
Abstract
The Gene Ontology (GO) is widely recognised as the gold standard bioinformatics resource for summarizing functional knowledge of gene products in a consistent and computable, information-rich language. GO describes cellular and organismal processes across all species, yet until now there has been a considerable gene annotation deficit within the neurological and immunological domains, both of which are relevant to Parkinson’s disease. Here we introduce the Parkinson’s disease GO Annotation Project, funded by Parkinson’s UK and supported by the GO Consortium, which is addressing this deficit by providing GO annotation to Parkinson’s-relevant human gene products, principally through expert literature curation. We discuss the steps taken to prioritise proteins, publications and cellular processes for annotation, examples of how GO annotations capture Parkinson’s-relevant information, and the advantages that a topic-focused annotation approach offers to users. Building on the existing GO resource, this project collates a vast amount of Parkinson’s-relevant literature into a set of high-quality annotations to be utilized by the research community.
Similar content being viewed by others


Introduction
Parkinson’s disease is the second most common neurodegenerative disorder after Alzheimer’s disease. An increased understanding of Parkinson’s disease has resulted from the identification and characterization of genes that cause or influence the risk of developing this condition (Gasser et al. 2011). Much of this knowledge has accumulated from ‘big data’ analyses, such as genome wide association (GWA) studies, large-scale sequencing, proteomics and transcriptomic analyses. Researchers have the increasingly complex task of evaluating these large datasets to detect relevant pathways and regulatory networks, supported by resources including the Gene Ontology (GO) (Gene Ontology Consortium 2015), Reactome (Croft et al. 2014), KEGG (Kotera et al. 2012) and protein interaction databases (Kerrien et al. 2012).
GO is widely recognised as the gold standard bioinformatics resource for summarizing functional knowledge of gene products across all kingdoms of life. GO contains three ontologies describing the molecular functions of a gene product, the processes those actions are part of, and the cellular location(s) of the gene product. GO terms are then associated with a gene product to create an ‘annotation’. GO annotations are linked to an underlying reference, and include an evidence code indicating the assay, algorithm or decision supporting the annotation. A combination of manual and computational approaches are taken to create these annotations, and include summarising published research, inference by homology to related annotated gene products, and mapping of annotations from other biological resources such as Reactome, KEGG, and InterPro (Mitchell et al. 2015).
GO is frequently used in the analysis of high-throughput datasets, particularly for enrichment analyses where a gene-set of interest is analysed for over-represented GO terms, as demonstrated by Manzoni and colleagues in their examination of the LRRK2 interactome (Manzoni et al. 2015). Meaningful interpretation of such analyses depends on the quality of annotations that are available for the identified gene products, and for Parkinson’s datasets this approach has been hampered by insufficient functional annotation of many of the key affected neurological and immunological pathways, both of which are relevant to Parkinson’s disease (Holmans et al. 2013).
Results and Discussion
The Parkinson’s GO Annotation Initiative began in January 2014 to address the neurological and immunological annotation deficit through the annotation of human proteins relevant to the risk and progression of Parkinson’s disease, principally by the expert curation of the biomedical literature. We aim to comprehensively curate the role of 800 human proteins within Parkinson’s-related processes by the end of 2016, and we are well on our way to our annotation target with over 1000 proteins already annotated, including 644 human proteins (as of August 2015). We consider a protein to be comprehensively annotated within a process when a curator is confident that the annotations cover all aspect of the protein’s function within that process. An overview of the full role of a protein within a process is typically gained from reading recent reviews. In practice, we have a limited time to annotate each protein, consequently we usually stop annotating a well-researched protein when annotation of additional papers leads to the repeated association of the same GO term to the protein. Proteins described in only a small number of papers will be annotated based on all available experimental evidence. Some proteins will be comprehensively annotated across multiple processes as we are taking a protein-based approach alongside our topic-based approach, as described below. One such fully-annotated protein is human PARK7 (DJ-1, Q99497), which (as of 6th August 2015) has 226 annotations in total, 135 of which were created by our project (Table 1 provides a selection of these annotations, note UniProtKB/Swiss-Prot protein IDs are used throughout this article). Furthermore, 22 PARK7 annotations were assigned semi-automatically by the Ensembl Compara pipeline (Cunningham et al. 2015) and eight of these derive from our manual annotations to the orthologous mouse Park7 entry (Q99LX0). Thus our project is increasing numbers of both manual and electronic annotations to improve coverage.
