
Abstract
Several preclinical and clinical studies have attempted to elucidate the pathophysiological mechanism associated with spinal cord injury. However, investigations have been unable to define the precise related mechanisms, and this has led to the lack of effective therapeutic agents for the condition. Neuroinflammation is one of the predominant processes that hinder spinal cord injury recovery. Resveratrol is a compound that has several biological features, such as antioxidation, antibacterial, and antiinflammation. Herein, we reviewed preclinical and clinical studies to delineate the role of toll-like receptors, nod-like receptors, and astrocytes in neuroinflammation. In particular, the alteration of astrocytes in SCI causes glial scar formation that impedes spinal cord injury recovery. Therefore, to improve injury recovery would be to prevent the occurrence of this process. Resveratrol is safe and effective in the significant modulation of neuroinflammatory factors, particularly those mediated by astrocytes. Thus, its potential ability to enhance the injury recovery process and ameliorate spinal cord injury.
Similar content being viewed by others

Introduction
When any part of the spinal cord is injured, it causes either permanent loss or reduction of physical function below the damaged area [1]. According to present data, the annual incidence of spinal cord injury (SCI) is approximately 54 cases per million people in the United States, or about 17,810 new SCI cases each year [2]. SCI profoundly imperils the physiological systems, such as cardiovascular and immune systems [3]. The clinical complications of SCI include urinary tract infections, autonomic dysreflexia, sudden hypertension, and other symptoms caused by neuroinflammation [4,5,6,7]. Although SCI is a severe condition, its complete remedy is still lacking [8] because of the complex injury mechanisms. Neuroinflammation is not the only basis of secondary SCI but also one of the main reasons underlying the impediment process to SCI repair [9].
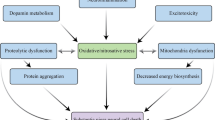
Neuroinflammatory responses in the secondary SCI can induce a series of cellular and molecular events, including activation of microglia/astrocyte, infiltration of macrophages from peripheral blood [10], imbalance of pro-inflammatory and antiinflammatory responses, abnormal mitochondrial activity, oxidative stress [11], abnormal protein aggregation, and free radical toxicity. Inflammasomes are closely related to neuroinflammation [12]. Inflammasomes are large multiprotein complexes that mediate neuroinflammatory responses [13]. Classical inflammasomes are composed of pattern-recognition receptors (PRRs), the adaptor protein known as apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), and the effector protein, pro-caspase-1 [14,15,16]. Pyrin domain-containing related nod-like receptor protein family (NLRP) is a sensor of inflammasomes [17]. The damage-associated molecular patterns (DAMPs) and adenosine triphosphate (ATP) triggers the NLRP2. Moreover, the human astrocytes express the NLRP2 [18]. Activated NLRP2 recruits ASC and pro-caspase-1. During the recruitment process, the complex oligomerizes and assembles to NLRP2 inflammasome. After central nervous system (CNS) injury, especially SCI, ATP released by apoptosis–activated astrocytes lead to inflammatory responses and induces the assembly of the NLRP2 inflammasomes. NLRP2 inflammasome cleaves the pro-caspase-1 and secretes caspase-1, which triggers interleukin-1β (IL-1β) to produce mature IL-1β.
Nuclear factor kappa-B (NF-κB), a typical pro-inflammatory signaling pathway, is closely related to the inflammatory response. In secondary SCI, neuroinflammation triggers the activation of the NF-κB signaling pathway through the classical pathway. The activated NF-κB signaling pathway expresses immature IL-1β, with caspase-1 modifying it to mature IL-1β and contributing to inflammatory responses. Presently, some glucocorticoids are commonly used in the clinic to prevent this mechanism of neuroinflammation caused by secondary SCI. However, long-term clinical use of these agents diminishes their effectiveness, coupled with significant side effects [19]. Therefore, modern interventional agents having minimal to no side effects concomitant with favorable therapeutic effects are needed.
Resveratrol (RSV), as a component of traditional Chinese medicine, is a natural polyphenol and is present in grapes, berries, peanuts, and wine. It has characteristics of antiproliferation, antiangiogenesis, antiinflammation, and antioxidation [20,21,22]. RSV can scavenge the generation of free radicals, inhibit lipid peroxidation, and regulate the activities of oxidation-related enzymes. Studies have shown that RSV can regulate the F1 subunit of ATP synthase [23] and reactive oxygen species (ROS) levels [24]. ATP is closely associated with the activation process of NLRP2 in astrocytes. In addition, RSV can effectively inhibit the activation of the NF-κB signaling pathway by suppressing the activity of inhibitor protein, κB kinase (Iκκ) [25]. RSV can also reduce ATP production and ROS levels to hinder the formation of NLRP2 inflammasome in astrocytes. Further, RSV can inhibit the production of pro-IL-1β by repressing the NF-κB signaling pathway. These inhibitory effects can reduce the occurrence and development of inflammatory response after SCI and accelerate the repair process.
Inflammatory Response After SCI
SCI neuroinflammatory response is relatively complex and encompasses the interaction between the nervous and immune systems. The activation of inflammasomes and inflammatory signaling pathways is a vital factor that causes the over-activation of astrocytes. In addition, some risk-related factors can bind to receptors on astrocytes and lead to their activation.
The Mechanism of Inflammatory Response
Inflammation is a defensive response to stimuli characterized by redness, swelling, fever, and pain. Generally, inflammation is beneficial for the human body; however, over-inflammation can damage tissues of the body to aggravate the disease. Inflammation in the brain and spinal cord is neuroinflammation. The definition of neuroinflammation is broad, as it includes two complex fields: nervous and immune systems [26]. Neuroinflammatory response mediates the expressions of cytokines, chemokines, secondary messengers, and ROS. Most of these factors come from microglia and astrocyte in the CNS. In the wake of CNS injury, the primary stage of neuroinflammation has a positive effect. During this stage, the inflammatory response triggers the immune response to play a protective role. When neuroinflammation develops to the second stage (i.e., chronic stage), instigation of cells of the CNS arises, resulting in aggravated inflammatory factors that impact the process of injury repair. Several neurodegenerative diseases (like Alzheimer’s disease, Parkinson’s disease, traumatic brain injury (TBI), and secondary SCI) have a close association with the chronic developmental stage of neuroinflammation [27, 28].
In a physiological state, microglia, a type of permanent immune cell in the brain, is an ineffective phenotype [27]. There are some risk signal factors in the microenvironment under pathological conditions. DAMP is a risk-related factor released by cells or tissues after receiving external stimuli. The non-self-factors or pathogenic microorganisms in the microenvironment are the pathogen-associated molecular patterns (PAMPs). PRRs are responsible for sensing PAMPs and DAMPs [29]. The main subfamilies of PRRs include toll-like receptors (TLRs), nod-like receptors (NLRs), RIG-like receptors (RLRs), AIM2-like receptors (ALRs), and C-type lectin receptors. NLRs form inflammasomes [29, 30]. The three known NLRs in the CNS are NLRP1, NLRP2, and NLRP3 [31,32,33]. Once PRRs on the microglia detect either DAMPs or PAMPs, microglia immediately switch to activated phenotype, phagocytized injury-related factors, and forms elements that affect astrocytes and neurons, leading to neuroinflammation. In addition, astrocytes and neurons can identify some DAMPs and PAMPs to mediate neuroinflammation. These neuroinflammatory responses could further transmit to the immune system and promote tissue repair. In most cases, these inflammatory responses are transient and would disappear once infection or injury is repaired [26].
