
Abstract
Mast cells (MCs) are commonly recognized for their crucial involvement in the pathogenesis of allergic diseases, but over time, it has come to light that they also play a role in the pathophysiology of non-allergic disorders including atherosclerosis. The involvement of MCs in the pathology of atherosclerosis is supported by their accumulation in atherosclerotic plaques upon their progression and the association of intraplaque MC numbers with acute cardiovascular events. MCs that accumulate within the atherosclerotic plaque release a cocktail of mediators through which they contribute to neovascularization, plaque progression, instability, erosion, rupture, and thrombosis. At a molecular level, MC-released proteases, especially cathepsin G, degrade low-density lipoproteins (LDL) and mediate LDL fusion and binding of LDL to proteoglycans (PGs). Through a complicated network of chemokines including CXCL1, MCs promote the recruitment of among others CXCR2+ neutrophils, therefore, aggravating the inflammation of the plaque environment. Additionally, MCs produce extracellular traps which worsen inflammation and contribute to atherothrombosis. Altogether, evidence suggests that MCs actively, via several underlying mechanisms, contribute to atherosclerotic plaque destabilization and acute cardiovascular syndromes, thus, making the study of interventions to modulate MC activation an interesting target for cardiovascular medicine.
Similar content being viewed by others

Introduction
Atherosclerosis Pathology and Microenvironment of Plaques
Atherosclerosis is an inflammatory lipid-driven pathology underlying many cardiovascular and metabolic diseases. Its complex pathology includes lesion initiation, progression, rupture or erosion, healing, and consolidation [1]. Plaques are dynamic entities in terms of cytoarchitecture and molecular interactions. The presence of lipid metabolites and events such as foam cell formation, accumulation of immune cells, and the release of proinflammatory mediators establish an inflammatory environment in the plaques [2]. Rupture of a plaque may lead to severe consequences of which stroke and myocardial infarction have a high medical significance [3]. The microenvironment of plaques is populated by a variety of cell types including endothelial cells, smooth muscle cells (SMCs), macrophages, lymphocytes, and MCs. These cells produce and release chemokines; cytokines; proinflammatory mediators such as IFN-γ, IL6, and TNF [4]; and proteases that may contribute to atherosclerotic lesion development [5, 6].
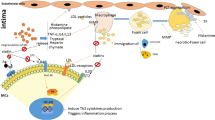
Atherosclerosis is triggered by endothelial damage of arteries and the deposition of low-density lipoproteins (LDL) [7]. Additionally, monocytes that enter the plaque differentiate into macrophages, which later play a key role in the pathology of the disease [8]. Endothelial cells transfer oxLDL to the intima by expressing lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) and exposing macrophages to oxidized low-density lipoprotein (oxLDL) [9]. This process drives the formation of foam cells leading to the initiation of atherosclerotic plaque formation [3]. Foam cells contribute to establishing the plaques by ingesting exceeding levels of lipids [10]. Moreover, macrophages present the peptide and lipid fragments to the surrounding T cells via MHC /TCR or NKTs through CD-1d /TCR interactions respectively [10,11,12]. Th1 cells act as proatherogenic cells by releasing IL-12 and IFNγ. Tregs are also held to play an atheroprotective role, by releasing IL-10 [13, 14]. The consequence of NKT activation in response to the stimuli is the release of cytokines including atherogenic Th1 cytokines such as IFNγ and TNFα as well as the Th2 cytokines IL-4 and IL-13 [13]. B cells can be, depending on their subset, either proatherogenic or atheroprotective. Their depletion may affect splenocyte proliferation in response to oxidized LDL suggesting a role for B cells in the presentation of lipid antigens to T cells [15]. Several autoantibodies such as anti-heat shock protein 60 (HSP-60) and anti-oxLDL have been reported to contribute to the pathology of atherosclerosis [15] (Fig. 1).

Microenvironment of atherosclerotic plaques and interactions between residing immune cells in brief. B cells become activated and produce antibodies including anti-oxLDL and anti-HSP-60 Abs. Th1 and Th2 cells produce IL-12, IFN-γ, and IL-4 respectively. Oxidized LDL is taken up through LOX-1 receptors expressed on endothelium and then is accumulated inside the macrophages to form foam cells. Th2 cells play an important role as atheroprotective immune cells. Th2-produced IL-4 counteracts the production of IFN-γ which is a proatherogenic cytokine; therefore, differentiation in favor of Th2 leads to modulating the production of atherogenic cytokines. NKTs recognize lipids presented by CD1d-expressing APCs that become activated and release IFN-γ, TNFα, IL-4, and IL-13. Macrophages act as APCs and present lipoproteins to CD4+ T cells. MCs accumulate within plaques during atherosclerosis and become activated and release proteases including cathepsin G that targets LDL. Tryptase and chymase released by MCs play a role in instability and rupture of the plaques. They release cytokines and recruit further immune cells into the plaque microenvironment. Additionally, mediators released by MCs inhibit the production of ECM components by fibroblasts and the proliferation of fibroblasts and induce the apoptosis of smooth muscle cells. The proteases released by MCs convert pro-MMPs to active MMPs which contribute to ECM degradation. MCs also act as the source of MMP
The histo-architecture of atherosclerotic plaques shows that they contain accumulations of SMCs and densely organized collagens covering the area with necrotic fragments, inflammatory cells, cholesterol clefts, and deposition of calcium [16]. Accumulation of immune cells and secretion of cytokines in favor of inflammation are of the key factors bridging the above-mentioned features. In this regard, macrophages accumulate and release inflammatory cytokines mainly TNF, IL-1β, and IL-6; induce the necrosis of cells within plaques; and produce matrix metalloproteinases in particular MMP-9 which degrade the extracellular matrix (ECM) and weaken the plaque stability [17, 18]. The stability of plaque can be affected by features especially the presence of large necrotic lipid-rich core, a thin fibrous cap, extension of vasculature, and hemorrhage that together make the plaque prone to rupture [17, 18]. The occurrence of atherosclerotic plaque rupture may result in arterial thrombosis, myocardial infarction, and stroke [16].
Considering our aim to highlight the role of MCs in the pathology of atherosclerosis, in the next paragraph, we review and highlight specific aspects of the disease and the main mechanisms of MC involvement in reshaping the atherosclerotic plaque microenvironment.
MCs in Plaque Microenvironment
Investigation of the role of MCs in atherosclerosis goes back to the early 1950s, when Constantinides, based on previous studies showing heparin’s capability to remove lipoproteins from the blood of individuals with atherosclerosis and prevent lipidemia and that MCs are of the sources of heparin, focused on these cells to reveal the possible new aspect of pathophysiology of atherosclerosis [19].
From a histologic point of view, MCs are found in both the intima and perivascular tissue of atherosclerotic plaques [20]. Very early studies showed the presence of MCs in coronary arteries and provided information regarding their subtypes using monoclonal Abs against tryptase and chymase [6].
Kaartinen et al. showed that in the normal coronary intimas, 0.1% of all nucleated cells were MCs. The fatty streaks showed a higher MC density (ninefold), and in the cap, core, and shoulder regions of atheromas, MC density reached 5-, 5-, and tenfold, respectively when compared to normal coronary intimas. Using specific anti-tryptase Abs, they detected an average of 1 MC/mm2 in the normal intima, while a higher number of MCs (5 MC/mm2) was detected in intimas from individuals with atherosclerosis. Their results showed that the fatty streaks of normal intimas and the shoulder of atheromas contain a population of MCs of which an average of 35–40% was chymase+, whereas cap and core regions were populated by an average of 53% and 45%, respectively [6].
Considering that SMCs produce ECM components, MC-released chymase may induce the apoptosis of SMCs and also production and activation of TGF-β in these cells which in turn inhibits their proliferation through which may play a role in predisposing the plaque to weakening and later to rupture [21].
Moreover, microscopic observation showed that the number of activated MCs with signs of degranulation was significantly higher in the intimal areas (92% and 85% in the core and shoulder regions of atheromas respectively) which were degranulated, this being a sign of their activation, featuring the progression of atherosclerosis including fatty streaks, cap, core, and shoulder compared to normal intimal areas [6].
Investigation of MC presence in specimens collected from postmortem coronary arteries showed that MCs were accumulated at immediate site of rupture and formed 6% of nucleated cells, while their density was lower at adjacent atheromatous area (1%) or at intact intimal (0.1%).
Moreover, this study showed that a large percentage of MCs in each anatomical site was degranulated. Degranulated MCs were, for example, found at the site of erosion or rupture (86%), while 63% in the adjacent atheromatous area. The non-ruptured intima had the lowest percentage of degranulated MCs (27%) [22].
In another study, performed on 44 sections of aortas collected from autopsies using anti-tryptase and anti-chymase antibodies, the following results were obtained: (1) chymase+ MCs were more abundant in the non-elderly group, (2) a positive correlation between the number of chymase+ MCs and percentage of collagen (rS = 0.115, P = 0.006) and also between number of tryptase+ MCs and percentage of collagen (rS = 0.111, P = 0.008) was found at the site, and (3) a positive correlation between lipidosis and the number of tryptase+ MCs (rS = 0.117; P = 0.004) was reported [23].