Given the vast amount of literature available on Parkinson’s disease, our first task was to define an annotation priority set, and below we discuss the steps taken to prioritise proteins, publications and cellular processes. Our high priority protein set (https://www.ucl.ac.uk/functional-gene-annotation/neurological/projects/high-priority-genes) currently includes 48 proteins encoded either by familial Parkinson’s disease genes or by candidate genes identified in GWA studies. An extended priority list (http://www.ebi.ac.uk/QuickGO/GProteinSet?id=ParkinsonsUK-UCL) also contains proteins that interact with the high priority set, as identified by IntAct (Porras et al. 2015), and proteins that participate in Parkinson’s-relevant processes, as described below.
A list of pathways and processes relevant to Parkinson’s disease was assembled based on publications and discussions with researchers. We took a process-based approach to annotation alongside our protein-based strategy, to ensure that all proteins within a Parkinson’s-relevant process are annotated, and not just proteins with an already established link to the disease. The process priority list includes mitochondrial processes (such as mitophagy and mitochondrial fusion and fission), response to cellular stresses (including oxidative stress, endoplasmic reticulum (ER) stress and nitrosative stress), Wnt signaling, lysosomal pathways, dopamine metabolism, dopamine transport, autophagy and synaptic transmission. We are annotating all key players in these processes to capture multiple aspects of Parkinson’s disease and allow examination of the cellular networks that underlie the condition. We define a key player as a protein that has been shown to be critical to a process or regulation of a process, information we acquire primarily through literature curation but also from attending meetings in the field and communicating with researchers. In addition, we are prioritising curation of Parkinson’s UK-funded research articles. Furthermore, our priority lists remain dynamic and are expanded to reflect new knowledge and scientific advancements.
Thus far, we have manually curated nearly 300 publications, and generated over 4000 manual annotations, including over 2700 annotations to human proteins (as of 6th August 2015). Our annotation focuses on human proteins, but given the enormous knowledge contribution from model organism data (Magen and Chesselet 2010; Pienaar et al. 2010), we also annotate other organisms with a priority on mouse, rat and fly. GO data is then transferred to orthologous human proteins through the Ensembl-Compara pipeline or, in the absence of automated methods, we manually transfer GO annotations between orthologs using the ISS (Inferred from Sequence or structural Similarity) evidence code.
The role of a gene product can vary depending on its environment or interactors, and in a GO Consortium-led approach (Huntley et al. 2014), we extend the core GO annotation to accommodate additional context, such as the cell type the process occurs in, or the substrate of a catalytic activity. For example, we capture that PML (P29590) and PARK7 (DJ-1, Q99497) interact under conditions of oxidative stress (Kim et al. 2012) by extending the ‘protein binding’ (GO:0005515) annotation with the statement “happens_during cellular response to oxidative stress (GO:0034599)”. By referencing the cell-type and anatomy ontologies (Bard et al. 2005; Mungall et al. 2012) we annotate that HERPUD1 (HERP, Q15011) is located in the ER of a neuron in the substantia nigra (Slodzinski et al. 2009) by including “part_of CL:0000540, part_of UBERON:0002038” in the annotation. One of the biggest benefits of providing this contextual information is the ability to record the target of a protein’s activity, e.g. capturing that murine Nfe2l2 (Nrf2, Q60795) is a phosphorylation substrate of Eif2ak3 (Perk, Q9Z2B5) during the Unfolded Protein Response (UPR) (Cullinan et al. 2003). In this way we can link annotations together to provide the full picture of a pathway or process, and in the future GO browsers will support more sophisticated queries and analyses allowing the extraction of, for example, all EIF2AK3 (PERK) substrates.
To ensure annotations are of a high quality, we annotate to GO Consortium guidelines that have been rigorously developed by trained curators. Internal and Consortium quality-checking procedures, including peer-review, ensure that annotations conform to these rules and are appropriate to the taxon of the organism being annotated (Deegan née Clark et al. 2010). A long-term approach to maintaining annotation quality comes from the Phylogenetic Annotation Inference Tool (PAINT) project (Gaudet et al. 2011) whereby annotations are inferred through the phylogenetic tree. Curator input into the annotation propagation ensures consistent annotation across orthologs and highlights any potential anomalies or errors. Our users and authors offer another layer of quality control; we correspond with authors during curation to clarify ambiguities (for example the species origin of construct sequences). Furthermore, when paper annotation is complete, we regularly notify the authors who can check that our annotations accurately represent the published data.
