
Abstract
Although the incidence of acute rheumatic fever and rheumatic heart disease has decreased significantly in regions of the world where antibiotics are easily accessible, there remains a high incidence in developing nations as well as in certain regions where there is a high incidence of genetic susceptibility. These diseases are a function of poverty, low socioeconomic status, and barriers to healthcare access, and it is in the developing world that a comprehensive prevention program is most critically needed. Development of group A streptococcal vaccines has been under investigation since the 1960s and 50 years later, we still have no vaccine. Factors that contribute to this lack of success include a potential risk for developing vaccine-induced rheumatic heart disease, as well as difficulties in covering the many serological subtypes of M protein, a virulence factor found on the surface of the bacterium. Yet, development of a successful vaccine program for prevention of group A streptococcal infection still offers the best chance for eradication of rheumatic fever in the twenty-first century. Other useful approaches include continuation of primary and secondary prevention with antibiotics and implementation of health care policies that provide patients with easy access to antibiotics. Improved living conditions and better hygiene are also critical to the prevention of the spread of group A streptococcus, especially in impoverished regions of the world. The purpose of this article is to discuss current and recent developments in the diagnosis, pathogenesis, and management of rheumatic fever and rheumatic heart disease.
Similar content being viewed by others

Introduction
Rheumatic fever is a multi-organ-system disease involving chronic inflammatory changes occurring in response to a group A streptococcal infection. Rheumatic fever most commonly occurs after group A streptococcal pharyngitis. Involvement of the heart, joints, nervous system [1], skin, and immune systems is common, although all of these are not universally seen in all patients. The incidence of rheumatic fever and rheumatic heart disease has decreased significantly in most developed countries in the Western world over the past half of a century [2, 3]. This trend has been attributed to the widespread use of antibiotics in these regions to combat group A streptococcal infections, as well as other measures that decreased the spread of this infectious agent, such as better living conditions, better access to care, and better hygiene. Even the severity of the disease is believed to have changed since the pre-antibiotic era. By 1960, Bland had already written that “the clinical features of severe rheumatic fever of three and four decades ago, characterized so frequently by nodules, pericarditis, pleuritis, pneumonitis, and congestive heart failure, bear only a faint resemblance to those of its milder counterpart in the present era” [4].
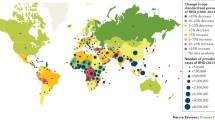
Unfortunately, this is not true in the developing world, where rheumatic fever is still one of the major killers of young people. Moreover, there has been recent indication that the disease may be making a comeback in certain areas of the developed world, with reports of such trends emerging from Italy [5] or the intermountain regions of Utah in the USA [6, 7]. There is also evidence that acute rheumatic fever occurs in higher frequency in certain ethnic or regional populations, such as in the Pacific Islanders or Australian aborigines. The reasons for this are not completely clear but may involve genetic differences in the host and differences in prevalence of specific highly virulent strains of the infectious agent. This will be discussed in detail in this paper as it is critically relevant to our ultimate quest for a vaccine for group A streptococcus, an endeavor that has been ongoing for as long as antibiotics have been available. An illustration of the patterns of worldwide prevalence of group A streptococcus infection is shown in Fig. 1. This figure is cross-referenced to Table 1, which shows prevalence or incidence of rheumatic heart disease in different countries or regions. The reliability of the data in these studies on prevalence may be limited to a variable extent by experimental design, patient selection variability, differences in the definition of rheumatic fever and inadequate statistical analysis.

Worldwide changes in the prevalence of rheumatic fever and rheumatic heart disease. Red squares, regions currently with a high incidence of rheumatic fever. Green down arrow, regions which have experienced a significant decline in rheumatic fever over the past few decades. Red dots, areas in which a resurgence of rheumatic fever has been observed. Numbers cross-referenced to Table 1
The successful development of a vaccine group A streptococcus has been considered, whether accurately or not, to be the holy grail of eradication of rheumatic fever. However, other cutting edge issues that need to be addressed include the changing global epidemiology of the disease, the effect of other preventative measures on the spread of group A streptococcus, and recent and future developments that promise improvements in the diagnosis of management of the disease.