After accumulation in the adventitial tissue and plaques [24], MCs act as inflammatory cells by releasing cocktails of proinflammatory cytokines [5]. Investigation of MC density (MCD) in human atherosclerotic plaques revealed that MCD was higher in unstable plaques than in stable plaques [25]. Mekke et al. studied human carotid atherosclerotic plaques and found that the highest MC numbers per square millimeter were observed in the symptomatic culprit lesion and the lowest in the symptomatic more distant segment, demonstrating a positive correlation between MC accumulation, symptomatic plaque segment, neovascularization, and subsequent inflammatory cell recruitment [26]. Interestingly, postmortem studies of individuals with coronary artery atherosclerosis showed that immunoglobulin E (IgE) and tryptase levels were elevated when compared to control deceased individuals who died because of non-cardiac reasons [5]. This is in line with studies performed by Willems et al., in which circulating tryptase levels were of predictive value for future cardiovascular events. Investigation of MC number in human carotid atherosclerotic lesions revealed high numbers, which correlated with plaque microvessel density and with future cardiovascular events [27].
Postinflammation plaque calcification is commonly reported in end-stage atherosclerosis. Osteogenic differentiation of arterial SMC is linked to vascular calcification. Since MCs interact with SMCs, their effects on phenotype reprogramming of SMCs have recently been investigated by Skenteris et al. They reported the colocalization of tryptase+ MCs and α-SMA/SOX9+ SMCs or α-SMA/ RUNX2+ SMCs. Next, they investigated the MC activation capacity of plaque medium on bone marrow–derived MC culture and found that, unlike supernatants of calcified plaques, the supernatant collected from highly calcified plaques could not induce MC activation and reduced the MC releasing of MIP-1α(CCL3) suggesting that calcification inhibits MC activation, while both resting or activated MCs induce SMC calcification [28].
Interaction of MCs and Immune Cells in Atherosclerotic Plaques
MC Interaction with T Cells
CD4+ T Cells
Early investigations of the atherosclerotic plaque microenvironment revealed the presence of activated T cells and antigen presenting cells (APCs) including macrophages that strongly express HLA class II molecules [29]. These plaque CD4+ T cells become activated and proliferate once exposed to for example oxLDL (a potential autoantigen found in atherosclerotic plaques) and produce IFN-γ and IL-4 [29]. IFN-γ activates macrophages, thus supporting inflammation, while IL-4 acts as a driver of B cell differentiation and antibody production [29, 30].
Upregulation of MHCII molecules in MCs has been documented in the presence of IL-4 and IFN-γ (Fig. 2a). Ldlr−/− mice with western-type diet (WTD) were reported to have increased levels of cholesterol, peritoneal MC (PMC) numbers, and upregulation of functional MHCII molecules as compared to mice on a low-cholesterol diet.
Besides, coculturing OT-II CD4+ T cells with WTD MCs loaded with OVA could induce the activation and proliferation of CD4+ T cells [7]. Investigation of the interaction of costimulatory molecules OX40L/OX40 expressed on MCs and T cells, respectively, in atherosclerosis provided another line of evidence for the role of MCs in the pathology of the disease. Treatment of mice with anti-OX40L (RM134L) Abs decreased the Th2 response, the levels of circulating IL-4 and IgE, and MC numbers in the plaque. Ldlr−/−mice receiving WTD treated with anti-OX40L Ab showed regression of atherosclerosis. This was linked to the production of IL-33 by APCs, which mediated the production of the atheroprotective cytokine IL-5 and oxLDL-specific IgM by T and B1 cells, respectively [31].
NKTs
MCs can present lipid antigens through their surface-expressed MHC class I–related molecule CD1d to CD1d-restricted natural killer T cells (NKTs), which activates them and induces the release of an array of cytokines [10, 32]. A recent study explored the role of the MC-CD1d/TCR-NKT axis in the pathology of atherosclerosis [10]. Kritikou and colleagues reconstituted MC-deficient apoE−/−KitW−sh/W−sh mice with either CD1d−/− or wild-type CD1d +/+ MCs and put both groups on an atherogenic diet. Their results showed that (1) apoE−/−KitW−sh/W−sh mice reconstituted with CD1d−/−MCs developed larger plaques when compared with apoE−/−KitW−sh/W−sh mice being reconstituted with WT MCs and (2) the density of intraplaque CD4+ T cells was higher in CD1d−/−MCs reconstituted apoE−/−KitW−sh/W−sh. They concluded that the disruption of the MC-CD1d/TCR-NKT axis aggravates the progression of atherosclerosis [10] (Fig. 2b).

a Molecular mechanism of activation, releasing profile, and crosstalk of immune cells upon being exposed to oxLDL. CD4 + T cells become activated, proliferate, and release IFN-γ and IL-4 upon exposure to oxLDL. IFN-γ drives the activation of macrophages, therefore, supporting the inflammation, whereas, IL-4 induces B cell differentiation and antibody production. b Investigation of the role of CD1d − expressing or knockout MCs in mice on the atherogenic diet, reconstitution of apoE −/− Kit W−sh/W−sh mice with either CD1d −/− or wild-type CD1d +/+ MCs after being on an atherogenic diet showed that apoE −/− Kit W−sh/W−sh mice receiving CD1d −/− MCs develop larger plaques and have more intraplaque CD4 T cells. c An illustrated model of “granule-mediated uptake of LDL,” an interaction of MC-macrophage is based on exocytosis of granules from MCs which lose their soluble mediators and the remnants provide a scaffold to binding LDL by which ApoB will be detached from LDL and the remaining molecule fuse with other similar ones and eventually will be phagocytosed by macrophages. After trapping into lysosomes, LDL will be hydrolyzed and a final process of esterification of LDL-derived cholesterol occurs in the cytoplasm and the final lipid cargo is stored in macrophage and transformed into foam cells
Neutrophils, Monocytes, Macrophages, and Foam Cells
Plaque-residing MCs have been reported to have a role in recruitment of other inflammatory cells to the cite. MCs degranulate after being activated by ROS released by the extravasated neutrophils. MCs then induce the recruitment of neutrophils and T cells by releasing TNF. Moreover, MC-released MCP-1 induces the recruitment of monocytes which later may develop in macrophage or foam cells and aggravate atherosclerosis [33].
Studies of the cellular architecture of atherosclerotic plaques showed a correlation between (a) the number of MCs with the percentage of the plaque area populated with macrophages (r = 0.156, P = 0.011) and (b) the number of degranulating MCs and the percentage of plaque residing macrophages (r = 0.310, P = 0.002) [27]. MC activation may contribute to the formation of foam cells. In line with this the term “granule-mediated uptake of LDL” was already introduced to picture how IgE-dependent MC activation results in the release of mediators that boost the macrophage LDL uptake. Granule-mediated uptake of LDL is defined as a process in which MC granules placing a variety of MC mediators promote after being released bind to LDL and phagocytosed by macrophages. The uptake of mediator LDL by macrophages results in enhancing the extent of cholesteryl ester synthesis in these cells and thus accumulation of cholesteryl esters [34].
In this model described by Dr. Petri Kovanen in 1991, two cells are playing crosstalk: on the one hand, MCs degranulate IgE dependently and release granules that their soluble content releases upon exocytosis. Then, LDL attaches the glycosaminoglycan chains in remnant structure afterward, hydrophilic segments of apolipoprotein B are detached under influence of chymase and carboxypeptidase, and then proteolyzed LDLs fuse and phagocytosed by macrophage. Phagolysosomes of macrophages hydrolyze the complex and finally, esterification of LDL-derived cholesterol is completed in the cytoplasm [35, 36] (Fig. 2c). In a study performed by Ma and Kovanen, male Wistar rats were immunized with ovalbumin and Bordetella pertussis vaccine (as adjuvant) to induce the production of anti-ovalbumin IgE, after which peritoneal MCs and macrophages were harvested from ovalbumin-treated rats and controls to analyze macrophage LDL uptake and formation of foam cells. They prepared a monolayer culture of peritoneal macrophages, added ovalbumin-sensitized and non-sensitized-MCs, and then added ovalbumin plus [14C] sucrose-LDL to track the process of LDL uptake. Moreover, they measured histamine in the supernatant. They found out that the presence of ovalbumin-sensitized MCs boosts the capacity of macrophages to take up LDL. However, macrophage LDL uptake was not significantly changed in non-immunized conditions. They concluded that the interaction of MCs and macrophages to form foam cells is IgE dependent which results in degranulation of MCs. Liberation of histamine from the macrophage-phagocytosed granules induces the macrophage LDL uptake and mediates their transformation into foam cells [37] (Fig. 3a). On the other hand, activated MCs have been shown to induce macrophage apoptosis and plaque instability in advanced mouse atherosclerotic plaques, which was suggested to be mediated by MC proteases and histamine [38]. Both MC-macrophage interactions suggest that stabilizing MCs may provide a therapeutic approach to control atherosclerosis.
Apart from the in vivo models to study crosstalk between MCs and macrophage foam cells, in vitro studies have been described to study their potential interactions.
Plotkin et al. considering the anti-inflammatory effects of fullerene derivatives (FDs) investigated the effects of these water-soluble carbon spheres both on MC activation and foam cell (CD36+) formation from monocyte-derived macrophages (CD68+). The authors reported that FDs may act through the NF-κB pathway to render biologic effects including preventing the lipoprotein-induced release of TNF-α, upregulation of foam cell marker CD36, and C5a-mediated activation of MCs [3]. The expression of CD36, CD68, and scavenger receptor class A (SRA) enables macrophages to recognize dying cells and different forms of LDL mainly oxLDL [39]. Upon exposure to TNF-α, monocytes become activated and show signs of clumping and lipid uptake to form foam cells. In their experiment, PMA-treated cells clumped and showed a significant reactivity to Oil-Red-O staining. Application of FDs reverted these features in treated U937 cells [3]. Assessment of β-hexosaminidase release and TNF production in C5a-activated connective tissue MCs before and after FD treatment showed that FDs inhibited C5a-induced degranulation and cytokine production in MCs [3] (Fig. 3b).