Ontology Improvements
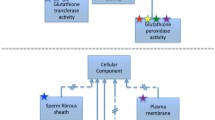
Ontology development goes hand-in-hand with annotation, thus GO is being continually updated and expanded. A considerable benefit of a topic-based curation approach is that expert curators can apply their accumulated knowledge of a biological research area to improve the ontology terms and structure. To date, our project has contributed over 240 new GO terms across all three ontology nodes including ‘CHOP-C/EBP complex’ (GO:0036488), ‘ERAD pathway’ (GO:0036503), ‘chaperone-mediated autophagy’ (GO:0061684), ‘L-dopa biosynthetic process’ (GO:1903185) and ‘tyrosine 3-monooxygenase activator activity’ (GO:0036470), thereby enabling more descriptive annotations to be created. Using the UPR as an example; the endoplasmic reticulum UPR is one pathway by which cells deal with unfolded and misfolded proteins in the ER to restore the cell to its resting state. ER stress has extensive links to Parkinson’s disease (Varma and Sen 2015) and is therefore a project priority topic. To expand the ontology in this node, we created new terms to represent the three main branches of the mammalian UPR (Fig. 1). Previously, the transcription factor ATF4 (P18848) had been annotated to ‘endoplasmic reticulum unfolded protein response’ (GO:0030968). While this annotation remains true, it does not capture how ATF4 fits into this process. With our creation of new UPR terms, we were able to associate the more descriptive term ‘PERK-mediated unfolded protein response’ (GO:0036499) with ATF4. GO terms mentioning gene products are the exception but where they exist, the name reflects the gene or protein name most commonly used in the research field (e.g. PERK) and is designed to be species-neutral wherever possible. Synonyms including species-specific nomenclature (e.g. EIF2AK3) are added when required to capture additional information and improve search capabilities. Ontology construction requires considerable curator input, consequently we have created over 180 of our new GO terms using the TermGenie tool (Dietze et al. 2014), which enables formally specified design patterns and templates to appropriately place and define each new term.

Representation of the Unfolded Protein Response (UPR) in the Gene Ontology. To better represent the endoplasmic reticulum UPR (GO:0030968: in green) within the Gene Ontology, we created new terms (denoted by red stars) for ‘IRE1-mediated unfolded protein response’ (GO:0036498), ‘PERK-mediated unfolded protein response’ (GO:0036499) and ‘ATF6-mediated unfolded protein response’ (GO:0036500), covering the three main branches of the mammalian UPR. GO:0070055 (in yellow) describes IRE1-mediated non-spliceosomal cleavage of the XBP1 mRNA in mammals and Ire1p-mediated cleavage of the HAC1 mRNA in yeast. Additional terms exist in this node but are not shown for conciseness. Black arrows denote is_a relationships between terms and blue arrows denote part_of relationships between terms. Image adapted from QuickGO
Call for Community Contributions
To make the greatest impact in the area of Parkinson’s disease, it is important that we consult experts in the field to ensure our GO annotations and terms are accurately capturing current knowledge. Thus we have established a scientific advisory panel for consultation and we interact with researchers and authors during curation, as described above. When curating a topic, we also refer to Reactome (Croft et al. 2014) and the Parkinson’s disease map (Fujita et al. 2014) to align our efforts with these manually-curated resources of cellular pathways and Parkinson’s-related mechanisms, respectively. We have held discussions with the scientists responsible for these resources to standardise annotations and Parkinson’s-related GO terms, and we plan to create further reciprocal links as our project progresses. We welcome feedback from the community, and there are many additional ways in which researchers can be involved with the project, or be kept informed about developments. Please contact us (goannotation@ucl.ac.uk) to:
-
i)
Subscribe to our quarterly newsletter.
-
ii)
Extend our gene priority lists by supplying us with details of key proteins.
-
iii)
Suggest gene annotations that are currently missing, or literature that requires curation.
Furthermore, our project progress can be followed on our Twitter account (@UCLgene) and the go-friends email list (http://geneontology.org/page/go-mailing-lists) can be subscribed to stay informed about all GO projects.
