
Abstract
Background and aims
Cholestatic liver disease is a leading referral to pediatric liver transplant centers. Inherited disorders are the second most frequent cause of cholestasis in the first month of life.
Methods
We retrospectively characterized the genotype and phenotype of 166 participants with intrahepatic cholestasis, and re-analyzed phenotype and whole-exome sequencing (WES) data from patients with previously undetermined genetic etiology for newly published genes and novel candidates. Functional validations of selected variants were conducted in cultured cells.
Results
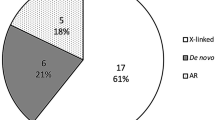
Overall, we identified disease-causing variants in 31% (52/166) of our study participants. Of the 52 individuals, 18 (35%) had metabolic liver diseases, 9 (17%) had syndromic cholestasis, 9 (17%) had progressive familial intrahepatic cholestasis, 3 (6%) had bile acid synthesis defects, 3(6%) had infantile liver failure and 10 (19%) had a phenocopy of intrahepatic cholestasis. By reverse phenotyping, we identified a de novo variant c.1883G > A in FAM111B of a case with high glutamyl transpeptidase (GGT) cholestasis. By re-analyzing WES data, two patients were newly solved, who had compound heterozygous variants in recently published genes KIF12 and USP53, respectively. Our additional search for novel candidates in unsolved WES families revealed four potential novel candidate genes (NCOA6, CCDC88B, USP24 and ATP11C), among which the patients with variants in NCOA6 and ATP11C recapitulate the cholestasis phenotype in mice models.
Conclusions
In a single-center pediatric cohort, we identified monogenic variants in 22 known human intrahepatic cholestasis or phenocopy genes, explaining up to 31% of the intrahepatic cholestasis patients. Our findings suggest that re-evaluating existing WES data from well-phenotyped patients on a regular basis can increase the diagnostic yield for cholestatic liver disease in children.





Similar content being viewed by others

References
Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the evaluation of cholestatic jaundice in infants: joint recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017;64:154–168
Vaisitti T, Peritore D, Magistroni P, Ricci A, Lombardini L, Gringeri E, et al. The frequency of rare and monogenic diseases in pediatric organ transplant recipients in Italy. Orphanet J Rare Dis. 2021;16:374
Karpen SJ, Kamath BM, Alexander JJ, Ichetovkin I, Rosenthal P, Sokol RJ, et al. Use of a comprehensive 66-gene cholestasis sequencing panel in 2171 cholestatic infants, children, and young adults. J Pediatr Gastroenterol Nutr. 2021;72:654–660
Wang NL, Lu Y, Gong JY, Xie XB, Lin J, Abuduxikuer K, et al. Molecular findings in children with inherited intrahepatic cholestasis. Pediatr Res. 2020;87:112–117
Shagrani M, Burkholder J, Broering D, Abouelhoda M, Faquih T, El-Kalioby M, et al. Genetic profiling of children with advanced cholestatic liver disease. Clin Genet. 2017;92:52–61
Maddirevula S, Shamseldin HE, Sirr A, AlAbdi L, Lo RS, Ewida N, et al. Exploiting the autozygome to support previously published mendelian gene-disease associations: an update. Front Genet. 2020;11: 580484
Wang C, Han Y, Zhou J, Zheng B, Zhou W, Bao H, et al. Splicing characterization of CLCNKB variants in four patients with type iii Barter syndrome. Front Genet. 2020;11:81
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424
Zhu YJ, Crawford SE, Stellmach V, Dwivedi RS, Rao MS, Gonzalez FJ, et al. Coactivator PRIP, the peroxisome proliferator-activated receptor-interacting protein, is a modulator of placental, cardiac, hepatic, and embryonic development. J Biol Chem. 2003;278:1986–1990