a Induction of foam cell formation using rat peritoneal MCs and macrophages. Peritoneal MCs and macrophages from immunized male Wistar rats with ovalbumin and Bordetella pertussis vaccine (as adjuvant) were harvested from treated and controls. Ovalbumin plus [ 14 C] sucrose-LDL were added to a monolayer culture of peritoneal macrophages and histamine levels in supernatant were measured. The results showed that the presence of ovalbumin-sensitized MCs boosts the capacity of macrophages to uptake LDL. In case MCs were not from immunized rats, the LDL uptake by macrophage was not significantly changed. Immunized MCs degranulated and released significantly higher levels of histamine and could induce the formation of foam cells more effectively. b Induction of foam cell formation from human myelomonocytic cell line in vitro: exposure of U937 cells to PMA followed by oxLDL resulted in the formation of foam cells. Addition of FDs inhibits the foam cell formation, TNFα release from U937 cells, cell adhesion, and lipid uptake. Β-Hexosaminidase and TNFα assessment in C5a-activated MCs and comparing the results with those obtained from C5a-activated MCs exposed to FDs revealed that FDs inhibit the degranulation and cytokine release. c Role of MC-released mediators in angiogenesis and lymphangiogenesis: MC-released VEGF-A and B induce angiogenesis, while VEGF-C and D induce lymphangiogenesis upon release in response to non-IgE/FcεRI-dependent activation
The Role of MCs in Neoangiogenesis and Vascular Remodeling in Atherosclerosis
MCs contribute actively to the processes of angiogenesis and lymphangiogenesis by releasing proangiogenic VEGF-A and VEGF-B, and prolymphangiogenic VEGF-C and VEGF-D, respectively [40, 41] (Fig. 3c). The production and release of VEGF are not IgE/FcεRI dependent (classic pathway of degranulation [42]) necessarily and MC-released IL-6 may induce the production of VEGF in an autocrine manner without inducing MC degranulation [43]. The role of MCs in the regulation of angiogenesis within atherosclerotic plaques was in the focus of Kaartinen and colleagues. They stained human coronary intima samples using Elastica-van Gieson to assess the formation of the plaques and applied anti-von Willebrand factor to screen the process of angiogenesis. Additionally, they benefited from monoclonal antibodies targeting MC tryptase and chymase to study MCs [44]. They reported the accumulation of MCs in areas with neoangiogenesis in the proximity of microvessels. Released tryptase and chymase from activated MCs and their angiogenic role are linked to the pathology of atherosclerosis [44]. Considering that coronary intima lacks the common capillary blood circulation, the invasion of microvessels to the deep regions of the atheromas supplies the plaques with necessary blood components and supports their progression [44]. Similarly, a significant association between mast cell numbers and microvessel density was shown in human carotid arteries, in which mast cells were shown in close proximity to intraplaque neovessels. Indeed, one could speculate that mast cells, upon activation, promote intraplaque hemorrhage due to the induction of microvessel leakiness [27]. Mast cells can also promote neovascularization upon the induction of ischemia, as was shown in a mouse model of hind limb ischemia [45].
Chillo and colleagues proposed a novel mechanism through which MCs may participate in arteriogenesis.
They considered that fluid shear stress, which drives the force behind arteriogenesis, is only directly detectable by vascular endothelial cells, but not by perivascular cells. They focused on platelets which sense the fluid shear stress and can adhere to endothelium-expressed von Willebrand factor (vWF) upon fluid shear stress increasing through platelet glycoprotein Ib (GPIb) receptor [33]. In their mouse model, blockage of the platelet receptor GPIba as well as genetic ablation of the ectodomain of GPIba in transgenic IL4-R/Iba mice inhibited MC degranulation to a level comparable to cromolyn treatment suggesting a functional link between platelet receptor GPIba and MC activation. Moreover, they found that in addition to platelet receptor GPIba, urokinase plasminogen activator (uPA), a serine protease that plays important roles in cancer invasion and cell migration [46] mediated extravasation of neutrophils [33]. Neutrophils after extravasation were shown to produce reactive oxygen species (ROS) by neutrophil-expressed Nox2 and then release these active molecules which in turn activate MCs [33]. They applied different techniques to deplete neutrophils (using Ly-6G (1A8) antibody), deficiency of Nox2, and blocking ROS production to investigate the effect of neutrophils and their produced ROS in activation of MCs and concluded that neutrophil-produced ROS mediate MC activation in their mouse model. Additionally, MCs activate matrix metalloproteinases (MMPs, zinc-containing endopeptidases involved in ECM turnover [47, 48]) by releasing proteases and play a role in vascular remodeling upon arteriogenesis [33].
Role of MCs in Atherosclerosis Plaque Calcification
Intimal calcification is a clinical marker of atherosclerosis and although it has been recognized as an active vascular inflammatory response and remodeling, its role remains unclear in terms of plaque stability and prognosis due to differences in size and morphology [49]. Previous studies provided a line of evidence on MC involvement in the process. As an example, a study on 250 samples of atherosclerotic lesions in carotid arteries using anti-tryptase IHC investigated the association of MCs, macrophages, SMCs, and elastin with the extent of calcification. The researchers found that MCs and their released tryptase were largely associated with areas of early calcification (including stippling and morula-type calcifications) although MC accumulation and release of tryptase were observed in some late-stage solid calcifications. Additionally, the tryptase was associated with local matrix disruption and oedema at the matrix–calcification interface [50].
The study by Skenteris et al. demonstrated that unstable carotid atherosclerotic plaques and vascular lesions are abundant with activated MCs and that the average number of MCs was correlated negatively with the calcification content [51].
Engaged MC Expressed Receptors in Atherosclerosis
FcεRI
IgE/FcεRI accounts for the main mechanism of MC activation, where FcεRI molecules are engaged with allergen-bound IgE [52, 53]. This crosslink triggers the activation of protein tyrosine kinase Syk which leads to downstream events including the phosphorylation of several targets including TRAPs and LAT. “Phospholipase Cγ” (PLCγ) after anchoring and becoming activated catalyzes the “phosphatidylinositol 4,5-biphosphate” (PIP2) hydrolysis to form second messengers “diacylglycerol” (DAG) and inositol 1,4,5, -triphosphate (IP3). IP3 binds to its receptors on the endoplasmic reticulum (ER) and promotes Ca2+ efflux from the ER that results in MC degranulation [54]. IgE-FcεRI network triggers the release of MC mediators including histamine, tryptase, and chymase which play a role in the progression of plaques and are discussed in the following section. In human atherosclerotic plaques, MCs have been shown to express FcεRI, and a subset of these MCs had IgE molecules bound in their surface, suggesting that this may be an important activation pathway within the plaque. The local antigen(s) however, have not been identified [63].
According to the literature, there are studies that associate above-normal IgE levels with endothelial dysfunction and CVD in humans and animals [55]. In line with this, atopic patients with no sign of atherosclerosis with higher IgE levels had notably lower coronary flow reserve (CFR) which is used as an endothelial dysfunction biomarker [56]. In another study, higher levels of serum IgE levels were found in patients with acute ischemic stroke [57]. Moreover, an association between higher IgE levels with cardiovascular mortality was reported [58].
Interestingly, Wilson et al., after evaluating the total and specific IgE to a food allergen (α-Galactose) in 118 subjects, found a correlation between total IgE and α-Gal specific IgE levels and between α-Gal-specific IgE levels and the atheroma burden [59].
Another aspect of IgE in exacerbation of atherosclerosis was reported when natural secreted IgM (sIgM) was studied. In the context of atherosclerosis, these Abs target oxidation-specific epitopes found on OxLDL; therefore, functionally anti-OxLDLsIgM was suggested to be protective in atherosclerosis and CVD mainly by neutralizing proatherogenic effects of OxLDL. Investigation of B cell generation in sIgM−/−and Ldlr−/−sIgM−/− revealed that these cells have an impaired generation; therefore, the IgE clearance mediated by their expressed low-affinity IgE receptor (CD23) was not performed normally resulted in an increase in IgE levels. Application of IgE-neutralizing Ab (R1E4) reduced vascular inflammation and limited atherosclerotic lesion formation. Among the results reported in this study, MCs in Ldlr−/−sIgM−/− mice had a higher rate of activation visualized by chloroacetate esterase staining when compared to Ldlr−/− mice probably due to higher levels of IgE [60].
Bruton’s tyrosine kinase (BTK) is expressed in B cells and MCs. In atherosclerosis, follicular B cells are described to exert atherogenic functions, such as proinflammatory cytokine production, IgG antibody secretion, and regulating T cell responses. In MCs, upon IgE to FcεRI binding, downstream proteins Lyn and Syk become phosphorylated, resulting in the phosphorylation of BTK. Phosphorylated BTK leads to phosphorylation of PLCγ2 and eventually leads to mast cell degranulation and production of inflammatory cytokines.