Conclusions
With this project we are providing an integrated, comprehensive set of annotations to describe proteins implicated, or predicted to play a role, in Parkinson’s disease. Our project will specifically build on the efforts of the Toronto Alzheimer’s disease annotation project (Patel et al. 2015), which is focused on using GO to identify proteins involved in Alzheimer’s disease through imaging genetics studies. To enhance the functionality of GO in their analyses, they created over 30 new relevant GO terms, and nearly 200 GO annotations to 15 human proteins (http://www.ebi.ac.uk/QuickGO/GAnnotation?tax=9606&source=Alzheimers_University_of_Toronto) with some notable overlap in prioritized proteins (e.g. Tau). There are a growing number of focused GO annotation projects, such as those covering the cardiovascular and transcription domains (Alam-Faruque et al. 2011; Tripathi et al. 2013), and these combined efforts are, together with the major GO Consortium members groups, improving the breadth and depth of the GO annotations available.
Previous GO Consortium projects have demonstrated the benefit of a topic-based annotation approach with notable improvements of the interpretation of relevant datasets (Alam-Faruque et al. 2014), and we envisage that our project will have a similar impact on the analysis of many neurobiological datasets, not only those related to Parkinson’s disease. Many neurodegenerative diseases share underlying cellular and molecular mechanisms, and the majority of the processes we have prioritised for annotation are relevant to other diseases. Disruption of the autophagy machinery, for example, can lead to a number of neurological disorders (Nah et al. 2015) including Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, Amyotrophic lateral sclerosis (ALS) and Multiple sclerosis, and beyond neurodegeneration autophagy has been linked to cancer, aging, liver and muscle disorders, and infection by pathogens (Shintani and Klionsky 2004). Dys-regulated Wnt signalling has also been suggested to underlie many neurological conditions including Alzheimer’s disease (Sadigh-Eteghad et al. 2015), autism and schizophrenia (reviewed in Berwick and Harvey 2014), and abnormal CDK5 signaling has a role in the pathogenesis of various neurodegenerative disorders including Parkinson’s and Alzheimer’s diseases, disrupting proteins including α-synuclein (ASN) and β-amyloid peptide (Aβ) (Wilkaniec et al. 2015). Lysosomal transport was prioritised for curation based on its relevance to Parkinson’s disease but it has wider implications, with lysosomal storage disorders and neurodegenerative disorders sharing a dysfunctional cellular transport network (Boland and Platt 2015).
Stress-insult is a common theme across human health, with the antioxidant PARK7 (DJ-1) playing a central role in protecting against oxidative stress in cancer, cardiovascular disease and neurodegeneration (Chan and Chan 2015). ER-stress and particularly ERAD is linked to more than 60 diseases (Guerriero and Brodsky 2012; Yoshida 2007), including Cystic fibrosis (Kerbiriou et al. 2007) and bipolar disorder (Pfaffenseller et al. 2014). Our prioritized proteins are also implicated in additional disorders; a search in OMIM (http://omim.org/) reveals that mutations in MAPT encoding the microtubule-associated protein Tau are linked to a number of neurological disorders including Frontotemporal dementia (FTD), Progressive supranuclear palsy 1 (PSNP1), and of course Alzheimer’s disease.
These examples are by no means exhaustive but aim to demonstrate how our improved annotations and additions to the GO structure will feed into the interpretation of datasets across a wide range of diseases, not only within the neurodegenerative spectrum, but also extending to other areas of health and disease. More manual annotations will mean that users will get more meaningful functional data analysis, with potentially more specific terms being significantly enriched in transcriptomic or proteomic studies. Furthermore, GO is already being used to improve GWAS identification of risk SNPs (Holmans et al. 2013), and improving GO for Parkinson’s disease is likely to further aid this approach, for a range of neurodegenerative diseases.
Combining GO with external resources further enhances its applications: Cytoscape (Shannon et al. 2003) enables our improved GO annotations to be combined with interaction data captured by IntAct, so researchers can visualize and manipulate protein interaction networks overlaid with significantly enriched GO processes. These networks may be used to predict ‘hubs’, key positions at which the whole network may be perturbed, and thus inform future research. Tools are currently being developed which will allow users to create complex queries based on the cross-links between GO and the chemical ontology ChEBI (Hill et al. 2013; Mungall et al. 2011), and the Parkinson’s-relevant annotations. Thus it will be possible to identify all gene products involved in dopamine processes in unrelated parts of the GO graph, such as dopamine transport, dopamine metabolism and dopamine binding, or all proteins involved in the metabolism of dopaminergic agents. We hope that our resource will benefit neurobiology researchers and scientists, and we encourage you to contact us with your questions or suggestions.