The persistent neuroinflammatory response is an indication of physiological mechanisms not being controlled. This leads to the over-activation of glial cells and neurons that produces neurotoxic factors, which aggravates the disease state. The accumulation of signal and endogenous factors (such as inflammasome polymers) can cause a persistent neuroinflammatory response. Infectious signaling factors (such as Gram-negative bacteria, viral double-stranded RNA, and bacterial lipopolysaccharide) are identified by TLRs, with endogenous signaling factors (including IL-1, tumor necrosis factor (TNF), and ATP) detected by NLRs in the PRR family. Presently, most CNS cells express TLRs. TLRs pronounced entirely in the microglia enhance its abilities to monitor the microenvironment. Astrocytes and neurons also express TLRs. Under pathological conditions, astrocytes may upregulate TLR3 [34], TLR2, and TLR4 [34, 35] expressions. These recognized PRRs stimulate the activation of the signal transduction pathway by regulating transcription and posttranscription processes. Notably, TLRs either recruit adapter protein MyD88 or send signals through the TRIF-dependent pathway, leading to transcription of downstream κB kinase and mitogen-activated protein kinase (MAPK). Activated downstream kinases regulate various inflammatory signaling pathways, including NF-κB and activator protein 1 (AP-1). These inflammatory signaling pathways highly express the precursors of interleukin and interferon (such as pro-IL-1) to exacerbate the occurrence of inflammatory response. These factors need further processing and modification to play their role, and caspase-1 is significant in this process. Activated inflammasomes express caspase-1, and NLRs play a vital role as the upstream factors of inflammasomes assembly and activation. NLRs recognize DAMPs and recruits ASC and pro-caspase-1 to further assemble into inflammasomes. Caspase-1 combines with precursors of interleukin and interferon to co-express inflammatory factors [36]. Both TLRs and NLRs cooperate to control immune response dysfunction (Fig. 1). This immune response is usually a feed-forward loop, i.e., the ultimate expressed inflammatory factors are delivered back to the initial recognition receptor as risk signal factors. This further aggravates the process of inflammatory responses. Although some inflammatory responses have beneficial effects and are closely related to the tissue repair process, uncontrolled inflammatory reactions can trigger secondary damage and hinder this repair process.

Combined TLRs and NLRs regulate inflammation
Astrocyte Activation and Inflammasome Formation
Astrocytes are abundant and complex in the mammalian brain, accounting for about 19%–40% of human brain cells [37]. Mature astrocytes have complex structures that interact with synapses to support neurons. CNS injury causes activation of astrocytes. Reactive astrocytes show alterations of morphology and metabolism, including cellular hypertrophy and upregulated expressions of glial fibrillary acidic protein (GFAP) and Nestin.
DAMPs stimulate human astrocytes to generate NLRP2 inflammasomes, which further induces inflammatory responses to form caspase-1 and IL-1β. In addition, NLRP2 inflammasome is significantly involved in immune response and disease occurrence. Therefore, NLRP2 inflammasome, as the core of inflammatory response, may provide a new target for the treatment of various inflammatory diseases. ATP is a representative of DAMP released from injured or dead cells after tissue trauma and can stimulate the activation of inflammasomes [36]. Studies have demonstrated the activation of several NLRs family proteins to be closely associated with the P2X7 receptor and Pannexin 1 channel [38]. Recombinant purinergic receptor P2X, ligand-gated ion channel 7 (P2X7), and Pannexin 1 of membrane proteins are ATP gating proteins [39]. A recent study evidenced that the interaction between NLRP2 inflammasome in astrocytes and P2X7 receptor and pannexin-1 channel protein on the cell membrane can accelerate inflammatory response process and promote caspase-1 expression [32, 36]. In addition, the P2X4 receptor may interact with these two gating proteins and control ATP to stimulate cells [40]. However, there is limited information regarding the expression and stress response of NLRP2 inflammasome in astrocytes after tissue trauma. The activation of NLRP2 inflammasome in astrocytes is related to P2X4/P2X7/Pannexin 1 channel; nonetheless, the specific molecular mechanism remains to be clarified.
After SCI, reactive astrocytes not only express NLRP2 inflammasomes but also migrate and gather at the center of the lesion, which helps repair the damaged tissue. However, excessive cell accumulation can cause scar formation and eventually create a glial scar. A glial scar can inhibit axonal regeneration and intricate injury recovery. Studies have shown that reactive astrocytes in glial scars secrete chondroitin and keratin sulfate proteoglycans, which are involved in inhibiting axonal regeneration [41]. Also, signal transducer and activator of transcription-3 (STAT3) was found in reactive astrocytes after SCI and participated in cell migration and aggregation processes [42].
In the wake of SCI, astrocytes change from primitive state to reactive state to scar-forming astrocytes, which is usually irreversible (Fig. 2). Among them, neuroinflammation caused by reactive astrocytes is beneficial to injury recovery to a certain extent. However, glial scars affect injury recovery. Thus, after SCI, inhibiting the occurrence and development of reactive astrocytes before glial scar formation can influence the injury recovery process.

The alteration of astrocytes from primitive state to reactive state to scar-forming astrocytes after SCI
RSV Can Attenuate Neuroinflammation
Several studies have reported the immense potential of RSV in the treatment of inflammatory diseases. RSV can act on multiple signaling pathways to inhibit inflammatory responses. For instance, RSV degrades intracellular and extracellular risk-related factors and inhibits κB kinase activity. In the wake of SCI, RSV mitigates inflammatory responses by inhibiting the activations of inflammasomes and NF-κB, thereby creating a conducive microenvironment for axonal recovery.
The Biological Functions of RSV
In recent years, RSV has gained the attention of scientists due to its beneficial outcomes in infectious, neurodegenerative, metabolic, and autoimmune diseases, while relieving aging and prolonging life (Table 1). RSV can reduce the concentration of low-density lipoprotein and inhibit cyclooxygenase activity on the cardiovascular system [59, 60]. More so, RSV can reduce oxidative stress via its regulation of ROS [61]. For instance, He LN et al. [62] showed that RSV could inhibit the oxidative stress of rat pulmonary artery endothelial cells under hypoxia by blocking hypoxia-inducible factor 1α (HIF-1α)/Nicotinamide Adenine Dinucleotide Phosphate Oxidase 4 (NOX4)/ROS signaling pathway. More so, RSV inhibited cancer growth by scavenging free radicals on the cell surface. Specifically, RSV induced the death of human ovarian cancer cells by inhibiting Notch1/phosphatase and tensin homolog deleted on chromosome ten (PTEN)/protein kinase B (Akt) signaling pathway mediated by ROS [63].
Concerning neuroinflammatory responses, RSV can prevent these reactions by inhibiting the activation of the inflammatory signaling pathway, such as NF-κB, or reducing the formation of inflammatory factors. Some studies have analyzed the interaction between RSV and vital enzymes, including Iκκ, cyclooxygenase 2 (COX‐2), and TNF receptor-associated factor-associated NF-κB activator binding kinase 1 (TBK1), in the TLR4 pathway [64], which showed the molecular mechanism of RSV in regulating inflammation. Also, studies have revealed that RSV could inhibit inflammation by regulating several inflammatory pathways: TLR4/NF-κB/STAT3, TLR4/Akt/ forkhead box protein O1 (FoxO1), nuclear factor erythroid 2-related factor 2 (Nrf2)/TLR4/NF-κB, and sirtuin1 (Sirt-1)/FoxO1 [65,66,67,68]. All these studies evidence RSV potential as an interventional agent for the treatment of inflammatory-associated diseases.