Considering the crucial role of FcεRI signaling in the responsiveness of MCs to IgE, Hemme et al. applied a Bruton tyrosine kinase inhibitor (acalabrutinib) to study the significance of the FcεRI receptor signaling in progression of atherosclerosis in Ldlr− /− mice on high-fat diet (HFD) 2 months while taking acalabrutinib. They observed an impaired B cell maturation (as BTK is a necessary factor in the maturation of B cells [61]), a significant increase in splenic immature follicular II B cells in acalabrutinib-treated mice, and a decrease in mature follicular I B cells, whereas MC numbers and activation remained unaffected, and the size of plaques was not altered [62].
These data confirm that acalabrutinib successfully inhibited BTK in vivo and that BTK inhibition leads to a shift towards less B cell maturation in atherosclerosis. Despite the effect on B cell maturation, circulating antibody levels remained unaffected. They concluded that BTK inhibition alone did not affect MC activation in early and advanced stage atherosclerosis, despite the systemic biological effect on follicular B cell maturation [62].
IgE/FcεRI-mediated activation of MCs in plaques has been also reported and rationalizes a molecular mechanism involved in the pathology of Kounis syndrome [63] in which cardiovascular symptoms occur secondary to allergic or hypersensitivity insults. In this unrecognized or undiagnosed cardiovascular pathology due to its broad clinical manifestations, the symptoms are initiated by a number of reasons mainly drugs, environmental exposures, nutrients, and coronary stents. Pathologic events such as coronary spasm, acute myocardial infarction, and stent thrombosis, accompanied by activation of MCs and platelets and the presence of inflammatory cells are the main findings of Kounis syndrome [64, 65].
Complement Receptor
The activation of the complement system has been documented in atherosclerosis, and high levels of C5a have been reported in individuals suffering from the disease. MCs become activated and degranulate upon C5a/CD88 binding [3]. In an in vivo experiment, C5a-mediated MC activation was shown to enhance atherosclerotic vein graft disease in a MC-dependent fashion [66].
TLRs
MCs express both endosomal and membrane toll-like receptors (TLRs) [67] of which TLR-4 responds to pathogens by sensing their LPS. It is now evident that LPS activation of MCs induces the release of chymase and IL-6 that destabilize plaques and induce the apoptosis of vascular SMCs in plaques, respectively [68].
Neuropeptide Receptors
Among the activating receptors expressed on MCs, MRGPRX2 makes MCs responsive to neuropeptides including substance P (SP) [69]. However, the expression of MRGPRX2 awaits to be explored on cardiac MCs, and responsiveness of MCs could be due to the engagement of other neuropeptide receptors. Neurokinin-1 receptor (NK1R) acts as a receptor for SP and is expressed on MCs. The presence of plaque-residing MCs in close vicinity of nerve fibers capable of releasing SP inspired Bot et al. to inspect the SP-mediated MC activation in a SP-treated WTD fed apoE−/− mice after 6 weeks of placing semiconstrictive collars at the carotid arteries. They reported that SP treatment increases adventitial MC numbers, activates MCs, and promotes intraplaque hemorrhage. Application of neurokinin-1 receptor antagonist spantide I could prevent the effects suggesting that SP induces MC activation [70].
Lysophosphatidic Acid (LPA) Receptor
It is evident that lysophosphatidic acid (LPA), a MC activator, progressively piles up in plaques during the progression of atherosclerosis and in vitro investigation of its potency on degranulation of a variety of MC types including MC/9 cells, BMMCs, and mouse PMCs. In vitro investigation showed that LPA induces β-hexosaminidase and tryptase release from these cells. Additionally, intradermally injection of LPA or LPA-activated MCs could induce vascular leakage, which destabilizes plaque, while this effect and the release of tryptase were abolished when LPA was injected to KitW−sh/W−sh mice. Application of this in vitro model of LPA-mediated MC activation to a mouse in vivo model provided a link to study the effects of LPA accumulation over time on activation of plaque-residing MCs and then the study of LPA-MC axis in atherosclerosis. Bot and colleagues then applied LPA perivascularly using a gel at the collar-induced carotid artery lesion in apoE−/− mice. Although no plaque size variation was observed between the LPA-challenged and control mice after 3 days, the number of activated MCs was notably higher (P < 0.05) in the LPA-challenged mice when cromolyn was applied [71].
Involvement of MC-Released Mediators in Atherosclerosis
Histamine
One mechanism by which cellular cholesterol homeostasis is maintained is reverse cholesterol transport (RCT). During this process, cellular cholesterol is transferred from peripheral organs to the liver to undergo fecal excretion as bile acid [72, 73]. MC-released histamine induces macrophage RCT in an H1R-mediated pathway. Via this receptor. histamine can disrupt the endothelial barrier and facilitate the transfer of atheroprotective HDL into interstitial fluid and then the liver [72].
Protease
Activated MCTCs release tryptase and chymase and have been reported to accumulate at the site of rupture [38]. Moreover, MCs contribute to the aggravation of the inflammation and destabilization of coronary plaques by releasing TNF-α [38]. Cathepsin G possesses serine protease activity and is produced mainly by MCs and neutrophils [74]. It induces the elastin solubilizing activity of elastase and activation of several MMPs including MMP-1, MMP-2, MMP-3, and MMP-9 [74, 75]. Investigating the role of cathepsin G in the induction of atherosclerosis in Ldlr−/− mice on WTD showed that cathepsin G acts as an atherogenic factor in the early stages of the disease; however, it degrades LDL (having no effect on HDL or triglyceride content) through which it plays a protective role in the progression of atherosclerosis [75]. Maaninka et al. studied the role of MC-released proteases in the pathology of atherosclerosis. They incubated LDL with MC-conditioned medium prepared by activating human MCs with calcium ionophore and studied the binding of modified LDL to isolated aortic proteoglycans or to atherosclerotic plaques ex vivo. Their results showed that of the released proteases including tryptase, chymase, carboxypeptidase A3 (CPA3), granzyme B (GrB), and cathepsin G, only the latter showed degradation of apoB-100, induction of LDL molecule fusion, and promoting the binding of LDL to aortic proteoglycans and lesions [76].
MC-released chymase is capable of proteolytically cleaving the carboxyl terminal of apolipoprotein A-I (apoA-I). This capability of chymase makes apoA-I unable to bind to coronary artery endothelial cells (ECs). Additionally, other outcomes of enzymatic effects of chymase on apoA-I include suppression of nuclear factor-κB (NF-κB)–dependent upregulation of vascular cell adhesion molecule-1 (VCAM)- and abrogation of apoA-I anti inflammatory activities such as downregulation of the expression of TNF-α, IL-1β, IL-6, and IL-8 in foam cells and inhibition of ROS formation in neutrophils [77]. A Langendorff heart perfusion system-based study showed that compound 48/80 activated rat cardiac MC release chymase that degrades apoA-I at Tyr192 and Phe229 sites. Interestingly, under hypoxia-mediated MC activation, chymase was reported to cleave 243-amino acid intact apoA-I only at Tyr192 site [78]. The Tyr192-truncated fragment in proportion to the intact apoA-I showed significantly lower ability to mediate the migration of cultured human coronary artery endothelial cells during in vitro wound-healing test [78].
SMCs play a crucial role in the production of matrix and their loss is a feature that predisposes the plaque to weakening and later to rupture. The results of in vitro studies showed that MC-produced chymase could inhibit the collagen type I and III mRNA expression SMCs in cultured rat and human coronary arterial SMCs. Moreover, chymase induces the production and activation of TGF-β in SMCs which in turn inhibits their proliferation and growth through arresting G0/G1 → S transition. Application of anti-TGF-β antibody could partially reverse the inhibitory effect [21]. MC-produced chymase can directly induce apoptosis in growth-arrested rat arterial SMCs treated for 4 h with chymase. The mechanism is based on the disruption of NF-κB signaling resulting in bcl-2 mRNA downregulation, influencing the expression of protein, and leakage of cytochrome c [79].
Mouse MC protease-4 (mMCP-4) is the mouse protease mostly related to human chymase and its involvement in the progression of atherosclerosis has been studied. The results of Houde et al. on ApoE−/−, mMCP-4−/−, and ApoE−/−mMCP-4−/− mice in different stages of atherosclerosis showed that mMCP-4 inhibition may reduce plaque progression in the earlier phases of atherosclerosis and stabilize the advanced plaques [80].
Cytokines and Chemokines
Additionally, MCs contain a complex array of chemokines that mediate the recruitment of immune cells, of which, for example, neutrophils aggravate the inflammatory status of the plaques. For instance, (MC-released) CXCL1/(neutrophil expressed) CXCR2 interaction provides a mechanism through which neutrophils may be recruited into the plaque environment [20]. Mouse MCs released chemokine KC (ortholog of human IL-8, also known as a cytokine-induced neutrophil chemoattractant (CINC), growth-related protein α (Gro-α), and CXCL1 [81]) acts through monocyte receptors CXCR2 and VLA-4 and plays a role in the recruitment of monocytes to the plaques [38].
Results obtained from mouse models revealed that MC-derived IL-6 and IFNγ play a role in the progression of atherogenesis [82]. The ability of plaque-residing MCs in the production of proinflammatory cytokines was also reported in human studies too. Kaartinen et al., after studying the TNF positivity in atheromas obtained from 37 postmortem collected coronary arteries, concluded that MCs were the main cells producing TNF and were distributed differently in atheromas in which above half of the TNF-positive MCs were found in the shoulder region, 35% in the cap, and 10% were in the core regions. Visualization of TNF in MCs showed that this proinflammatory mediator is placed within the granules which enables MCs to release it upon degranulation [83] (Fig. 4a).