Information Sharing Statement
All of the annotations associated with our prioritised Parkinson’s-relevant proteins are freely available for viewing or download in QuickGO (http://www.ebi.ac.uk/QuickGO/, RRID:SCR_004608) (Binns et al. 2009) using the ‘Gene Product ID’ filter ‘ParkinsonsUK-UCL’ or in the GO Consortium browser, AmiGO (http://amigo.geneontology.org/, RRID:SCR_002143) (Carbon et al. 2009). In addition, these annotations and ontologies are uploaded to major biological knowledgebases such as Ensembl, NCBIGene and UniProtKB, and are available for download and local installation via ftp sites. Users can perform enrichment analysis on neurological datasets using a number of GO-supported enrichment tools (http://geneontology.org/page/go-enrichment-analysis).
References
Alam-Faruque, Y., Huntley, R. P., Khodiyar, V. K., Camon, E. B., Dimmer, E. C., Sawford, T., et al. (2011). The impact of focused gene ontology curation of specific mammalian systems. PLoS One, 6(12), e27541. doi:10.1371/journal.pone.0027541.
Alam-Faruque, Y., Hill, D. P., Dimmer, E. C., Harris, M. A., Foulger, R. E., Tweedie, S., et al. (2014). Representing kidney development using the gene ontology. PLoS One, 9(6), e99864. doi:10.1371/journal.pone.0099864.
Bard, J., Rhee, S. Y., & Ashburner, M. (2005). An ontology for cell types. Genome Biology, 6(2), R21. doi:10.1186/gb-2005-6-2-r21.
Berwick, D. C., & Harvey, K. (2014). The regulation and deregulation of Wnt signaling by PARK genes in health and disease. Journal of Molecular Cell Biology, 6(1), 3–12. doi:10.1093/jmcb/mjt037.
Binns, D., Dimmer, E., Huntley, R., Barrell, D., O’Donovan, C., & Apweiler, R. (2009). QuickGO: a web-based tool for gene ontology searching. Bioinformatics (Oxford, England), 25(22), 3045–3046. doi:10.1093/bioinformatics/btp536.
Boland, B., & Platt, F. M. (2015). Bridging the age spectrum of neurodegenerative storage diseases. Best practice & research. Clinical Endocrinology & Metabolism, 29(2), 127–143. doi:10.1016/j.beem.2014.08.009.
Carbon, S., Ireland, A., Mungall, C. J., Shu, S., Marshall, B., Lewis, S., et al. (2009). AmiGO: online access to ontology and annotation data. Bioinformatics (Oxford, England), 25(2), 288–289. doi:10.1093/bioinformatics/btn615.
Chan, J. Y. H., & Chan, S. H. H. (2015). Activation of endogenous antioxidants as a common therapeutic strategy against cancer, neurodegeneration and cardiovascular diseases: a lesson learnt from DJ-1. Pharmacology & Therapeutics. doi:10.1016/j.pharmthera.2015.09.005.
Croft, D., Mundo, A. F., Haw, R., Milacic, M., Weiser, J., Wu, G., et al. (2014). The reactome pathway knowledgebase. Nucleic Acids Research, 42(Database issue), D472–D477. doi:10.1093/nar/gkt1102.
Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J., & Diehl, J. A. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and Cellular Biology, 23(20), 7198–7209.
Cunningham, F., Amode, M. R., Barrell, D., Beal, K., Billis, K., Brent, S., et al. (2015). Ensembl 2015. Nucleic Acids Research, 43(Database issue), D662–D669. doi:10.1093/nar/gku1010.
Deegan née Clark, J. I., Dimmer, E. C., & Mungall, C. J. (2010). Formalization of taxon-based constraints to detect inconsistencies in annotation and ontology development. BMC Bioinformatics, 11, 530. doi:10.1186/1471-2105-11-530.
Dietze, H., Berardini, T. Z., Foulger, R. E., Hill, D. P., Lomax, J., Osumi-Sutherland, D., et al. (2014). TermGenie - a web-application for pattern-based ontology class generation. Journal of Biomedical Semantics, 5, 48. doi:10.1186/2041-1480-5-48.
Fujita, K. A., Ostaszewski, M., Matsuoka, Y., Ghosh, S., Glaab, E., Trefois, C., et al. (2014). Integrating pathways of Parkinson’s disease in a molecular interaction map. Molecular Neurobiology, 49(1), 88–102. doi:10.1007/s12035-013-8489-4.