The Epidemiology of Acute Rheumatic Fever
Rheumatic fever is a complication of group A streptococcal infections, the vast majority following pharyngitis (so-called “strep throat”). In 1956, it was estimated that up to 3% of untreated strep infections in the general population will result in acute rheumatic fever [8]. It is estimated that there are about 600 million cases of pharyngitis in the world per year. It is not known how many of these cases are untreated. A systematic overview of the literature on prospective population based studies revealed that the overall global incidence of first attack rheumatic fever is five to 51 per 100,000 people with a mean of 19 per 100,000 [9]. Estimates of the overall incidence of rheumatic fever vary, but figures range from 282,000 to 471,000 new cases per year and between 233,000 and 500,000 rheumatic fever related deaths per year globally [10, 11]. The prevalence of rheumatic heart disease is between 15 and 20 million cases globally. About two million cases require repeated hospitalization and one million cases may need a heart transplant in 5–20 years. Rheumatic heart disease is diagnosed by echocardiographic screening in 2–3% of school aged children in Mozambique and Cambodia. Echocardiography has proven to be a valuable tool in the diagnosis of rheumatic heart disease. There are approximately an additional 250,000 deaths per year from other complications of group A streptococcal infections.
The incidence of rheumatic fever varies widely throughout the globe and is a constantly changing landscape. Prior to the 1940s, when antibiotics were first discovered, group A streptococcal disease was a major cause of morbidity and mortality in all parts of the world. Now, the highest rates of rheumatic heart disease appear to be in the Indian subcontinent, other parts of Asia, the Mediterranean and Middle East, the Pacific Islands and Australia, and in Latin America and the Caribbean. Overall, 95% of cases of rheumatic fever and rheumatic heart disease occur in the developing world.
About 60% of the new cases of rheumatic fever per year develop rheumatic heart disease. The economic burden of this disease is immense. It is estimated that the cost of group A streptococcal disease in the USA alone is between $250 to $400 million, and the cost worldwide is much greater. Ongoing treatment with antibiotics, cardiac surgery, and close physician monitoring contribute to this financial burden. Other complications from group A streptococcal infections, including glomerulonephritis and neurologic sequelae raise these figures considerably. The economic impact of group A streptococcal infection is discussed in more detail below. The global burden of group A streptococcal disease is summarized in Table 2.
The USA and Western Europe
The impact of antibiotics and better living conditions, among other improvements in health care, is evident in that the incidence of rheumatic fever in the USA and Western Europe has dropped to a low of 0.5–3 cases/100,000 per year. By the early 1980s, the incidence in Denmark had dropped to 0.3 per 100,000 [12].
Between 1960 and 1964, there were 13.3 cases per 100,000 per year in the USA. Between 1968 and 1970, the rate had decreased to 10.2 per 100,000 per year, and by the 1980s, the rate had further declined to 2 per 100,000 per year [13]. Currently, the incidence is less than one per 100,000 per year in the USA. However, periodically there have been persistent pockets of resurgences of rheumatic fever in the USA, particular in Western Pennsylvania [14, 15] and in the aforementioned intermountain states [7]. Incidence rates in Hawaii have also been found to be higher than the continental USA, more closely resembling the rates seen in the Pacific Islands. The rates also did not appear to drop between the late 1960s and the late 1980s, and remained fairly constant on the island of Oahu at a rate of 12.4 per 100,000. Interestingly, Hawaiians, part Hawaiian, and Somoans had the highest rates of rheumatic fever, and Polynesian children had a markedly higher risk of developing carditis [16]. Conditions of close quartering such as those seen in military facilities have also been found to present potential risks for high rates of the disease [17, 18].
Eastern Europe
The incidence in Eastern Europe may vary and the rates are frequently higher than what one would expect based on national GDP and standard of living. For example, in Italy in 2007–2008, the rates were 23–27 per 100,000 population per year in the 5–15 year age group [19]. Polyarthritis, carditis, and chorea were the most common manifestations. Carditis was detected even in the absence of clinical cardiac manifestations, prompting the authors to advocate for an increased role of echocardiography in suspected cases. Prevalence rates in the remainder of Eastern Europe are less readily available, but in 1995, a study in Ankara, Turkey, in which schoolchildren between the ages of 6 and 17 were screened for rheumatic fever, revealed an overall prevalence of 3.7 per 1000 [20].
