):100–6.
Turner NA, Moake J. Assembly and activation of alternative complement components on endothelial cell-anchored ultra-large von Willebrand factor links complement and hemostasis-thrombosis. PLoS ONE. 2013;8(3): e59372. https://doi.org/10.1371/journal.pone.0059372 (Epub 2013 Mar 29).
Jose RJ, Manuel A. COVID-19 cytokine storm: the interplay between inflammation and coagulation. Lancet Respir Med. 2020;8(6):e46–7. https://doi.org/10.1016/S2213-2600(20)30216-2 (Epub 2020 Apr 27).
Starke R, Ferraro F, Paschalaki KE, Dryden NH, McKinnon TA, Sutton RE, et al. Endothelial von Willebrand factor regulates angiogenesis. Blood. 2011;117:1071–80.
Randi AM, Laffan MA. Von Willebrand factor and angiogenesis: basic and applied issues. J Thromb Haemost. 2017;15(1):13–20. https://doi.org/10.1111/jth.13551.
Acknowledgements
We dedicate this review to the late professor Theodore S. Zimmerman of Scripps Clinic and Research Foundation, who initiated his research career in complement followed by VWF in the early 1970’s, and subsequently invited both of us to join his research on the structure and function relationships of VWF in 1984. This work was supported in part by the Ministry of Health, Labour and Welfare of Japan (MHLW) Research Program on Rare and Intractable Diseases Grant Number JPMH20FC1024 (Director: Prof. Eriko Morishita of Kanazawa University School of Medicine).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
YF is a recipient of patent royalty for ADAMTS13 activity-ELISA from Alfresa Co. and has received consulting fees from Kainos Co. and Alexion Inc. He also serves as a senior scientist of Japanese Red Cross Kinki Block Blood Center. LZH declares no conflicts of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Fujimura, Y., Holland, L.Z. COVID-19 microthrombosis: unusually large VWF multimers are a platform for activation of the alternative complement pathway under cytokine storm. Int J Hematol 115, 457–469 (2022). https://doi.org/10.1007/s12185-022-03324-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-022-03324-w