Gasser, T., Hardy, J., & Mizuno, Y. (2011). Milestones in PD genetics. Movement Disorders: Official Journal of the Movement Disorder Society, 26(6), 1042–1048. doi:10.1002/mds.23637.
Gaudet, P., Livstone, M. S., Lewis, S. E., & Thomas, P. D. (2011). Phylogenetic-based propagation of functional annotations within the gene ontology consortium. Briefings in Bioinformatics, 12(5), 449–462. doi:10.1093/bib/bbr042.
Gene Ontology Consortium (2015). Gene ontology consortium: going forward. Nucleic Acids Research, 43(Database issue), D1049–D1056. doi:10.1093/nar/gku1179.
Guerriero, C. J., & Brodsky, J. L. (2012). The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiological Reviews, 92(2), 537–576. doi:10.1152/physrev.00027.2011.
Hill, D. P., Adams, N., Bada, M., Batchelor, C., Berardini, T. Z., Dietze, H., et al. (2013). Dovetailing biology and chemistry: integrating the gene ontology with the ChEBI chemical ontology. BMC Genomics, 14, 513. doi:10.1186/1471-2164-14-513.
Holmans, P., Moskvina, V., Jones, L., Sharma, M., International Parkinson’s Disease Genomics Consortium, Vedernikov, A., et al. (2013). A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson’s disease. Human Molecular Genetics, 22(5), 1039–1049. doi:10.1093/hmg/dds492.
Huntley, R. P., Harris, M. A., Alam-Faruque, Y., Blake, J. A., Carbon, S., Dietze, H., et al. (2014). A method for increasing expressivity of gene ontology annotations using a compositional approach. BMC Bioinformatics, 15, 155. doi:10.1186/1471-2105-15-155.
Kerbiriou, M., Le Drévo, M.-A., Férec, C., & Trouvé, P. (2007). Coupling cystic fibrosis to endoplasmic reticulum stress: differential role of Grp78 and ATF6. Biochimica Et Biophysica Acta, 1772(11–12), 1236–1249. doi:10.1016/j.bbadis.2007.10.004.
Kerrien, S., Aranda, B., Breuza, L., Bridge, A., Broackes-Carter, F., Chen, C., et al. (2012). The IntAct molecular interaction database in 2012. Nucleic Acids Research, 40(Database issue), D841–D846. doi:10.1093/nar/gkr1088.
Kim, S.-J., Park, Y.-J., Hwang, I.-Y., Youdim, M. B. H., Park, K.-S., & Oh, Y. J. (2012). Nuclear translocation of DJ-1 during oxidative stress-induced neuronal cell death. Free Radical Biology & Medicine, 53(4), 936–950. doi:10.1016/j.freeradbiomed.2012.05.035.
Kotera, M., Hirakawa, M., Tokimatsu, T., Goto, S., & Kanehisa, M. (2012). The KEGG databases and tools facilitating omics analysis: latest developments involving human diseases and pharmaceuticals. Methods in Molecular Biology (Clifton, N.J.), 802, 19–39. doi:10.1007/978-1-61779-400-1_2.
Magen, I., & Chesselet, M.-F. (2010). Genetic mouse models of Parkinson’s disease the state of the art. Progress in Brain Research, 184, 53–87. doi:10.1016/S0079-6123(10)84004-X.
Manzoni, C., Denny, P., Lovering, R. C., & Lewis, P. A. (2015). Computational analysis of the LRRK2 interactome. PeerJ, 3, e778. doi:10.7717/peerj.778.
Mitchell, A., Chang, H.-Y., Daugherty, L., Fraser, M., Hunter, S., Lopez, R., et al. (2015). The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Research, 43(Database issue), D213–D221. doi:10.1093/nar/gku1243.
Mungall, C. J., Bada, M., Berardini, T. Z., Deegan, J., Ireland, A., Harris, M. A., et al. (2011). Cross-product extensions of the gene ontology. Journal of Biomedical Informatics, 44(1), 80–86. doi:10.1016/j.jbi.2010.02.002.
Mungall, C. J., Torniai, C., Gkoutos, G. V., Lewis, S. E., & Haendel, M. A. (2012). Uberon, an integrative multi-species anatomy ontology. Genome Biology, 13(1), R5. doi:10.1186/gb-2012-13-1-r5.