Asia, the Middle East, and Australia
In the Middle East, Asia, and Australia, the incidence may be still as high as 100–200 per 100,000 per year. A recent paper from Israel reported that the incidence of rheumatic fever in a population of 450,000 members was 3.2 per 100,000 a year in patients under 30 years of age [21]. There was a male predominance of 2:1. The incidence was found to be higher in rural areas, and the peak incidence of 7.5 pre 100,000 per year occurred in children between 5–14 years of age. Carditis was found to be the most common sign. The results of this study differed from previous studies in Israel in that the incidence in the more recent study was higher and also that carditis and not reactive polyarthritis was the most common manifestation. The authors postulated that the lower incidence in previous studies may reflect an underreporting of the disease, and that the higher incidence of carditis reflected a higher use of echocardiography in the latter study.
Rheumatic fever has been extensively studied in India. In 1996, 15080 children in the Shimla town area were screened for rheumatic heart disease by a trained cardiologist. The overall prevalence was found to be 2.98 per 1000 children. The rate in rural schools was significantly higher than that in urban schools (4.8 vs 1.98 per thousand), and other risk factors included overcrowding and poor housing conditions [22]. In 2003, a study in Southern India suggested that the rate of rheumatic fever and rheumatic heart disease had not decreased in the preceding 10-year period [23]. However, this was a retrospective examination of case reports and not all records were available. Carditis was the most frequent Jones Criteria found, followed by arthritis.
The higher incidence of rheumatic fever in Pacific Islanders and indigenous Australian children warrants discussion [24]. Australia currently has one of the highest incidence of acute rheumatic fever and rheumatic heart disease in the world [25, 26]. The highest rates appear to occur in the Northern Territory, where there is a high rate of indigenous people. In this area, rates in 2010 were still reported to be as high as 150 to 380 per 100,000 people in the 5–14 years age group, which is associated with the highest risk. Other risk factors associated with the high risk in this population include the remoteness of the region, high mobility, poor health care access, poverty, lack of adequate follow-up, high turnover rates of healthcare staff, and lack of knowledge of acute rheumatic fever and rheumatic heart disease among health staff, patients, and the community.
Another population with some of the highest incidences of rheumatic fever are the Pacific Islanders, where the rates can be as high as 650/100,000 per year. The risk factors for acute rheumatic fever in New Zealand between 1996 and 2005 include household overcrowding with an odd ratio of 10.7 attributed to this risk factor. Other risk factors include those children of Maori descent or Pacific Islanders with an increased risk ratio of 28.65 (95% confidence interval). Pacific Islanders from the Northern Mariana Islands have also been found to be more susceptible to development of rheumatic fever [27]. The incidence of rheumatic fever in the Federated States of Micronesia has been estimated to be 50–134 per 100,000. A primary prevention program is in effect to reduce the prevalence of group A streptococcus [28]. In Fiji, the overall annual incidence of invasive group A streptococcal infection was 13.9 per 100,000, but when stratified to consider only the indigenous Fijian population, the rate was an even higher 19.8 per 100,000 [29].
Latin America
An observational study of 3,150 randomly selected children aged 5 to 15 years in rural and urban areas of Leon, Nicaragua, identified, using Doppler echocardiographic criteria, the prevalence of rheumatic heart disease to be 48 per 1000. The rate was significantly higher in rural areas (80 compared with 35 per 1,000). The investigators also studied 489 randomly selected adults from age 20 to 35 years, and identified, again using Doppler echocardiographic criteria, a prevalence of rheumatic heart disease of 22 in 1,000.
Africa
Rheumatic fever is a serious problem in sub-Saharan Africa. In Kinsasha, the prevalence was found to be much greater in the slums than in urban areas (22.2 vs 4.0 per 1,000), perhaps reflecting the difference in access to care [30]. In Tanzania, rates of acute rheumatic fever are unknown, but are believed to be high. The country is setup for high rates of untreated group A streptococcal infection, with only 822 physicians for a population of 42 million, poor living conditions, lack of ability to test for group A streptococcus, poor education, and low prioritization of rheumatic fever all presenting barriers to care [31]. While rates appear to be uniformly high in sub-Saharan Africa, reliable data are scarce. The prevalence of rheumatic fever in Northern Africa is also quite high, with data from Egypt being the most abundant. A recent study concluded that the prevalence of rheumatic heart disease in Egyptian students was 6.2 per 1000 [32]. The authors identified similar risk factors to those described above.