Naik J, de Waart DR, Utsunomiya K, Duijst S, Mok KH, Oude Elferink RP, et al. ATP8B1 and ATP11C: two lipid flippases important for hepatocyte function. Dig Dis. 2015;33:314–318
Ananthanarayanan M, Li Y, Surapureddi S, Balasubramaniyan N, Ahn J, Goldstein JA, et al. Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am J Physiol Gastrointest Liver Physiol. 2011;300:G771-781
Siggs OM, Schnabl B, Webb B, Beutler B. X-linked cholestasis in mouse due to mutations of the P4-ATPase ATP11C. Proc Natl Acad Sci USA. 2011;108:7890–7895
de Waart DR, Naik J, Utsunomiya KS, Duijst S, Ho-Mok K, Bolier AR, et al. ATP11C targets basolateral bile salt transporter proteins in mouse central hepatocytes. Hepatology. 2016;64:161–174
Ham H, Huynh W, Schoon RA, Vale RD, Billadeau DD. HkRP3 is a microtubule-binding protein regulating lytic granule clustering and NK cell killing. J Immunol. 2015;194:3984–3996
Girard M, Bizet AA, Lachaux A, Gonzales E, Filhol E, Collardeau-Frachon S, et al. DCDC2 mutations cause neonatal sclerosing cholangitis. Hum Mutat. 2016;37:1025–1029
Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet. 2017;49:269–273
Zhang L, Lubin A, Chen H, Sun Z, Gong F. The deubiquitinating protein USP24 interacts with DDB2 and regulates DDB2 stability. Cell Cycle. 2012;11:4378–4384
Maddirevula S, Alhebbi H, Alqahtani A, Algoufi T, Alsaif HS, Ibrahim N, et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet Med. 2019;21:1164–1172
Feldman AG, Sokol RJ. Neonatal cholestasis: emerging molecular diagnostics and potential novel therapeutics. Nat Rev Gastroenterol Hepatol. 2019;16:346–360
Chen HL, Li HY, Wu JF, Wu SH, Chen HL, Yang YH, et al. Panel-based next-generation sequencing for the diagnosis of cholestatic genetic liver diseases: clinical utility and challenges. J Pediatr. 2019;205(153–159): e156
Togawa T, Sugiura T, Ito K, Endo T, Aoyama K, Ohashi K, et al. Molecular genetic dissection and neonatal/infantile intrahepatic cholestasis using targeted next-generation sequencing. J Pediatr. 2016;171(171–177):e171-174
Lipinski P, Ciara E, Jurkiewicz D, Pollak A, Wypchlo M, Ploski R, et al. Targeted next-generation sequencing in diagnostic approach to monogenic cholestatic liver disorders-single-center experience. Front Pediatr. 2020;8:414
Chen HL, Wu SH, Hsu SH, Liou BY, Chen HL, Chang MH. Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J Biomed Sci. 2018;25:75
Esteve C, Francescatto L, Tan PL, Bourchany A, De Leusse C, Marinier E, et al. Loss-of-function mutations in UNC45A cause a syndrome associating cholestasis, diarrhea, impaired hearing, and bone fragility. Am J Hum Genet. 2018;102:364–374
Shaheen R, Alsahli S, Ewida N, Alzahrani F, Shamseldin HE, Patel N, et al. Biallelic mutations in tetratricopeptide repeat domain 26 (intraflagellar transport 56) cause severe biliary ciliopathy in humans. Hepatology. 2020;71:2067–2079
Pham DH, Kudira R, Xu L, Valencia CA, Ellis JL, Shi T, et al. Deleterious variants in ABCC12 are detected in idiopathic chronic cholestasis and cause intrahepatic bile duct loss in model organisms. Gastroenterology. 2021;161(287–300): e216
Sultan M, Rao A, Elpeleg O, Vaz FM, Abu-Libdeh B, Karpen SJ, et al. Organic solute transporter-beta (SLC51B) deficiency in two brothers with congenital diarrhea and features of cholestasis. Hepatology. 2018;68:590–598
Luan W, Hao CZ, Li JQ, Wei Q, Gong JY, Qiu YL, et al. Biallelic loss-of-function ZFYVE19 mutations are associated with congenital hepatic fibrosis, sclerosing cholangiopathy and high-GGT cholestasis. J Med Genet. 2021;58:514–525