Studies are currently exploring the positive role of RSV in bacterial infections. For instance, Al Azzaz J et al. [69] found RSV to promote heterogeneous phagocytosis and enhance the clearance of two invasive bacteria (Salmonella typhimurium and invasive Escherichia coli). Similarly, RSV has extensive antiviral activities, mainly in Epstein-Barr virus (EBV), HIV infections, and the Middle East Respiratory Syndrome Coronavirus (MERS-CoV)) [70,71,72]. In particular, RSV suppressed nucleocapsid (N) protein expression, leading to the inhibition of MERS-CoV infection. While the biological absorption rate of RSV in clinical trials has been low, the biological efficacy of RSV derivatives and nano-scale RSV are immensely high [73], implying that RSV effectiveness in the clinical setting may be pronounced following modification.
RSV Attenuates Inflammatory Responses of Diseases
The occurrence of inflammation further induces various diseases, including atherosclerosis, rheumatism, rash, Alzheimer’s disease, tumor, and various secondary symptoms [74,75,76]. RSV has diverse biological effects and is less toxic to the body [77]. It has been employed in various medical conditions, such as inflammatory bowel disease (IBD), nervous system inflammation, eye inflammation, and gynecological inflammation (Table 2).
The antiinflammatory effects of RSV have been attributed to the cascade effects of disparate mechanisms, which lead to the inhibition of both the inducer and intermediate enzyme that mediate the occurrence of inflammatory responses and eventually hindering the production of inflammatory factors. Generally, DAMPs and PAMPs trigger inflammation. RSV curtails DAMPs by scavenging reactive substances and ROS through its antioxidative action [94, 95], while it reduces PAMPs via its antibacterial effect [96, 97], thereby decreasing the incidence of inflammatory responses. Additionally, TLRs and pro-inflammatory cytokines, such as TNF and IL-1, trigger the classical pathway that instigates p65 activation [98]. Noteworthy is that RelA regulates the expression of pro-inflammatory and cell survival genes, and its activation promotes the assembly of inhibitors of NF-κB (IκB). The activation of the NF-κB signaling pathway typically occurs after Iκκ phosphorylates IκB [99]. RSV acts on the intermediate enzyme of the NF-κB signaling pathway to block the transmission of information by the inflammatory pathway. All these functions of RSV aid repress the instigation and development of inflammation. Moreover, studies have shown that RSV could improve autophagy [93] when inflammation occurs.
RSV Curtails Inflammasome Formation After Nervous System Injury
The inflammasome is a multiprotein complex that plays a vital role in the immune system. In general, the recognition of PRRs with PAMPs or DAMPs is the first step in triggering the development of neuroinflammation. Identified PAMPs or DAMPs by PRRs of the inflammasome leads to the recruitment and activation of caspase-1. Activated Caspase-1 can modify the precursor substances of IL-1 or IL-18 to form cytokines that cause an inflammatory response. Significant mitigation of the recognition of risk-related factors and PRRs can decrease inflammasome formation. Therefore, to obviate inflammation is to block the initial recognition mechanism.
The above evidence shows that the development of inflammatory responses requires the cooperation of TLRs with NLRs. The activation of TLRs can stimulate the NF-κB inflammatory signaling pathway, and NLRs activation can trigger inflammasomes. TLRs are present in a variety of cells and are often associated with inflammatory responses. Presently, there are ten different TLRs (TLR1-10) in human cells and twelve in mice [100]. Ahmad SF et al. [101] indicated that RSV could reduce the expression of TLRs/NF-κB/COX-related factors and improve the deterioration of neuro-immune diseases. In this study, RSV effectively downregulated the expressions of TLR2, TLR3, and TLR4. The inhibition of TLR2, TLR3, and TLR4 is a treatment strategy for various nervous system diseases, including neuroinflammation. For instance, Kwilasz AJ et al. [102] showed TLR2/TLR4 antagonists to effectively alleviate neuroinflammation and memory decline caused by experimental autoimmune diseases. The suppression of TLRs expression affects the activation of inflammatory pathways and the secretion of related cytokines, thereby curtailing the inflammatory response. Previous studies have shown RSV to repress the expression of NLRP3 inflammasome [56, 103]. However, the study did not show RSV to directly act on inflammasome, but rather through an indirect effect. The activation of inflammasome was caused by NLRs recognizing risk signaling factors. Therefore, a vital step in inflammatory signaling pathways and inflammasome activation may be the recognition of PRRs with risk-related factors. The discerning of these injury factors by PRRs may further promote the inflammatory response that could lead to injury aggravation [104]. Also, RSV can directly act on these risk-related factors, resulting in the downregulated expression of these factors and significantly hindering the assembly process of inflammasomes (Fig. 3).

RSV inhibits the inflammatory response through multiple pathways. RSV hinders assembly and stimulation of TLRs by oxidizing degradation of risk factors associated with the activation of inflammatory responses, DAMPs and PAMPs, such as ATP and ROS. Furthermore, the inhibition of DAMPs and PAMPs prevents the activation of P2X4, P2X7, and Pannexin 1, thereby indirectly inhibiting the activation of inflammasomes, NLRP2. In addition, RAV is an inhibitor of κB kinase activation, which blocks the NF-κB signaling pathway by suppressing p65 activation. RSV significantly reduces the inflammatory response and repress astrocyte activation through the above-mentioned mechanisms
RSV Can Improve the Repair of SCI by Inhibiting Inflammatory Responses
The occurrence of inflammatory response is an inevitable secondary symptom of SCI. However, neuroinflammation has a two-way effect on SCI repair: positive inflammatory response is beneficial to tissue regeneration, while negative inflammatory response inhibits the injury repair process. In secondary SCI, the activation of glial cells and subsequent release of inflammatory factors and interferon accelerate neuronal death and induce vascular endothelial cells to express various cell adhesion and chemotactic molecules that attract more inflammatory factors [105]. The intensification of inflammatory response is the primary cause of secondary SCI and is also a considerable hindrance in the SCI repair process. Chronic inflammation instigates the over-activation of motor neuronal and glial cells that leads to CNS dysfunction and form scar tissue, which impedes the process of SCI repair. Hence, improving the immune microenvironment of the spinal cord during secondary SCI is one of the prime treatment goals [106]. It is, therefore, necessary to find an effective treatment that can inhibit the inflammatory response and promote SCI repair. Studies at the cellular level have shown extracellular vesicles of mesenchymal stromal cells filled with at the injured area of the spinal cord to significantly alleviate inflammatory response and inhibit glial scar formation [107]. Also, another study at the molecular level showed progranulin deficiency to promote neuroinflammation and apoptosis and aggravate SCI [108].
Currently, increased attention is being afforded to the considerable impact of Chinese medicinal extracts for neuroinflammation management induced by SCI. In particular, the antiinflammatory and antioxidative effects of RSV play an immense role in secondary SCI. Prior studies have shown RSV to hinder inflammatory response in SCI by repressing and triggering the NF-κB inflammatory signaling pathway and Sirt-1 signaling pathway, respectively [109]. Moreover, Menghay et al. [110] showed that RSV could regulate adenosine 5′-monophosphate-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) signaling pathway to improve neuroprotective functions after SCI. Another study also demonstrated that RSV promoted autophagy and recovery of motor neurons by regulating the Sirt-1/AMPK signaling pathway after SCI [111]. In recent years, study outcomes concerning the employment of RSV have been encouraging and have boosted its position as a potential interventional agent for alleviation of secondary SCI.