a Role AQ of MC-released mediators in plaque microenvironment. MC-released chymase degrades ApoA-I and prevents its binding to endothelial cells. Both neutrophils and MCs produce cathepsin G that activates elastase which in turn solubilizes the elastin and activates MMPs. Both tryptase and chymase destabilize the plaque and contribute to rupture. Additionally, MCs play a role in recruiting immune cells into plaques., i.e., chemokine KC (CXCL1) in mice acts through CXCR2 and VLA-4 and recruits the monocytes into plaques, and CXCL1 released from MCs acts through neutrophil expressed CXCR2 and recruits them. b Investigation of the plaque progression in Apoe −/− mice and Apoe −/− Kit W−sh/W−sh after a fat-enriched diet showed that Apoe −/− Kit W−sh/W−sh develop smaller lesions. c To explore whether α7 nicotinic acetylcholine receptor (and binding to nicotine) plays a role in the development of plaques, Apoe −/− Kit W−sh/W−sh mice were reconstituted with MCs isolated from Apoe −/− α7nAChR −/− mice and compared with Apoe −/− Kit W−sh/W−sh mice as controls after putting them on an atherogenic diet. No significant difference was found between the size of the plaques between the two groups showing that deficiency in α7 nicotinic acetylcholine receptor hampers the atherogenic effects of nicotine on the progression of plaques. d Comparing the effects of induction of MC activation and stabilization using C48/80 and cromolyn in an Ldlr −/− mouse model fed an atherogenic diet showed that MC activation results in larger lesions and increased levels of cholesterol, LDL and triglycerides, while application of MC stabilizer showed converse results
Application of Mouse Models in Studying the MC Involvement in Atherogenesis: Lessons from Animal Models
Nicotine is an addicting component in cigarettes and has atherogenic properties. Wang et al. provided a MC-dependent link between smoking and atherosclerosis. Nicotine possesses atherogenic properties, and exposure to nicotine may increase the size of plaques and increases the levels of proinflammatory cytokines including IL-4, IL-6, TNF-α, and IFN-γ. They compared the progression of plaques between apolipoprotein E-deficient mice (Apoe−/−) and MC-deficient Apoe−/−KitW−sh/W−sh mice after they were fed a fat-enriched diet and received nicotine for 12 weeks and showed that the latter had decreased atherosclerotic lesion size (Fig. 4b). They concluded that MCs may aggravate atherosclerosis and that nicotine not only increases the density of MC density but also activates them. Additionally, their experiment on activation of bone marrow–derived MCs by nicotine in vitro showed that the α7 nicotinic acetylcholine receptor may be the receptor through which nicotine activates MCs. To test this mechanism, they reconstituted Apoe−/−KitW−sh/W−sh mice with MCs of Apoe−/−α7nAChR−/− mice and reported no alteration in the size of plaques suggesting that MC activation by nicotine is achieved by engaging α7 nicotinic acetylcholine receptors [84] (Fig. 4c).
One important aspect of studying the role of MCs in the pathology of atherosclerosis could be monitoring the progression of the disease in the presence of MC stabilizers and secretagogues. Wang et al. monitored atherosclerosis in Ldlr−/− mice fed an atherogenic diet who received either the MC secretagogue compound 48/80 (C48/80) or the MC stabilizer cromolyn. They found that the application of C48/80 could cause histologic alterations including increasing aortic arch intima and total lesion areas and biochemistry changes such as elevated levels of total cholesterol, LDL, and triglyceride. As expected, cromolyn acted conversely and lowered the lipid deposition of thoracic-abdominal aortas and prevented the activation of MCs [85] (Fig. 4d). Sun and colleagues studied the microenvironment of atheromata in Ldlr–/–KitW−sh/W−sh mice and reported decreased lesion size, reduction in lipid deposition, and low numbers of macrophages and T cells. To elucidate the role of MC-produced mediators in the pathology of atherosclerosis, they used an adoptive transfer strategy using MCs obtained from either syngeneic WT, IL-6−/−, IFNγ−/−, or TNFα−/− mice to restore atherogenesis in Ldlr–/–KitW−sh/W−sh mice. They concluded that MC-derived IL-6 and IFNγ actively participate in the progression of atherogenesis in mice [82].
Lagraauw and colleagues reported that acute stress may have a role in the activation of MCs. Using a stress protocol, they studied the outcomes in apoE−/− mice and reported that (a) circulatory leukocytes shift in favor of neutrophils, (b) glucocorticoid levels increase, (c) perivascular MCs under stress become activated and degranulate, and (d) the levels of MC-derived mediator including β-hexosaminidase increase and correlate with the number of activated MCs [86].
Unmet Questions in MC Involvement in Atherosclerosis
In this section, we summarize a list of unmet questions with the corresponding rationale for MC involvement in the pathology of atherosclerosis that need further investigations (Table 1).
Discussion and Conclusion
Although accumulation and then further chemical modification of lipids in arteries account for the main features of atherosclerosis, the crosstalk of cells of innate and adaptive immunity and the immune responses they orchestrate contribute to the establishment and progression of atherosclerotic plaques [10]. Studying the role of tissue-resident cell types including MCs in human atherosclerosis has its difficulties. The investigation of samples from aortic plaques was largely restricted to postmortem samples obtained by autopsy. However, recently, this problem has been partially solved using carotid endarterectomy tissues to study plaques from cardiovascular patients undergoing surgery in which MCs were detected [133]. Additionally, the emergence of methods based on obtaining tissues from patients or animals, enzymatically digesting the plaques, preparation of single-cell suspension, and then studying the MCs using flow cytometry could provide further data on the role of MCs in atherosclerosis [89]. Involvement in atherosclerosis and the mechanisms through which MCs contribute to the progression and instability of the plaques are partially revealed; stimuli capable of activating these cells locally in the plaque are up to date not well-determined. Cigarette smoke and allergy are two predisposing factors of atherosclerosis, and their mechanisms of action are now partially understood. However, since cigarette smoke contains 5000–7000 chemicals [134] including nicotine [135], it is expected that more xenobiotics may possess atherogenic properties and they may engage further receptors to activate MCs. In this regard, it has been reported that cigarette smoke may activate MCs and induce histamine release which acts through endothelial-expressed H1Rs and increase the inflammation and upregulate TLR2/TLR4 and nicotinic acetylcholine receptors (NAChR) expression [136]. Besides, recent findings link allergy to atherosclerosis according to the ox-LDL potency of coactivation of both MCs and macrophages to release histamine and TNF-α respectively and contribution to the initiation and progression of plaques through endothelial activation and monocyte adhesion in atopic individuals with a high-lipid diet [137]. Investigation of the role of micro-RNAs involved in atherogenesis may elucidate new aspects of MC involvement in atherosclerosis of which miR-223 and its lower levels are associated with the progression of the plaques and worsening of the patient’s conditions [138]. miR-223 overexpression has been reported to decrease the MC production of proinflammatory cytokines mainly IL-6 [139]. Additionally, it induces autophagy in vascular SMCs which prevents their transformation to foam cells the master cells of atherosclerosis [140]. Developing ex vivo culture models may be a promising method to investigate the cytoarchitecture dynamic of plaques during different phases and study the microenvironment of the lesions with MCs therein. Two models of such ex vivo systems were developed by Lebedeva et al. and Vorobyova and colleagues by culturing circular segments of plaques on collagen rafts at the medium–air interface to supply them with oxygen that could preserve the majority of cells including fibroblasts, lymphocytes, macrophages, and SMCs [141, 142]. Human MCs release an array of proteases of which cathepsin G reacts with LDL more effectively and degrades apoB-100 (mediates the LDL uptake upon binding to LDL receptor [143]) and mediates LDL molecules fusion and increases their size from 22 to 30 nm [76]. Investigations using mouse models showed that atopic mice were more predisposed to develop atherosclerosis [4]. Promising effects of anti-allergic medication (such as MC stabilizers) in mice with atherosclerosis provided a line of evidence that MCs play a role in the pathology of the disease and our current knowledge in allergy may be useful to target MCs in the treatment of atherosclerosis [4]. Finally, we address and summarize the main features and involved molecules having a role in the MC-dependent reshaping of atherosclerotic plaques in Table 2.
Looking at the literature regarding the role of MCs in the pathology of atherosclerosis, one can easily find out that the basic and fundamental cellular findings regarding the presence of MCs in the pathology of atherosclerosis in terms of accumulation, activation, and protease content was acquired in the 1990s; however, the molecular involvement of MCs and their crosstalk with other immune and non-immune cells were revealed later. During the past decades using MC stabilizers and secretagogue agents, development of strategies to simulate the development of plaques in mouse models by putting them on fat-rich diets and later developing MC-deficient mice alone or ApoE knockdown mice or combined (ApoE−/−KitW−sh/W−sh) provided an opportunity to link genetics and nutrition for having a better understanding of MC involvement in the diseases. In recent years, the emergence of new technology to study the MCs in plaques of patients such as tissue microarrays [28] and recently the introduction of monoclonal antibodies to suppress or deplete MCs brought us to the idea that it is time to rethink the role of these cells in the pathology of atherosclerosis. The Investigation the role of MCs in pathogenesis of atherosclerosis was speeded up owing to the following elements: (1) introduction of IgE-independent MC activation pathway(s); (2) highlighting the role of nerve-MC axis through results of stress/atherosclerosis in mice and human; (3) the possibility of profiling plaque-accumulated MCs using flow cytometry rather than IHC and therefore the possibility of determining the mediator and cytokine secretion profile, expressed surface markers, and crosstalk of MCs with other cells; (4) analysis of gene expression profile using single-cell RNA sequencing (scRNA); and (5) introduction of new MC suppressor or depleting agents for human model (investigated in patients with MC-driven pathologies) to study the microenvironment of plaques with or without the role of MCs. Despite all the above-mentioned achievements, we believe there are different aspects of MCs in the initiation, development, and instability of plaques in atherosclerosis addressed in Table 1 that can be considered for further studies.