Pan Q, Luo G, Qu J, Chen S, Zhang X, Zhao N, et al. A homozygous R148W mutation in Semaphorin 7A causes progressive familial intrahepatic cholestasis. EMBO Mol Med. 2021;13: e14563
Kazmierczak M, Harris SL, Kazmierczak P, Shah P, Starovoytov V, Ohlemiller KK, et al. Progressive hearing loss in mice carrying a mutation in Usp53. J Neurosci. 2015;35:15582–15598
Zhang J, Yang Y, Gong JY, Li LT, Li JQ, Zhang MH, et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: clinical, histological and ultrastructural characterization. Liver Int. 2020;40:1142–1150
Vij M, Sankaranarayanan S. Biallelic mutations in ubiquitin-specific peptidase 53 (USP53) Causing progressive intrahepatic cholestasis. Report of a case with review of literature. Pediatr Dev Pathol. 2022;25:207–212
Shatokhina O, Semenova N, Demina N, Dadali E, Polyakov A, Ryzhkova O. A two-year clinical description of a patient with a rare type of low-GGT cholestasis caused by a novel variant of USP53. Genes (Basel). 2021;12:1618
Unlusoy Aksu A, Das SK, Nelson-Williams C, Jain D, OzbayHosnut F, EvirgenSahin G, et al. Recessive mutations in KIF12 cause high gamma-glutamyl transferase cholestasis. Hepatol Commun. 2019;3:471–477
Stalke A, Sgodda M, Cantz T, Skawran B, Lainka E, Hartleben B, et al. KIF12 variants and disturbed hepatocyte polarity in children with a phenotypic spectrum of cholestatic liver disease. J Pediatr. 2022;240(284–291): e289
Khumalo NP, Pillay K, Beighton P, Wainwright H, Walker B, Saxe N, et al. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br J Dermatol. 2006;155:1057–1061
Mercier S, Kury S, Salort-Campana E, Magot A, Agbim U, Besnard T, et al. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10:135
Seo A, Walsh T, Lee MK, Ho PA, Hsu EK, Sidbury R, et al. FAM111B mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas. 2016;45:858–862
Takimoto-Sato M, Miyauchi T, Suzuki M, Ujiie H, Nomura T, Ikari T, et al. Case report: hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis (POIKTMP) presenting with liver cirrhosis and steroid-responsive interstitial pneumonia. Front Genet. 2022;13: 870192
Mahajan MA, Das S, Zhu H, Tomic-Canic M, Samuels HH. The nuclear hormone receptor coactivator NRC is a pleiotropic modulator affecting growth, development, apoptosis, reproduction, and wound repair. Mol Cell Biol. 2004;24:4994–5004
Li Q, Chu MJ, Xu J. Tissue- and nuclear receptor-specific function of the C-terminal LXXLL motif of coactivator NCoA6/AIB3 in mice. Mol Cell Biol. 2007;27:8073–8086
Arashiki N, Takakuwa Y, Mohandas N, Hale J, Yoshida K, Ogura H, et al. ATP11C is a major flippase in human erythrocytes and its defect causes congenital hemolytic anemia. Haematologica. 2016;101:559–565
Funding
This work was supported by grants from the National Natural Science Foundation of China (no. 81873542) and Nanjing Medical Science and technique Development Foundation, Nanjing Department of Health (no. YKK19107).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Yucan Zheng, Hongmei Guo, Leilei Chen, Weixia Cheng, Kunlong Yan, Zhihua Zhang, Mei Li, Yu Jin, Guorui Hu, Chunli Wang, Chunlei Zhou, Wei Zhou, Zhanjun Jia, Bixia Zheng, and Zhifeng Liu disclose no conflicts.
Ethical approval
The study was approved by the Ethics Committee of Children’s Hospital of Nanjing Medical University (no.202012090-1). A written informed consent was obtained from all patients or their guardians.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zheng, Y., Guo, H., Chen, L. et al. Diagnostic yield and novel candidate genes by next generation sequencing in 166 children with intrahepatic cholestasis. Hepatol Int 18, 661–672 (2024). https://doi.org/10.1007/s12072-023-10553-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-023-10553-6