Clinical Features and Diagnosis of Acute Rheumatic Fever
The diagnosis of acute rheumatic fever is a clinical one, and is inherently susceptible to the interpretation of the treating physician. Although rheumatic fever is relatively rare under 3 years of age, clinical findings in this age group tend to be less specific. To circumvent this problem, various criteria have been developed to assist in this diagnosis, the most recent being the modified Jones criteria. The modified Jones criteria define major criteria that include carditis, migratory polyarthritis, subcutaenous nodules, Sydenham's chorea and erythema marginatum, as well as minor criteria that include fever, elevated erythrocyte sedimentation rate, and arthralgias. There must also be positive evidence of recent group A streptococcal infection by culture, rapid Strep tests, or DNAse or ASO serology. While polyarthritis is the most common symptom, pancarditis is the most serious and can lead to premature death and significant morbidity. The most common chronic heart sequelae are mitral and aortic insufficiency. A summary of the modified Jones criteria is shown in Table 3.
The development of rheumatic heart disease appears to be a risk factor for recurrent episodes [33] which are possibly related to repeated strep infections. Besides cardiac disease, the most severe long-term sequelae of group A streptococcal infections are glomerulonephritis and Sydenham’s chorea (Table 4). The carditis associated with rheumatic heart disease is a pancarditis, but the valvular involvement generally represents the highest morbidity. The valvular involvement is generally manifested clinically as a heart murmur, due to mitral insufficiency and to a lesser extent, aortic insufficiency. Therefore, auscultation of the heart sounds is of critical importance. If there is myocardial damage, one might also detect tachycardia and if valvular insufficiency is severe, an S3 gallop may be present indicating heart failure.
Echocardiography is probably the most valuable tool to diagnose and monitor progression of rheumatic heart disease. Laboratory monitoring of patients with rheumatic heart disease can be done by measurement of creatine kinase MB isoenzyme to detect myocardial damage. It has been suggested that measurement of serum cardiac troponin T in children is a more sensitive and specific marker of cardiac damage because it normally does not exist in peripheral blood, and it may be useful to rule out significant pathology [34].
Sydenham’s chorea is the most famous of the neuropsychiatric conditions related to rheumatic fever. It is also called “St. Vitus dance”, based on the manic dancing that took place during the feast of St. Vitus, the patron saint of dancing. A latency period of 2–6 months may occur between infection and onset of symptoms. Clinically, the patients present with purposeless movements of all extremities, which becomes more severe over time. Late manifestations include a speech impediment and lack of coordination. Sydenham’s chorea may or may not resolve spontaneously.
It is often said that group A streptococcal infection lead to “neuropyschiatric” sequelae, because in addition to Sydenham’s chorea, many patients with acute rheumatic fever develop an emotional lability and/or obsessive–compulsive disorder.
The M serotypes that are associated with the development of glomerulonephritis are different from the rheumatogenic strains. Post-streptococcal glomerulonephritis may have a different mechanism of action in that it is generally accepted to be a type III hypersensitivity, or immune complex disease. On the other hand, autoantibodies to laminin in the glomerular basement membrane that cross-react with streptococcal M proteins have been reported. Post-streptococcal glomerulonephritis may present with hematuria or edema, and can have serious consequences, including hypertension, acute renal necrosis, and anemia. Diagnosis is made by renal biopsy. Complement levels may be decreased.
A post-reactive polyarthritis can occur as a result of group A streptococcus. This usually presents as a migratory polyarthritis. The most commonly involved joints are the medium-sized joints such as the elbows, knees, wrists, and ankles. The polyarthritis is usually self-limiting, generally resolving within a month. The most common symptom is pain at the joint, which has been described as being out of proportion with the amount of swelling.
Recurrent Rheumatic Fever
After the initial attack of acute rheumatic fever, repeated group A streptococcal infections can lead to rheumatic recurrences [35]. It was recognized early on that those patients who had rheumatic heart disease as a manifestation of their rheumatic fever developed recurrences more frequently than did those with no cardiac involvement [33]. Prevention of recurrent rheumatic fever is currently by antibiotic prophylaxis, and this will be discussed in detail later in the paper.