Data Availability
No data are associated with this article.
Abbreviations
- APC:
-
Antigen presenting cell
- CKD:
-
Chronic kidney disease
- CPA3:
-
Carboxypeptidase A3
- CVD:
-
Cardiovascular disease
- ECM:
-
Extracellular matrix
- GPIb:
-
Glycoprotein Ib
- GrB:
-
Granzyme B
- HFD:
-
High fat diet
- HSP-60:
-
Heat shock protein-60
- LDL:
-
Low-density lipoproteins
- LOX-1:
-
Lectin-like oxLDL receptor-1
- LPA:
-
Lysophosphatidic acid
- MC:
-
Mast Cell
- MCD:
-
MC density
- MCps:
-
Mast cell progenitors
- MI:
-
Myocardial Infarction
- mMCP-4:
-
Mouse MC protease-4
- NETs:
-
Neutrophil extracellular traps
- NF-κB:
-
Nuclear factor-κB
- NK1R:
-
Neurokinin-1 receptor
- oxLDL:
-
Oxidized LDL
- PGs:
-
Proteoglycans
- PMCs:
-
Peritoneal mast cells
- RCT:
-
Reverse cholesterol transport
- ROS:
-
Reactive oxygen species
- SCF:
-
Stem cell factor
- sIgM:
-
Secreted IgM
- scRNA:
-
Single-cell RNA sequencing
- SM:
-
Systemic mastocytosis
- SMCs:
-
Smooth muscle cells
- SP:
-
Substance P
- SRA:
-
Scavenger receptor class A
- TKIs:
-
Tyrosine kinase inhibitors
- TLRs:
-
Toll-like receptors
- VCAM-1:
-
Vascular cell adhesion molecule-1
- VWF:
-
Von Willebrand factor
- WTD:
-
Western-type diet
References
Zuiderwijk M et al (2018) Leukocyte dynamics during the evolution of human coronary atherosclerosis: conclusions from a sevenfold, chromogen-based, immunohistochemical evaluation. Am J Pathol 188(7):1524–1529
van Dijk RA et al (2015) A change in inflammatory footprint precedes plaque instability: a systematic evaluation of cellular aspects of the adaptive immune response in human atherosclerosis. J Am Heart Assoc 4(4)
Plotkin JD et al (2017) NF-κB inhibitors that prevent foam cell formation and atherosclerotic plaque accumulation. Nanomedicine 13(6):2037–2048
Liu CL et al (2016) Allergic lung inflammation promotes atherosclerosis in apolipoprotein E-deficient mice. Transl Res 171:1–16
Lambert K et al (2019) Postmortem IgE determination in coronary artery disease. J Forensic Leg Med 62:1–6
Kaartinen M, Penttilä A, Kovanen PT (1994) Accumulation of activated mast cells in the shoulder region of human coronary atheroma, the predilection site of atheromatous rupture. Circulation 90(4):1669–1678
Kritikou E et al (2019) Hypercholesterolemia induces a mast cell-CD4(+) T cell interaction in atherosclerosis. J Immunol 202(5):1531–1539
Barrett TJ (2020) Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol 40(1):20–33
Kume N et al (2000) Inducible expression of LOX-1, a novel receptor for oxidized LDL, in macrophages and vascular smooth muscle cells. Ann N Y Acad Sci 902:323–327
Kritikou E et al (2019) Disruption of a CD1d-mediated interaction between mast cells and NKT cells aggravates atherosclerosis. Atherosclerosis 280:132–139
Moore KJ, Sheedy FJ, Fisher EA (2013) Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 13(10):709–721
Saigusa R, Winkels H, Ley K (2020) T cell subsets and functions in atherosclerosis. Nat Rev Cardiol 17(7):387–401
Nakai Y et al (2004) Natural killer T cells accelerate atherogenesis in mice. Blood 104(7):2051–2059
Libby P, Hansson GK (2019) From focal lipid storage to systemic inflammation: JACC review topic of the week. J Am Coll Cardiol 74(12):1594–1607
Major AS, Fazio S, Linton MF (2002) B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol 22(11):1892–1898
Qian C et al (2021) Comprehensive analysis of dysregulated genes associated with atherosclerotic plaque destabilization. Exp Biol Med (Maywood) 246(23):2487–2494
Liu Q et al (2022) Major vault protein prevents atherosclerotic plaque destabilization by suppressing macrophage ASK1-JNK signaling. Arterioscler Thromb Vasc Biol 42(5):580–596
Li T et al (2020) The role of matrix metalloproteinase-9 in atherosclerotic plaque instability. Mediators Inflamm 2020:3872367
Constantinides P (1953) Mast cells and susceptibility to experimental atherosclerosis. Science 117(3045):505–506
Wezel A et al (2015) Mast cells mediate neutrophil recruitment during atherosclerotic plaque progression. Atherosclerosis 241(2):289–296
Wang Y et al (2001) Mast cell chymase inhibits smooth muscle cell growth and collagen expression in vitro: transforming growth factor-beta1-dependent and -independent effects. Arterioscler Thromb Vasc Biol 21(12):1928–1933
Kovanen PT, Kaartinen M, Paavonen T (1995) Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation 92(5):1084–1088
Ramalho LS et al (2013) Role of mast cell chymase and tryptase in the progression of atherosclerosis: study in 44 autopsied cases. Ann Diagn Pathol 17(1):28–31
Munteanu AI, Raica M, Zota EG (2016) Immunohistochemical study of the role of mast cells and macrophages in the process of angiogenesis in the atherosclerotic plaques in patients with metabolic syndrome. Arkh Patol 78(2):19–28
Joo SP et al (2020) Vasa vasorum densities in human carotid atherosclerosis is associated with plaque development and vulnerability. J Korean Neurosurg Soc 63(2):178–187
Mekke JM et al (2021) Mast cell distribution in human carotid atherosclerotic plaque differs significantly by histological segment. Eur J Vasc Endovasc Surg 62(5):808–815
Willems S et al (2013) Mast cells in human carotid atherosclerotic plaques are associated with intraplaque microvessel density and the occurrence of future cardiovascular events. Eur Heart J 34(48):3699–3706
Skenteris NT et al (2023) Mast cells participate in smooth muscle cell reprogramming and atherosclerotic plaque calcification. Vascul Pharmacol 150:107167
Stemme S et al (1995) T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci U S A 92(9):3893–3897
Ribatti D, Tamma R, Komi DE (2019) The morphological basis of the development of the chick embryo immune system. Exp Cell Res 381(2):323–329
Foks AC et al (2013) Interruption of the OX40-OX40 ligand pathway in LDL receptor-deficient mice causes regression of atherosclerosis. J Immunol 191(9):4573–4580
VanderLaan PA et al (2019) Invariant natural killer T-cells and total CD1d restricted cells differentially influence lipid metabolism and atherosclerosis in low density receptor deficient mice. Int J Mol Sci 20(18)
Chillo O et al (2016) Perivascular mast cells govern shear stress-induced arteriogenesis by orchestrating leukocyte function. Cell Rep 16(8):2197–2207
Kokkonen JO, Kovanen PT (1987) Stimulation of mast cells leads to cholesterol accumulation in macrophages in vitro by a mast cell granule-mediated uptake of low density lipoprotein. Proc Natl Acad Sci U S A 84(8):2287–2291
Kovanen PT (1991) Mast cell granule-mediated uptake of low density lipoproteins by macrophages: a novel carrier mechanism leading to the formation of foam cells. Ann Med 23(5):551–559
Kovanen PT (1993) The mast cell–a potential link between inflammation and cellular cholesterol deposition in atherogenesis. Eur Heart J 14(Suppl K):105–17
Ma H, Kovanen PT (1995) IgE-dependent generation of foam cells: an immune mechanism involving degranulation of sensitized mast cells with resultant uptake of LDL by macrophages. Arterioscler Thromb Vasc Biol 15(6):811–819
Bot I et al (2007) Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation 115(19):2516–2525
Hung J et al (2020) Novel plaque enriched long noncoding RNA in atherosclerotic macrophage regulation (PELATON). Arterioscler Thromb Vasc Biol 40(3):697–713
McHale C, Mohammed Z, Gomez G (2019) Human skin-derived mast cells spontaneously secrete several angiogenesis-related factors. Front Immunol 10:1445
Komi DE, Khomtchouk K, Santa Maria PL (2020) A review of the contribution of mast cells in wound healing: involved molecular and cellular mechanisms. Clin Rev Allergy Immunol 58(3):298–312
Elieh Ali Komi D, Jalili A (2021) The emerging role of mast cells in skin cancers: involved cellular and molecular mechanisms. Int J Dermatol
McHale C et al (2018) Interleukin-6 potentiates FcεRI-induced PGD(2) biosynthesis and induces VEGF from human in situ-matured skin mast cells. Biochim Biophys Acta Gen Subj 1862(5):1069–1078
Kaartinen M, Penttilä A, Kovanen PT (1996) Mast cells accompany microvessels in human coronary atheromas: implications for intimal neovascularization and hemorrhage. Atherosclerosis 123(1–2):123–131
Bot I et al (2020) Local mast cell activation promotes neovascularization. Cells 9(3)
Cook AD et al (2010) Urokinase-type plasminogen activator and arthritis progression: role in systemic disease with immune complex involvement. Arthritis Res Ther 12(2):R37