Conclusion
SCI is a neurological disease that is difficult to treat, and chronic neuroinflammation and glial scar formation aggravate its recovery process. Current investigations are predominantly focusing on the treatment of neuroinflammation. In the wake of SCI, astrocyte-mediated neuroinflammation occurs within a short time. Transient inflammatory response is conducive to injury recovery. However, excessive neuroinflammation provokes cells to secrete detrimental factors that promote the over-activation of reactive astrocytes and further transforms to scar-forming astrocytes. The formation of glial scar complicates SCI recuperation. Therefore, it is imperative to find novel treatment strategies that suppress this process and promote injury repair.
In recent years, biological functions (such as antiinflammation and antioxidation) of RSV have had significant treatment effects on SCI. Specifically, RSV can downregulate the expression of injury-related factors, prevent cells that inhibit the inflammatory pathway, and reduce the materialization of inflammatory reactions. Although the antiinflammatory effect of RSV has been evident in experiments, its specific action mechanism is yet to be clarified. Nonetheless, RSV can minimize the activity of inflammatory factors through its antioxidant effect, and inhibit inflammatory signaling pathways through its antiinflammatory properties, thereby hindering astrocyte-mediated inflammatory response, curtailing glial scar formation, and promoting SCI repair. Studies addressing the precise mechanism of this process would provide the molecular basis for RSV employment in treating SCI. Recent completed clinical trial investigations have shown the significant effects of RSV [112,113,114]. For example, clinical studies have evinced the effectiveness of high-dose RSV with no serious adverse effects [115, 116]. Although these studies did show the efficacy of RSV, more clinical investigations are needed for a better understanding of its safety in the clinical setting (Tab. 1 and 2).
Data Availability
I confirm that I have included a citation for available data in my reference section.
References
Holmes D (2017) Spinal-cord injury: spurring regrowth. Nature 552(7684):S49. https://doi.org/10.1038/d41586-017-07550-9
Spinal Cord Injury Facts and Figures at a Glance. National Spinal Cord Injury Statistical Center. https://www.nscisc.uab.edu/Public/Facts%20and%20Figures%202020.pdf
Mironets E, Fischer R, Bracchi-Ricard V, Saltos TM, Truglio TS, O’Reilly ML, Swanson KA, Bethea JR, Tom VJ (2020) Attenuating Neurogenic Sympathetic Hyperreflexia Robustly Improves Antibacterial Immunity After Chronic Spinal Cord Injury. J Neurosci 40(2):478–492. https://doi.org/10.1523/JNEUROSCI.2417-19.2019
Skelton-Dudley F, Doan J, Suda K, Holmes SA, Evans C, Trautner B (2019) Spinal cord injury creates unique challenges in diagnosis and management of catheter-associated urinary tract infection. Top Spinal Cord Inj Rehabil 25(4):331–339. https://doi.org/10.1310/sci2504-331
Eldahan KC, Rabchevsky AG (2018) Autonomic dysreflexia after spinal cord injury: systemic pathophysiology and methods of management. Auton Neurosci 209:59–70. https://doi.org/10.1016/j.autneu.2017.05.002
Phillips AA, Matin N, Jia M, Squair JW, Monga A, Zheng MMZ, Sachdeva R, Yung A, Hocaloski S, Elliott S, Kozlowski P, Dorrance AM, Laher I, Ainslie PN, Krassioukov AV (2018) Transient Hypertension after Spinal Cord Injury Leads to Cerebrovascular Endothelial Dysfunction and Fibrosis. J Neurotrauma 35(3):573–581. https://doi.org/10.1089/neu.2017.5188
Goodes LM, King GK, Rea A, Murray K, Boan P, Watts A, Bardsley J, Hartshorn C, Thavaseelan J, Rawlins M, Brock JA, Dunlop SA (2020) Early urinary tract infection after spinal cord injury: a retrospective inpatient cohort study. Spinal Cord 58(1):25–34. https://doi.org/10.1038/s41393-019-0337-6
Hurlbert RJ, Hadley MN, Walters BC, Aarabi B, Dhall SS, Gelb DE, Rozzelle CJ, Ryken TC, Theodore N (2015) Pharmacological therapy for acute spinal cord injury. Neurosurgery 76(Suppl 1):S71-83. https://doi.org/10.1227/01.neu.0000462080.04196.f7
Wang C, Wang Q, Lou Y, Xu J, Feng Z, Chen Y, Tang Q, Zheng G, Zhang Z, Wu Y, Tian N, Zhou Y, Xu H, Zhang X (2018) Salidroside attenuates neuroinflammation and improves functional recovery after spinal cord injury through microglia polarization regulation. J Cell Mol Med 22(2):1148–1166. https://doi.org/10.1111/jcmm.13368
Fan B, Wei Z, Yao X, Shi G, Cheng X, Zhou X, Zhou H, Ning G, Kong X, Feng S (2018) Microenvironment Imbalance of Spinal Cord Injury. Cell Transplant 27(6):853–866. https://doi.org/10.1177/0963689718755778
Lv R, Du L, Zhang L, Zhang Z (2019) Polydatin attenuates spinal cord injury in rats by inhibiting oxidative stress and microglia apoptosis via Nrf2/HO-1 pathway. Life Sci 217:119–127. https://doi.org/10.1016/j.lfs.2018.11.053
Sun Z, Nyanzu M, Yang S, Zhu X, Wang K, Ru J, Yu E, Zhang H, Wang Z, Shen J, Zhuge Q, Huang L (2020) VX765 Attenuates Pyroptosis and HMGB1/TLR4/NF-κB Pathways to Improve Functional Outcomes in TBI Mice. Oxid Med Cell Longev 2020:7879629. https://doi.org/10.1155/2020/7879629
Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, Liu X, Huang YJ, Cai H, Zhan XY, Han QY, Wang H, Chen Y, Li HY, Li AL, Zhang XM, Zhou T, Li T (2017) NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell 68(1):185-197.e6. https://doi.org/10.1016/j.molcel.2017.08.017
Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 Inflammasome: an Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 20(13):3328. https://doi.org/10.3390/ijms20133328
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H, Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156(6):1193–1206. https://doi.org/10.1016/j.cell.2014.02.008
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J (2004) NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20(3):319–325. https://doi.org/10.1016/s1074-7613(04)00046-9
Poli G, Fabi C, Bellet MM, Costantini C, Nunziangeli L, Romani L, Brancorsini S (2020) Epigenetic mechanisms of inflammasome regulation. Int J Mol Sci 21(16):5758. https://doi.org/10.3390/ijms21165758
Zhang Q, Sun Y, He Z, Xu Y, Li X, Ding J, Lu M, Hu G (2020) Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain Behav Immun 88:471–481. https://doi.org/10.1016/j.bbi.2020.04.016