Nah, J., Yuan, J., & Jung, Y.-K. (2015). Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Molecules and Cells, 38(5), 381–389. doi:10.14348/molcells.2015.0034.
Patel, S., Park, M. T. M., Chakravarty, M. M., & Knight, J. (2015). Novel bioinformatics approach to investigate quantitative phenotype-genotype associations in neuroimaging studies (No. biorxiv;015065v1). http://biorxiv.org/lookup/doi/10.1101/015065. Accessed 5 November 2015
Pfaffenseller, B., Wollenhaupt-Aguiar, B., Fries, G. R., Colpo, G. D., Burque, R. K., Bristot, G., et al. (2014). Impaired endoplasmic reticulum stress response in bipolar disorder: cellular evidence of illness progression. The International Journal of Neuropsychopharmacology/Official Scientific Journal of the Collegium Internationale Neuropsychopharmacologicum (CINP), 17(9), 1453–1463. doi:10.1017/S1461145714000443.
Pienaar, I. S., Götz, J., & Feany, M. B. (2010). Parkinson’s disease: insights from non-traditional model organisms. Progress in Neurobiology, 92(4), 558–571. doi:10.1016/j.pneurobio.2010.09.001.
Porras, P., Duesbury, M., Fabregat, A., Ueffing, M., Orchard, S., Gloeckner, C. J., & Hermjakob, H. (2015). A visual review of the interactome of LRRK2: using deep-curated molecular interaction data to represent biology. Proteomics, 15(8), 1390–1404. doi:10.1002/pmic.201400390.
Sadigh-Eteghad, S., Askari-Nejad, M. S., Mahmoudi, J., & Majdi, A. (2015). Cargo trafficking in Alzheimer’s disease: the possible role of retromer. Neurological Sciences: Official Journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. doi:10.1007/s10072-015-2399-3.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. doi:10.1101/gr.1239303.
Shintani, T., & Klionsky, D. J. (2004). Autophagy in health and disease: a double-edged sword. Science (New York, N.Y.), 306(5698), 990–995. doi:10.1126/science.1099993.
Slodzinski, H., Moran, L. B., Michael, G. J., Wang, B., Novoselov, S., Cheetham, M. E., et al. (2009). Homocysteine-induced endoplasmic reticulum protein (herp) is up-regulated in parkinsonian substantia nigra and present in the core of Lewy bodies. Clinical Neuropathology, 28(5), 333–343.
Tripathi, S., Christie, K. R., Balakrishnan, R., Huntley, R., Hill, D. P., Thommesen, L., et al. (2013). Gene Ontology annotation of sequence-specific DNA binding transcription factors: setting the stage for a large-scale curation effort. Database: The Journal of Biological Databases and Curation, 2013 bat062. doi:10.1093/database/bat062.
Varma, D., & Sen, D. (2015). Role of the unfolded protein response in the pathogenesis of Parkinson’s disease. Acta Neurobiologiae Experimentalis, 75(1), 1–26.
Wilkaniec, A., Czapski, G. A., & Adamczyk, A. (2015). CDK5 at crossroads of protein oligomerization in neurodegenerative diseases: facts and hypotheses. Journal of Neurochemistry. doi:10.1111/jnc.13365.
Yoshida, H. (2007). ER stress and diseases. The FEBS Journal, 274(3), 630–658. doi:10.1111/j.1742-4658.2007.05639.x.
Acknowledgments
We would like to thank the GO editorial team for useful ontology discussions and implementation of many ontology changes. We are very grateful to Heiko Dietze and other contributors to the TermGenie tool. PD, RF, JH, MM, RL and TS are funded by Parkinson’s UK, grant G-1307. PD, RF and RL are also supported by the National Institute for Health Research University College London Hospitals Biomedical Research. RL received additional support as a British Heart Foundation (BHF) chair scholar and MM, RL and TS received funding by BHF grant RG/13/5/30112. TS is also supported by National Institute of Health NIH/NHGRI grant U41HG002273. JH is also supported by the M.J. Fox Pritzker Prize, and a Wellcome Trust/MRC Joint Call in Neurodegeneration award (WT089698) to the UK Parkinson’s Disease Consortium (UKPDC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Foulger, R.E., Denny, P., Hardy, J. et al. Using the Gene Ontology to Annotate Key Players in Parkinson’s Disease. Neuroinform 14, 297–304 (2016). https://doi.org/10.1007/s12021-015-9293-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12021-015-9293-2