Elahirad S et al (2021) Association of matrix metalloproteinase-2 (MMP-2) and MMP-9 promoter polymorphisms, their serum levels, and activities with coronary artery calcification (CAC) in an Iranian population. Cardiovasc Toxicol
Khadijeh N et al (2018) Investigation of serum levels and activity of matrix metalloproteinases 2 and 9 (MMP2, 9) in opioid and methamphetamine-dependent patients. Acta Medica Iranica 56(9)
Shi X et al (2020) Calcification in atherosclerotic plaque vulnerability: friend or foe? Front Physiol 11:56
Jeziorska M, McCollum C, Woolley DE (1998) Calcification in atherosclerotic plaque of human carotid arteries: associations with mast cells and macrophages. J Pathol 185(1):10–17
Skenteris N-T et al (2020) The role of mast cells in atherosclerotic plaque calcification. 1:250–251
Folkerts J et al (2020) Butyrate inhibits human mast cell activation via epigenetic regulation of FcεRI-mediated signaling. Allergy 75(8):1966–1978
Elieh Ali Komi D, Shafaghat F, Christian M (2020) Crosstalk between mast cells and adipocytes in physiologic and pathologic conditions. Clin Rev Allergy Immunol 58(3):388–400
Elieh Ali Komi D, Wöhrl S, Bielory L (2020) Mast cell biology at molecular level: a comprehensive review. Clin Rev Allergy Immunol 58(3):342–365
Geng C et al (2022) Allergic asthma aggravates angiotensin II-induced cardiac remodeling in mice. Transl Res 244:88–100
Unal D et al (2017) Impact of high serum immunoglobulin E levels on the risk of atherosclerosis in humans. Asia Pac Allergy 7(2):74–81
Jiang W et al (2022) A retrospective study of immunoglobulin E as a biomarker for the diagnosis of acute ischemic stroke with carotid atherosclerotic plaques. PeerJ 10:e14235
Min KB, Min JY (2019) Risk of cardiovascular mortality in relation to increased total serum IgE levels in older adults: a population-based cohort study. Int J Environ Res Pub Health 16(22)
Wilson JM et al (2018) IgE to the mammalian oligosaccharide galactose-α-1,3-galactose is associated with increased atheroma volume and plaques with unstable characteristics-brief report. Arterioscler Thromb Vasc Biol 38(7):1665–1669
Tsiantoulas D et al (2017) Increased plasma IgE accelerate atherosclerosis in secreted IgM deficiency. Circ Res 120(1):78–84
Middendorp S et al (2003) Function of Bruton’s tyrosine kinase during B cell development is partially independent of its catalytic activity. J Immunol 171(11):5988–5996
Hemme E et al (2023) Bruton’s tyrosine kinase inhibition by acalabrutinib does not affect early or advanced atherosclerotic plaque size and morphology in Ldlr(-/-) mice. Vascul Pharmacol 150:107172
Conti L et al (2019) Kounis syndrome uncovers severe coronary disease: an unusual case of acute coronary syndrome secondary to allergic coronary vasospasm. BMJ Case Rep 12(12)
Kounis NG (2016) Kounis syndrome: an update on epidemiology, pathogenesis, diagnosis and therapeutic management. Clin Chem Lab Med 54(10):1545–1559
Poggiali E et al (2022) Kounis syndrome: from an unexpected case in the emergency room to a review of the literature. Acta Biomed 93(1):e2022002
de Vries MR et al (2013) Complement factor C5a as mast cell activator mediates vascular remodelling in vein graft disease. Cardiovasc Res 97(2):311–320
Elieh Ali Komi D, Grauwet K (2018) Role of mast cells in regulation of T cell responses in experimental and clinical settings. Clin Rev Allergy Immunol 54(3):432–445
den Dekker WK et al (2012) Mast cells induce vascular smooth muscle cell apoptosis via a toll-like receptor 4 activation pathway. Arterioscler Thromb Vasc Biol 32(8):1960–1969
Raj S et al (2023) Substance P analogs devoid of key residues fail to activate human mast cells via MRGPRX2. Front Immunol 14:1155740
Bot I et al (2010) The neuropeptide substance P mediates adventitial mast cell activation and induces intraplaque hemorrhage in advanced atherosclerosis. Circ Res 106(1):89–92
Bot M et al (2013) Lysophosphatidic acid triggers mast cell-driven atherosclerotic plaque destabilization by increasing vascular inflammation. J Lipid Res 54(5):1265–1274
Kareinen I et al (2015) Enhanced vascular permeability facilitates entry of plasma HDL and promotes macrophage-reverse cholesterol transport from skin in mice. J Lipid Res 56(2):241–253
Jia Q et al (2019) Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARγ, LXRα and ABCA1. Int J Mol Med 44(3):893–902
Boudier C et al (1991) The elastolytic activity of cathepsin G: an ex vivo study with dermal elastin. Am J Respir Cell Mol Biol 4(6):497–503
Wang J et al (2014) Cathepsin G activity lowers plasma LDL and reduces atherosclerosis. Biochim Biophys Acta 1842(11):2174–2183
Maaninka K et al (2018) Human mast cell neutral proteases generate modified LDL particles with increased proteoglycan binding. Atherosclerosis 275:390–399
Nguyen SD et al (2016) Carboxyl-terminal cleavage of apolipoprotein A-I by human mast cell chymase impairs its anti-inflammatory properties. Arterioscler Thromb Vasc Biol 36(2):274–284
Kareinen I et al (2018) Chymase released from hypoxia-activated cardiac mast cells cleaves human apoA-I at Tyr(192) and compromises its cardioprotective activity. J Lipid Res 59(6):945–957
Leskinen MJ et al (2006) Mast cell chymase induces smooth muscle cell apoptosis by disrupting NF-kappaB-mediated survival signaling. Exp Cell Res 312(8):1289–1298
Houde M et al (2016) Endothelin receptor antagonist macitentan or deletion of mouse mast cell protease 4 delays lesion development in atherosclerotic mice. Life Sci 159:71–75
Ueland JM et al (2004) The chemokine KC regulates HGF-stimulated epithelial cell morphogenesis. Am J Physiol Renal Physiol 286(3):F581–F589
Sun J et al (2007) Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med 13(6):719–724
Kaartinen M, Penttilä A, Kovanen PT (1996) Mast cells in rupture-prone areas of human coronary atheromas produce and store TNF-alpha. Circulation 94(11):2787–2792
Wang C et al (2017) Nicotine accelerates atherosclerosis in apolipoprotein E-deficient mice by activating α7 nicotinic acetylcholine receptor on mast cells. Arterioscler Thromb Vasc Biol 37(1):53–65
Wang J et al (2013) Pharmaceutical stabilization of mast cells attenuates experimental atherogenesis in low-density lipoprotein receptor-deficient mice. Atherosclerosis 229(2):304–309
Lagraauw HM et al (2019) Stress-induced mast cell activation contributes to atherosclerotic plaque destabilization. Sci Rep 9(1):2134
Morici N et al (2016) Mast cells and acute coronary syndromes: relationship between serum tryptase, clinical outcome and severity of coronary artery disease. Open Heart 3(2):e000472
Lewicki Ł et al (2019) Mast cell derived carboxypeptidase A3 is decreased among patients with advanced coronary artery disease. Cardiol J 26(6):680–686
Kritikou E et al (2019) Flow cytometry-based characterization of mast cells in human atherosclerosis. Cells 8(4)
Wachter DL et al (2018) In-situ analysis of mast cells and dendritic cells in coronary atherosclerosis in chronic kidney disease (CKD). Histol Histopathol 33(8):871–886
Rohm I et al (2016) Increased number of mast cells in atherosclerotic lesions correlates with the presence of myeloid but not plasmacytoid dendritic cells as well as pro-inflammatory T cells. Clin Lab 62(12):2293–2303
Guo Y et al (2020) Shenlian extract against myocardial injury induced by ischemia through the regulation of NF-κB/IκB signaling axis. Front Pharmacol 11:134
Esfandyari-Manesh M et al (2020) S2P peptide-conjugated PLGA-maleimide-PEG nanoparticles containing imatinib for targeting drug delivery to atherosclerotic plaques. Daru 28(1):131–138
Ashry NA, Abdelaziz RR, Suddek GM (2020) The potential effect of imatinib against hypercholesterolemia induced atherosclerosis, endothelial dysfunction and hepatic injury in rabbits. Life Sci 243:117275
Pouwer MG et al (2018) The BCR-ABL1 inhibitors imatinib and ponatinib decrease plasma cholesterol and atherosclerosis, and nilotinib and ponatinib activate coagulation in a translational mouse model. Front Cardiovasc Med 5:55
Vorkapic E et al (2016) Imatinib treatment attenuates growth and inflammation of angiotensin II induced abdominal aortic aneurysm. Atherosclerosis 249:101–109
Kupreishvili K et al (2017) Mast cells are increased in the media of coronary lesions in patients with myocardial infarction and may favor atherosclerotic plaque instability. J Cardiol 69(3):548–554
Yu X et al (2018) Propofol attenuates myocardial ischemia reperfusion injury partly through inhibition of resident cardiac mast cell activation. Int Immunopharmacol 54:267–274