Stout A, Friedly J, Standaert CJ (2019) Systemic absorption and side effects of locally injected glucocorticoids. PM R 11(4):409–419. https://doi.org/10.1002/pmrj.12042
Ho Y, Sh Yang YC, Chin YT, Chou SY, Chen YR, Shih YJ, Whang-Peng J, Changou CA, Liu HL, Lin SJ, Tang HY, Lin HY, Davis PJ (2018) Resveratrol inhibits human leiomyoma cell proliferation via crosstalk between integrin αvβ3 and IGF-1R. Food Chem Toxicol 120:346–355. https://doi.org/10.1016/j.fct.2018.07.030
Popescu M, Bogdan C, Pintea A, Rugină D, Ionescu C (2018) Antiangiogenic cytokines as potential new therapeutic targets for resveratrol in diabetic retinopathy. Drug Des Devel Ther 12:1985–1996. https://doi.org/10.2147/DDDT.S156941
Jia R, Li Y, Cao L, Du J, Zheng T, Qian H, Gu Z, Jeney G, Xu P, Yin G (2019) Antioxidative, anti-inflammatory and hepatoprotective effects of resveratrol on oxidative stress-induced liver damage in tilapia (Oreochromis niloticus). Comp Biochem Physiol C Toxicol Pharmacol 215:56–66. https://doi.org/10.1016/j.cbpc.2018.10.002
Dadi PK, Ahmad M, Ahmad Z (2009) Inhibition of ATPase activity of Escherichia coli ATP synthase by polyphenols. Int J Biol Macromol 45(1):72–79. https://doi.org/10.1016/j.ijbiomac.2009.04.004
Zhu X, Lei X, Wang J, Dong W (2020) Protective effects of resveratrol on hyperoxia-induced lung injury in neonatal rats by alleviating apoptosis and ROS production. J Matern Fetal Neonatal Med 33(24):4150–4158. https://doi.org/10.1080/14767058.2019.1597846
Ren Z, Wang L, Cui J, Huoc Z, Xue J, Cui H, Mao Q, Yang R (2013) Resveratrol inhibits NF-kB signaling through suppression of p65 and IkappaB kinase activities. Pharmazie 68(8):689–694
DiSabato DJ, Quan N, Godbout JP (2016) Neuroinflammation: the devil is in the details. J Neurochem 139(Suppl 2):136–153. https://doi.org/10.1111/jnc.13607
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(6):918–934. https://doi.org/10.1016/j.cell.2010.02.016
Russo MV, McGavern DB (2016) Inflammatory neuroprotection following traumatic brain injury. Science 353(6301):783–785. https://doi.org/10.1126/science.aaf6260
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820. https://doi.org/10.1016/j.cell.2010.01.022
Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW (2014) Pattern recognition receptors and central nervous system repair. Exp Neurol 258:5–16. https://doi.org/10.1016/j.expneurol.2014.01.001
Rivero Vaccari JP, Brand F 3rd, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, Bullock MR, Dietrich WD, Keane RW (2016) Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem 136(Suppl 1):39–48. https://doi.org/10.1111/jnc.13036
Minkiewicz J, de Rivero Vaccari JP, Keane RW (2013) Human astrocytes express a novel NLRP2 inflammasome. Glia 61(7):1113–1121. https://doi.org/10.1002/glia.22499
Zhou K, Shi L, Wang Y, Chen S, Zhang J (2016) Recent Advances of the NLRP3 Inflammasome in Central Nervous System Disorders. J Immunol Res 2016:9238290. https://doi.org/10.1155/2016/9238290
Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM (2006) Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 53(7):688–695. https://doi.org/10.1002/glia.20328
Borysiewicz E, Fil D, Konat GW (2009) Rho proteins are negative regulators of TLR2, TLR3, and TLR4 signaling in astrocytes. J Neurosci Res 87(7):1565–1572. https://doi.org/10.1002/jnr.21968
Al Mamun A, Wu Y, Monalisa I, Jia C, Zhou K, Munir F, Xiao J (2020) Role of pyroptosis in spinal cord injury and its therapeutic implications. J Adv Res 28:97–109. https://doi.org/10.1016/j.jare.2020.08.004
von Bartheld CS, Bahney J, Herculano-Houzel S (2016) The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol 524(18):3865–3895. https://doi.org/10.1002/cne.24040
Murphy N, Cowley TR, Richardson JC, Virley D, Upton N, Walter D, Lynch MA (2012) The neuroprotective effect of a specific P2X7 receptor antagonist derives from its ability to inhibit assembly of the NLRP3 inflammasome in glial cells. Brain Pathol 22(3):295–306. https://doi.org/10.1111/j.1750-3639.2011.00531.x
Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, Muruve DA, McKay DM, Beck PL, Mawe GM, Thompson RJ, Sharkey KA (2012) Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med 18(4):600–604. https://doi.org/10.1038/nm.2679
Draganov D, Gopalakrishna-Pillai S, Chen YR, Zuckerman N, Moeller S, Wang C, Ann D, Lee PP (2015) Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via Ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci Rep 10(5):16222. https://doi.org/10.1038/srep16222
Silver J (2016) The glial scar is more than just astrocytes. Exp Neurol 286:147–149. https://doi.org/10.1016/j.expneurol.2016.06.018
Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H (2006) Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med 12(7):829–834. https://doi.org/10.1038/nm1425
Movahed A, Raj P, Nabipour I, Mahmoodi M, Ostovar A, Kalantarhormozi M, Netticadan T (2020) Efficacy and Safety of Resveratrol in Type 1 Diabetes Patients: A Two-Month Preliminary Exploratory Trial. Nutrients 12(1):161. https://doi.org/10.3390/nu12010161
Pannu N, Bhatnagar A (2020) Combinatorial therapeutic effect of resveratrol and piperine on murine model of systemic lupus erythematosus. Inflammopharmacology 28(2):401–424. https://doi.org/10.1007/s10787-019-00662-w
Chen Y, Zheng Z, Li C, Pan Y, Tang X, Wang XJ (2019) Synthetic Imine Resveratrol Analog 2-Methoxyl-3,6-Dihydroxyl-IRA Ameliorates Colitis by Activating Protective Nrf2 Pathway and Inhibiting NLRP3 Expression. Oxid Med Cell Longev 2019:7180284. https://doi.org/10.1155/2019/7180284
Wang D, Li SP, Fu JS, Zhang S, Bai L, Guo L (2016) Resveratrol defends blood-brain barrier integrity in experimental autoimmune encephalomyelitis mice. J Neurophysiol 116(5):2173–2179. https://doi.org/10.1152/jn.00510.2016
Wang X, Wu J, Ma S, Xie Y, Liu H, Yao M, Zhang Y, Yang GL, Yang B, Guo R, Guan F (2020) Resveratrol Preincubation Enhances the Therapeutic Efficacy of hUC-MSCs by Improving Cell Migration and Modulating Neuroinflammation Mediated by MAPK Signaling in a Mouse Model of Alzheimer’s Disease. Front Cell Neurosci 14:62. https://doi.org/10.3389/fncel.2020.00062
Xia D, Sui R, Zhang Z (2019) Administration of resveratrol improved Parkinson’s disease-like phenotype by suppressing apoptosis of neurons via modulating the MALAT1/miR-129/SNCA signaling pathway. J Cell Biochem 120(4):4942–4951. https://doi.org/10.1002/jcb.27769
Yun YC, Jeong SG, Kim SH, Cho GW (2019) Reduced sirtuin 1/adenosine monophosphate-activated protein kinase in amyotrophic lateral sclerosis patient-derived mesenchymal stem cells can be restored by resveratrol. J Tissue Eng Regen Med 13(1):110–115. https://doi.org/10.1002/term.2776
Li W, Li C, Ma L, Jin F (2020) Resveratrol inhibits viability and induces apoptosis in the small-cell lung cancer H446 cell line via the PI3K/Akt/c-Myc pathway. Oncol Rep 44(5):1821–1830. https://doi.org/10.3892/or.2020.7747
Xiao Y, Qin T, Sun L, Qian W, Li J, Duan W, Lei J, Wang Z, Ma J, Li X, Ma Q, Xu Q (2020) Resveratrol Ameliorates the Malignant Progression of Pancreatic Cancer by Inhibiting Hypoxia-induced Pancreatic Stellate Cell Activation. Cell Transplant 29:963689720929987. https://doi.org/10.1177/0963689720929987
Yuan L, Zhou M, Huang D, Wasan HS, Zhang K, Sun L, Huang H, Ma S, Shen M, Ruan S (2019) Resveratrol inhibits the invasion and metastasis of colon cancer through reversal of epithelial- mesenchymal transition via the AKT/GSK-3β/Snail signaling pathway. Mol Med Rep 20(3):2783–2795. https://doi.org/10.3892/mmr.2019.10528
Sun Y, Zhou QM, Lu YY, Zhang H, Chen QL, Zhao M, Su SB (2019) Resveratrol inhibits the migration and metastasis of MDA-MB-231 human breast cancer by reversing TGF-β1-induced epithelial-mesenchymal transition. Molecules 24(6):1131. https://doi.org/10.3390/molecules24061131
Zhou L, Long J, Sun Y, Chen W, Qiu R, Yuan D (2020) Resveratrol ameliorates atherosclerosis induced by high-fat diet and LPS in ApoE-/- mice and inhibits the activation of CD4+ T cells. Nutr Metab (Lond) 17:41. https://doi.org/10.1186/s12986-020-00461-z
Sarkar O, Li Y, Anand-Srivastava MB (2019) Resveratrol prevents the development of high blood pressure in spontaneously hypertensive rats through the inhibition of enhanced expression of Giα proteins 1. Can J Physiol Pharmacol 97(9):872–879. https://doi.org/10.1139/cjpp-2019-0040
Feng H, Mou SQ, Li WJ, Zhang N, Zhou ZY, Ding W, Bian ZY, Liao HH (2020) Resveratrol Inhibits Ischemia-Induced Myocardial Senescence Signals and NLRP3 Inflammasome Activation. Oxid Med Cell Longev 2020:2647807. https://doi.org/10.1155/2020/2647807
Di Fermo P, Di Lodovico S, Amoroso R, De Filippis B, D’Ercole S, Di Campli E, Cellini L, Di Giulio M (2020) Searching for New Tools to Counteract the Helicobacter pylori Resistance: The Positive Action of Resveratrol Derivatives. Antibiotics (Basel) 9(12):891. https://doi.org/10.3390/antibiotics9120891
Tang F, Li L, Meng XM, Li B, Wang CQ, Wang SQ, Wang TL, Tian YM (2019) Inhibition of alpha-hemolysin expression by resveratrol attenuates Staphylococcus aureus virulence. Microb Pathog 127:85–90. https://doi.org/10.1016/j.micpath.2018.11.027
Marques BCAA, Trindade M, Aquino JCF, Cunha AR, Gismondi RO, Neves MF, Oigman W (2018) Beneficial effects of acute trans-resveratrol supplementation in treated hypertensive patients with endothelial dysfunction. Clin Exp Hypertens 40(3):218–223. https://doi.org/10.1080/10641963.2017.1288741
Wiciński M, Socha M, Walczak M, Wódkiewicz E, Malinowski B, Rewerski S, Górski K, Pawlak-Osińska K (2018) Beneficial Effects of Resveratrol Administration-Focus on Potential Biochemical Mechanisms in Cardiovascular Conditions. Nutrients 10(11):1813. https://doi.org/10.3390/nu10111813
Keshtkar S, Kaviani M, Jabbarpour Z, Al-Abdullah IH, Aghdaei MH, Nikeghbalian S, Shamsaeefar A, Geramizadeh B, Azarpira N, Ghahremani MH (2020) Significant reduction of apoptosis induced via hypoxia and oxidative stress in isolated human islet by resveratrol. Nutr Metab Cardiovasc Dis 30(7):1216–1226. https://doi.org/10.1016/j.numecd.2020.04.011
He LN, Lan YR, He GM, Guo SJ, Wen FQ, Wang T (2020) Resveratrol inhibits hypoxia-induced oxidative stress and proliferation in pulmonary artery smooth muscle cells through the HIF-1α/NOX4/ROS signaling pathway. Sheng Li Xue Bao 72(5):551–558
Kim TH, Park JH, Woo JS (2019) Resveratrol induces cell death through ROS-dependent downregulation of Notch1/PTEN/Akt signaling in ovarian cancer cells. Mol Med Rep 19(4):3353–3360. https://doi.org/10.3892/mmr.2019.9962
Saqib U, Faisal SM, Saluja R, Baig MS (2019) Structural insights of resveratrol with its binding partners in the toll-like receptor 4 pathway. J Cell Biochem 120(1):452–460. https://doi.org/10.1002/jcb.27401
Zhang M, Xue Y, Chen H, Meng L, Chen B, Gong H, Zhao Y, Qi R (2019) Resveratrol inhibits MMP3 and MMP9 expression and secretion by suppressing TLR4/NF-κB/STAT3 activation in ox-LDL-treated HUVECs. Oxid Med Cell Longev 2019:9013169. https://doi.org/10.1155/2019/9013169
Xu X, Liu X, Yang Y, He J, Jiang M, Huang Y, Liu X, Liu L, Gu H (2020) Resveratrol exerts anti-osteoarthritic effect by inhibiting TLR4/NF-κB signaling pathway via the TLR4/Akt/FoxO1 axis in IL-1β-Stimulated SW1353 cells. Drug Des Devel Ther 14:2079–2090. https://doi.org/10.2147/DDDT.S244059
Li J, Li L, Wang S, Zhang C, Zheng L, Jia Y, Xu M, Zhu T, Zhang Y, Rong R (2018) Resveratrol alleviates inflammatory responses and oxidative stress in rat kidney ischemia-reperfusion injury and H2O2-induced NRK-52E cells via the Nrf2/TLR4/NF-κB pathway. Cell Physiol Biochem 45(4):1677–1689. https://doi.org/10.1159/000487735
Mo X, Wang X, Ge Q, Bian F (2019) The effects of SIRT1/FoxO1 on LPS induced INS-1 cells dysfunction. Stress 22(1):70–82. https://doi.org/10.1080/10253890.2018.1501022
Al Azzaz J, Rieu A, Aires V, Delmas D, Chluba J, Winckler P, Bringer MA, Lamarche J, Vervandier-Fasseur D, Dalle F, Lapaquette P, Guzzo J (2019) Resveratrol-Induced Xenophagy Promotes Intracellular Bacteria Clearance in Intestinal Epithelial Cells and Macrophages. Front Immunol 9:3149. https://doi.org/10.3389/fimmu.2018.03149
De Leo A, Arena G, Lacanna E, Oliviero G, Colavita F, Mattia E (2012) Resveratrol inhibits Epstein Barr Virus lytic cycle in Burkitt’s lymphoma cells by affecting multiple molecular targets. Antiviral Res 96(2):196–202. https://doi.org/10.1016/j.antiviral.2012.09.003
Zeng X, Pan X, Xu X, Lin J, Que F, Tian Y, Li L, Liu S (2017) Resveratrol Reactivates Latent HIV through Increasing Histone Acetylation and Activating Heat Shock Factor 1. J Agric Food Chem 65(22):4384–4394. https://doi.org/10.1021/acs.jafc.7b00418