Fuijkschot WW et al (2016) Orthopedic surgery increases atherosclerotic lesions and necrotic core area in ApoE-/- mice. Atherosclerosis 255:164–170
Lalmohamed A et al (2012) Timing of acute myocardial infarction in patients undergoing total hip or knee replacement: a nationwide cohort study. Arch Intern Med 172(16):1229–1235
Elieh Ali Komi D, Kuebler WM (2021) Significance of mast cell formed extracellular traps in microbial defense. Clin Rev Allergy Immunol 1–20
Pertiwi KR et al (2019) Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J Pathol 247(4):505–512
Indhirajanti S et al (2018) Systemic mastocytosis associates with cardiovascular events despite lower plasma lipid levels. Atherosclerosis 268:152–156
Bot I et al (2019) Reply to: “The “cholesterol paradox” in patients with mastocytosis”. Atherosclerosis 284:262–263
Battisha A et al (2020) Acute myocardial infarction in systemic mastocytosis: case report with literature review on the role of inflammatory process in acute coronary syndrome. Curr Cardiol Rev 16(4):333–337
Paratz ED, Khav N, Burns AT (2017) Systemic mastocytosis, kounis syndrome and coronary intervention: case report and systematic review. Heart Lung Circ 26(8):772–778
Komi DEA, Rambasek T, Wöhrl S (2018) Mastocytosis: from a molecular point of view. Clin Rev Allergy Immunol 54(3):397–411
Gonzalez Roldan N et al (2016) CD252 regulates mast cell mediated, CD1d-restricted NKT-cell activation in mice. Eur J Immunol 46(2):432–439
Tripolino C et al (2019) Acute coronary stent thrombosis: a case of type 3 Kounis syndrome. J Cardiol Cases 19(1):33–35
Priyankara WDD et al (2019) Cardiogenic shock due to Kounis syndrome following cobra bite. Case Rep Crit Care 2019:5185716
Madsen L et al (1999) Mice lacking all conventional MHC class II genes. Proc Natl Acad Sci U S A 96(18):10338–10343
Komi DE, Ribatti D (2019) Mast cell-mediated mechanistic pathways in organ transplantation. Eur J Pharmacol 857:172458
Elieh-Ali-Komi D et al (2023) Chronic urticaria and the pathogenic role of mast cells. Allergol Int
Kolkhir P et al (2022) Understanding human mast cells: lesson from therapies for allergic and non-allergic diseases. Nat Rev Immunol 22(5):294–308
Maurer M et al (2021) Biologics for the use in chronic spontaneous urticaria: when and which. J Allergy Clin Immunol Pract 9(3):1067–1078
Hans CP et al (2011) Opposing roles of PARP-1 in MMP-9 and TIMP-2 expression and mast cell degranulation in dyslipidemic dilated cardiomyopathy. Cardiovasc Pathol 20(2):e57-68
Zhu G et al (2022) Knockout and double knockout of cathepsin K and Mmp9 reveals a novel function of cathepsin K as a regulator of osteoclast gene expression and bone homeostasis. Int J Biol Sci 18(14):5522–5538
Mirabdaly S et al (2020) Effects of temozolomide on U87MG glioblastoma cell expression of CXCR4, MMP2, MMP9, VEGF, anti-proliferatory cytotoxic and apoptotic properties. Mol Biol Rep 47(2):1187–1197
Johnson JL (2017) Metalloproteinases in atherosclerosis. Eur J Pharmacol 816:93–106
Komi DEA, Redegeld FA (2020) Role of mast cells in shaping the tumor microenvironment. Clin Rev Allergy Immunol 58(3):313–325
Depuydt M et al (2022) Blockade of the BLT1-LTB4 axis does not affect mast cell migration towards advanced atherosclerotic lesions in LDLr mice. Sci Rep 12
Elieh Ali Komi D, Bjermer L (2019) Mast cell-mediated orchestration of the immune responses in human allergic asthma: current insights. Clin Rev Allergy Immunol 56(2):234–247
Bot I et al (2008) Abstract 3607: CCR3 antagonism inhibits adventitial mast cell accumulation and plaque development in ApoE deficient mice. Circulation 118(suppl_18):S_450–S_450
Xiang YK et al (2023) Chronic spontaneous urticaria: new evidences on the role of autoimmunity. Curr Opin Allergy Clin Immunol
Elieh Ali Komi D et al (2020) Mast cells and complement system: ancient interactions between components of innate immunity. Allergy 75(11):2818–2828
Martínez-López D et al (2020) Complement C5 protein as a marker of subclinical atherosclerosis. J Am Coll Cardiol 75(16):1926–1941
Nieto-Patlán A et al (2015) Recognition of Candida albicans by Dectin-1 induces mast cell activation. Immunobiology 220(9):1093–1100
Elieh Ali Komi D, Sharma L, Dela Cruz CS (2018) Chitin and its effects on inflammatory and immune responses. Clin Rev Allergy Immunol 4(2):213–223
Wang Y et al (2021) Fine particulate matter (PM2.5) promotes IgE-mediated mast cell activation through ROS/Gadd45b/JNK axis. J Dermatol Sci 102(1):47–57
Du L et al (2021) Association between Helicobacter pylori infection and carotid atherosclerosis in Chinese adults. Atheroscler Plus 44:25–30
Curtiss LK, Tobias PS (2009) Emerging role of Toll-like receptors in atherosclerosis. J Lipid Res 50(Suppl):S340-5
Kornstädt L et al (2020) Bacterial and fungal Toll-like receptor activation elicits type I IFN responses in mast cells. Front Immunol 11:607048
Depuydt MAC et al (2020) Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res 127(11):1437–1455
Kispert S et al (2017) In vivo effects of long-term cigarette smoke exposure on mammary tissue in mice. Am J Pathol 187(6):1238–1244
Kim SM et al (2018) The cigarette smoke components induced the cell proliferation and epithelial to mesenchymal transition via production of reactive oxygen species in endometrial adenocarcinoma cells. Food Chem Toxicol 121:657–665
Barua RS, Sharma M, Dileepan KN (2015) Cigarette smoke amplifies inflammatory response and atherosclerosis progression through activation of the H1R-TLR2/4-COX2 axis. Front Immunol 6:572
Chen C, Khismatullin DB (2015) Oxidized low-density lipoprotein contributes to atherogenesis via co-activation of macrophages and mast cells. PLoS ONE 10(3):e0123088
Vegter EL et al (2017) Low circulating microRNA levels in heart failure patients are associated with atherosclerotic disease and cardiovascular-related rehospitalizations. Clin Res Cardiol 106(8):598–609
Yang Q et al (2016) MicroRNA-223 affects IL-6 secretion in mast cells via the IGF1R/PI3K signaling pathway. Int J Mol Med 38(2):507–512
Wu W et al (2019) Overexpression of miR-223 inhibits foam cell formation by inducing autophagy in vascular smooth muscle cells. Am J Transl Res 11(7):4326–4336
Lebedeva A et al (2017) Ex vivo culture of human atherosclerotic plaques: a model to study immune cells in atherogenesis. Atherosclerosis 267:90–98
Vorobyova DA et al (2016) Immunological analysis of human atherosclerotic plaques in ex vivo culture system. Kardiologiia 56(11):78–85
Fernández-Higuero JA et al (2016) Structural changes induced by acidic pH in human apolipoprotein B-100. Sci Rep 6:36324
Zhi X et al (2013) Tryptase promotes atherosclerotic plaque haemorrhage in ApoE-/- mice. PLoS ONE 8(4):e60960
Lee-Rueckert M, Kovanen PT (2006) Mast cell proteases: physiological tools to study functional significance of high density lipoproteins in the initiation of reverse cholesterol transport. Atherosclerosis 189(1):8–18
Lee M, Lindstedt LK, Kovanen PT (1992) Mast cell-mediated inhibition of reverse cholesterol transport. Arterioscler Thromb 12(11):1329–1335
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
DEAK prepared the primary draft and designed and generated the figures. IB revised the clinical, pathological, and cellular sections of the manuscript. MRG revised the manuscript from mast cell biology point of view. MM revised the whole manuscript and provided input.
Corresponding author
Ethics declarations
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
No informed consent was required to prepare the manuscript.
Conflict of Interest
Marcus Maurer is or recently was a speaker and/or advisor for and/or has received research funding from Allakos, Amgen, Aquestive, Aralez, AstraZeneca, Astria, Bayer, BioCryst, Blueprint, Celldex, Celltrion, Centogene, CSL Behring, Evommune, GSK, Ipsen, KalVista, Kyowa Kirin, Leo Pharma, Lilly, Menarini, Mitsubishi Tanabe Pharma, Moxie, Noucor, Novartis, Pharvaris, Sanofi/Regeneron, Septerna, Takeda, Teva, Third Harmonic Bio, Valenza Bio, Yuhan Corporation, and Zurabio. I.B. is an established investigator of the Dutch Heart Foundation (2019T067). The other authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elieh-Ali-Komi, D., Bot, I., Rodríguez-González, M. et al. Cellular and Molecular Mechanisms of Mast Cells in Atherosclerotic Plaque Progression and Destabilization. Clinic Rev Allerg Immunol 66, 30–49 (2024). https://doi.org/10.1007/s12016-024-08981-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-024-08981-9