Lin SC, Ho CT, Chuo WH, Li S, Wang TT, Lin CC (2017) Effective inhibition of MERS-CoV infection by resveratrol. BMC Infect Dis 17(1):144. https://doi.org/10.1186/s12879-017-2253-8
Elgizawy HA, Ali AA, Hussein MA (2020) Resveratrol: isolation, and its nanostructured, inhibits cell proliferation, induces cell apoptosis in certain human cell lines carcinoma and exerts protective effect against paraquat-induced hepatotoxicity. J Med Food. https://doi.org/10.1089/jmf.2019.0286
Bäck M, Yurdagul A Jr, Tabas I, Öörni K, Kovanen PT (2019) Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16(7):389–406. https://doi.org/10.1038/s41569-019-0169-2
Nerurkar L, Siebert S, McInnes IB, Cavanagh J (2019) Rheumatoid arthritis and depression: an inflammatory perspective. Lancet Psychiatry 6(2):164–173. https://doi.org/10.1016/S2215-0366(18)30255-4
Mansurov A, Ishihara J, Hosseinchi P, Potin L, Marchell TM, Ishihara A, Williford JM, Alpar AT, Raczy MM, Gray LT, Swartz MA, Hubbell JA (2020) Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat Biomed Eng 4(5):531–543. https://doi.org/10.1038/s41551-020-0549-2
Novelle MG, Wahl D, Diéguez C, Bernier M, de Cabo R (2015) Resveratrol supplementation: where are we now and where should we go? Ageing Res Rev 21:1–15. https://doi.org/10.1016/j.arr.2015.01.002
Ding S, Wang H, Wang M, Bai L, Yu P, Wu W (2019) Resveratrol alleviates chronic “real-world” ambient particulate matter-induced lung inflammation and fibrosis by inhibiting NLRP3 inflammasome activation in mice. Ecotoxicol Environ Saf 182:109425. https://doi.org/10.1016/j.ecoenv.2019.109425
Shabani M, Sadeghi A, Hosseini H, Teimouri M, Babaei Khorzoughi R, Pasalar P, Meshkani R (2020) Resveratrol alleviates obesity-induced skeletal muscle inflammation via decreasing M1 macrophage polarization and increasing the regulatory T cell population. Sci Rep 10(1):3791. https://doi.org/10.1038/s41598-020-60185-1
Zhang G, Liu Y, Xu L, Sha C, Zhang H, Xu W (2019) Resveratrol alleviates lipopolysaccharide-induced inflammation in PC-12 cells and in rat model. BMC Biotechnol 19(1):10. https://doi.org/10.1186/s12896-019-0502-1
Jiang H, Duan J, Xu K, Zhang W (2019) Resveratrol protects against asthma-induced airway inflammation and remodeling by inhibiting the HMGB1/TLR4/NF-κB pathway. Exp Ther Med 18(1):459–466. https://doi.org/10.3892/etm.2019.7594
Chen M, Hou P, Zhou M, Ren Q, Wang X, Huang L, Hui S, Yi L, Mi M (2020) Resveratrol attenuates high-fat diet-induced non-alcoholic steatohepatitis by maintaining gut barrier integrity and inhibiting gut inflammation through regulation of the endocannabinoid system. Clin Nutr 39(4):1264–1275. https://doi.org/10.1016/j.clnu.2019.05.020
Gong W, Li J, Chen W, Feng F, Deng Y (2020) Resveratrol inhibits lipopolysaccharide-induced extracellular matrix accumulation and inflammation in rat clomerular mesangial cells by SphK1/S1P2/NF-κB pathway. Diabetes Metab Syndr Obes 13:4495–4505. https://doi.org/10.2147/DMSO.S278267
Brenjian S, Moini A, Yamini N, Kashani L, Faridmojtahedi M, Bahramrezaie M, Khodarahmian M, Amidi F (2020) Resveratrol treatment in patients with polycystic ovary syndrome decreased pro-inflammatory and endoplasmic reticulum stress markers. Am J Reprod Immunol 83(1):e13186. https://doi.org/10.1111/aji.13186
Zhang Y, Zhu XB, Zhao JC, Gao XF, Zhang XN, Hou K (2020) Neuroprotective effect of resveratrol against radiation after surgically induced brain injury by reducing oxidative stress, inflammation, and apoptosis through NRf2/HO-1/NF-κB signaling pathway. J Biochem Mol Toxicol 34(12):e22600. https://doi.org/10.1002/jbt.22600
Pujara N, Wong KY, Qu Z, Wang R, Moniruzzaman M, Rewatkar P, Kumeria T, Ross BP, McGuckin M, Popat A (2020) Oral delivery of β-lactoglobulin-nanosphere-encapsulated resveratrol alleviates inflammation in winnie mice with spontaneous ulcerative colitis. Mol Pharm. https://doi.org/10.1021/acs.molpharmaceut.0c00048
Shin JW, Lee HS, Na JI, Huh CH, Park KC, Choi HR (2020) Resveratrol Inhibits Particulate Matter-Induced Inflammatory Responses in Human Keratinocytes. Int J Mol Sci 21(10):3446. https://doi.org/10.3390/ijms21103446
Josifovska N, Albert R, Nagymihály R, Lytvynchuk L, Moe MC, Kaarniranta K, Veréb ZJ, Petrovski G (2020) Resveratrol as Inducer of Autophagy, Pro-Survival, and Anti-Inflammatory Stimuli in Cultured Human RPE Cells. Int J Mol Sci 21(3):813. https://doi.org/10.3390/ijms21030813
Jiang T, Gu J, Chen W, Chang Q (2019) Resveratrol inhibits high-glucose-induced inflammatory “metabolic memory” in human retinal vascular endothelial cells through SIRT1-dependent signaling. Can J Physiol Pharmacol 97(12):1141–1151. https://doi.org/10.1139/cjpp-2019-0201
Vairappan B, Sundhar M, Srinivas BH (2019) Resveratrol Restores Neuronal Tight Junction Proteins Through Correction of Ammonia and Inflammation in CCl4-Induced Cirrhotic Mice. Mol Neurobiol 56(7):4718–4729. https://doi.org/10.1007/s12035-018-1389-x
Gandy KAO, Zhang J, Nagarkatti P, Nagarkatti M (2019) Resveratrol (3, 5, 4’-Trihydroxy-trans-Stilbene) Attenuates a mouse model of multiple sclerosis by altering the miR-124/sphingosine kinase 1 axis in encephalitogenic T cells in the brain. J Neuroimmune Pharmacol 14(3):462–477. https://doi.org/10.1007/s11481-019-09842-5
Le K, Chibaatar Daliv E, Wu S, Qian F, Ali AI, Yu D, Guo Y (2019) SIRT1-regulated HMGB1 release is partially involved in TLR4 signal transduction: a possible anti-neuroinflammatory mechanism of resveratrol in neonatal hypoxic-ischemic brain injury. Int Immunopharmacol 75:105779. https://doi.org/10.1016/j.intimp.2019.105779
Wang Y, Shi Y, Huang Y, Liu W, Cai G, Huang S, Zeng Y, Ren S, Zhan H, Wu W (2020) Resveratrol mediates mechanical allodynia through modulating inflammatory response via the TREM2-autophagy axis in SNI rat model. J Neuroinflammation 17(1):311. https://doi.org/10.1186/s12974-020-01991-2
Singh AK, Vinayak M (2017) Resveratrol alleviates inflammatory hyperalgesia by modulation of reactive oxygen species (ROS), antioxidant enzymes and ERK activation. Inflamm Res 66(10):911–921. https://doi.org/10.1007/s00011-017-1072-0
Oliviero F, Zamudio-Cuevas Y, Belluzzi E, Andretto L, Scanu A, Favero M, Ramonda R, Ravagnan G, López-Reyes A, Spinella P, Punzi L (2019) Polydatin and Resveratrol Inhibit the Inflammatory Process Induced by Urate and Pyrophosphate Crystals in THP-1 Cells. Foods 8(11):560. https://doi.org/10.3390/foods8110560
Ben Lagha A, Andrian E, Grenier D (2019) Resveratrol attenuates the pathogenic and inflammatory properties of Porphyromonas gingivalis. Mol Oral Microbiol 34(3):118–130. https://doi.org/10.1111/omi.12260
Euba B, López-López N, Rodríguez-Arce I, Fernández-Calvet A, Barberán M, Caturla N, Martí S, Díez-Martínez R, Garmendia J (2017) Resveratrol therapeutics combines both antimicrobial and immunomodulatory properties against respiratory infection by nontypeable Haemophilus influenzae. Sci Rep 7(1):12860. https://doi.org/10.1038/s41598-017-13034-7
Ngo KA, Kishimoto K, Davis-Turak J, Pimplaskar A, Cheng Z, Spreafico R, Chen EY, Tam A, Ghosh G, Mitchell S, Hoffmann A (2020) Dissecting the Regulatory Strategies of NF-κB RelA Target Genes in the Inflammatory Response Reveals Differential Transactivation Logics. Cell Rep 30(8):2758-2775.e6. https://doi.org/10.1016/j.celrep.2020.01.108
Faria CP, Neves BM, Lourenço Á, Cruz MT, Martins JD, Silva A, Pereira S, Sousa MDC (2020) Giardia lamblia Decreases NF-κB p65RelA Protein Levels and Modulates LPS-Induced Pro-Inflammatory Response in Macrophages. Sci Rep 10(1):6234. https://doi.org/10.1038/s41598-020-63231-0
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34(5):637–650. https://doi.org/10.1016/j.immuni.2011.05.006
Ahmad SF, Ansari MA, Nadeem A, Alzahrani MZ, Bakheet SA, Attia SM (2018) Resveratrol Improves Neuroimmune Dysregulation Through the Inhibition of Neuronal Toll-Like Receptors and COX-2 Signaling in BTBR T+ Itpr3tf/J Mice. Neuromolecular Med 20(1):133–146. https://doi.org/10.1007/s12017-018-8483-0
Kwilasz AJ, Todd LS, Duran-Malle JC, Schrama AEW, Mitten EH, Larson TA, Clements MA, Harris KM, Litwiler ST, Wang X, Van Dam AM, Maier SF, Rice KC, Watkins LR, Barrientos RM (2021) Experimental autoimmune encephalopathy (EAE)-induced hippocampal neuroinflammation and memory deficits are prevented with the non-opioid TLR2/TLR4 antagonist (+)-naltrexone. Behav Brain Res 396:112896. https://doi.org/10.1016/j.bbr.2020.112896
Li A, Zhang S, Li J, Liu K, Huang F, Liu B (2016) Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol Cell Endocrinol 434:36–47. https://doi.org/10.1016/j.mce.2016.06.008
Albornoz EA, Woodruff TM, Gordon R (2018) Inflammasomes in CNS Diseases Exp Suppl 108:41–60. https://doi.org/10.1007/978-3-319-89390-7_3
Zeng H, Liu N, Yang YY, Xing HY, Liu XX, Li F, La GY, Huang MJ, Zhou MW (2019) Lentivirus-mediated downregulation of α-synuclein reduces neuroinflammation and promotes functional recovery in rats with spinal cord injury. J Neuroinflammation 16(1):283. https://doi.org/10.1186/s12974-019-1658-2
Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR (2004) Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J 4(4):451–464. https://doi.org/10.1016/j.spinee.2003.07.007
Romanelli P, Bieler L, Scharler C, Pachler K, Kreutzer C, Zaunmair P, Jakubecova D, Mrowetz H, Benedetti B, Rivera FJ, Aigner L, Rohde E, Gimona M, Strunk D, Couillard-Despres S (2019) Extracellular vesicles can deliver anti-inflammatory and anti-scarring activities of mesenchymal stromal cells after spinal cord injury. Front Neurol 10:1225. https://doi.org/10.3389/fneur.2019.01225
Wang C, Zhang L, Ndong JC, Hettinghouse A, Sun G, Chen C, Zhang C, Liu R, Liu CJ (2019) Progranulin deficiency exacerbates spinal cord injury by promoting neuroinflammation and cell apoptosis in mice. J Neuroinflammation 16(1):238. https://doi.org/10.1186/s12974-019-1630-1
Liu J, Yi L, Xiang Z, Zhong J, Zhang H, Sun T (2015) Resveratrol attenuates spinal cord injury-induced inflammatory damage in rat lungs. Int J Clin Exp Pathol 8(2):1237–1246
Meng HY, Shao DC, Li H, Huang XD, Yang G, Xu B, Niu HY (2018) Resveratrol improves neurological outcome and neuroinflammation following spinal cord injury through enhancing autophagy involving the AMPK/mTOR pathway. Mol Med Rep 18(2):2237–2244. https://doi.org/10.3892/mmr.2018.9194
Zhao H, Chen S, Gao K, Zhou Z, Wang C, Shen Z, Guo Y, Li Z, Wan Z, Liu C, Mei X (2017) Resveratrol protects against spinal cord injury by activating autophagy and inhibiting apoptosis mediated by the SIRT1/AMPK signaling pathway. Neuroscience 348:241–251. https://doi.org/10.1016/j.neuroscience.2017.02.0
Moussa C, Hebron M, Huang X, Ahn J, Rissman RA, Aisen PS, Turner RS (2017) Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflammation 14(1):1. https://doi.org/10.1186/s12974-016-0779-0
Timmers S, de Ligt M, Phielix E, van de Weijer T, Hansen J, Moonen-Kornips E, Schaart G, Kunz I, Hesselink MK, Schrauwen-Hinderling VB, Schrauwen P (2016) Resveratrol as Add-on Therapy in Subjects With Well-Controlled Type 2 Diabetes: a Randomized Controlled Trial. Diabetes Care 39(12):2211–2217. https://doi.org/10.2337/dc16-0499
Ávila-Gálvez MÁ, García-Villalba R, Martínez-Díaz F, Ocaña-Castillo B, Monedero-Saiz T, Torrecillas-Sánchez A, Abellán B, González-Sarrías A, Espín JC (2019) Metabolic Profiling of Dietary Polyphenols and Methylxanthines in Normal and Malignant Mammary Tissues from Breast Cancer Patients. Mol Nutr Food Res 63(9):e1801239. https://doi.org/10.1002/mnfr.201801239
Boocock DJ, Faust GE, Patel KR, Schinas AM, Brown VA, Ducharme MP, Booth TD, Crowell JA, Perloff M, Gescher AJ, Steward WP, Brenner DE (2007) Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev 16(6):1246–1252. https://doi.org/10.1158/1055-9965.EPI-07-0022
Anton SD, Embry C, Marsiske M, Lu X, Doss H, Leeuwenburgh C, Manini TM (2014) Safety and metabolic outcomes of resveratrol supplementation in older adults: results of a twelve-week, placebo-controlled pilot study. Exp Gerontol 57:181–187. https://doi.org/10.1016/j.exger.2014.05.015
Funding
This work was supported by the Natural Science Foundation of Zhejiang Province (Grant No. LY19H170001).
Author information
Authors and Affiliations
Contributions
XL designed the study. RF, YZ, BOAB, and XL prepared the first draft and revised the manuscript. All authors approved the final paper.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
All authors agreed to publish this article.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fan, R., Zhang, Y., Botchway, B.O.A. et al. Resveratrol Can Attenuate Astrocyte Activation to Treat Spinal Cord Injury by Inhibiting Inflammatory Responses. Mol Neurobiol 58, 5799–5813 (2021). https://doi.org/10.1007/s12035-021-02509-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02509-4