The Clinical Diagnosis of Group A Streptococcal Pharyngitis and the Accuracy of Rapid Strep Detection Tests
While certain clinical signs and symptoms may be more characteristic of a group A streptococcal rather than a viral pharyngitis, it is impossible to make a definitive diagnosis without laboratory testing. Typical features of viral and group A streptococcal infections are shown in Table 5, but it is important to mention that there is considerable variation in clinical presentation and that even in cases with the most typical presentation (fever, beefy red pharynx, abdominal pain, headache, and scarlatiniform rash), and in the best clinical diagnosticians hands, the accuracy of predicting which patient has group A streptococcus is less than 80% [36]. Since prompt treatment of group A streptococcal pharyngitis with antibiotics plays an important role in prevention of rheumatic fever, it is important that infectious agent be identified early in order to initiate treatment. The gold standard for the diagnosis of group A streptococcal pharyngitis remains a throat culture. However, throat cultures take time to complete (up to 48 h). Rapid antigen detection tests have been introduced that will give a result within 5 min, are cheap and can be performed in an office setting, thus allowing for more accurate diagnosis. The earliest rapid strep tests used a latex agglutination technique which was relatively insensitive and prone to errors. Now, there are many commercially available rapid tests. Studies of these tests, which now utilize various techniques including optical immunoassay and chemiluminescent DNA probes, lead to inconsistent results, with sensitivities ranging from 70–90%, even as recently as 2006 [37–39].
More recent rapid diagnostic tests show improved sensitivity and specificity. A study of 228 patients by Rogo et al. in 2010 compared the accuracy of three commercially available tests and found sensitivity and specificity for all three tests to be over 95% [40]. While the American Academy of Pediatrics, in their most recent edition of the Red Book, still recommends that offices who perform rapid strep tests back up their results by culture, this study suggests that rapid strep tests may be a sufficiently accurate test to use alone in the diagnosis of group A streptococcal pharyngitis. However, if clinical findings are highly suggestive of group A streptococcal infection, and the rapid strep test is negative, it is reasonable to obtain a back up culture. The pros and cons of rapid strep testing are shown in Table 6 [41]. It is important to realize that neither a throat culture nor a rapid antigen detection test can distinguish between an acute infection and a carrier state.
Pathogenesis of Acute Rheumatic Fever
Streptococcus refers to a genus of Gram-positive bacteria. The scientific classification is as follows: kingdom (Bacteria), phylum (Firmicutes), class (Bacilli), order (Lactobacillales), family (Streptococcaceae), and genus (Streptococcus). Streptococci are spherical, grow in groups of chains or pairs, are oxidase and catalase negative, and may be facultative anaerobes. The name “Streptos” means easily bent or twisted, i.e., chainlike, in Greek. Streptococcus can be associated with many infectious diseases, including pyoderma, pneumonia, meningitis, otitis media, sinusitis, osteomyelitis, vaginitis, septic arthritis, pharyngitis, endocarditis, erysipelas, and necrotizing fasciitis.
The labeling of Streptococcus as group A, B, C, D, or G is based on the Lancefield classification system of serotyping of a carbohydrate moiety, an N-acetyl glucosamine linked to a rhamnose polymer backbone, on the surface of the organism. Group A strep are β-hemolytic, based on their ability to completely rupture red blood cells when applied to a blood agar plate. Alpha hemolytic streptococcus exhibit partial hemolysis and gamma hemolytic streptococcus demonstrate no hemolysis on blood agar.
M Proteins
Group A streptococcus are further sub-typed according to a cell surface protein known as the M protein. M proteins are thought to be highly antigenic and play a pivotal role in the pathogenesis of diseases related to group A streptococcus. There are over 180 M serotypes, previously defined by conventional serologic procedures, but more recently by M protein gene (emm) typing. Group A streptococcus can cause infections at various sites in the human body, the most well-recognized being pharyngitis and impetigo, although other respiratory infections can occur as well. The M subtype generally influences the type of infection that occurs, which in turn determines the type of sequelae, namely acute rheumatic fever or post-streptococcal glomerulonephritis. While it is pharyngitis that is usually associated with acute rheumatic fever and rheumatic heart disease, pyoderma can be the predominant trigger for development of rheumatic fever in some Aboriginal communities in which pharyngitis is rare [42].
The M protein is believed to be directly involved in the pathogenesis of infection, with different M serotypes conferring different degrees of virulence. Other structures that determine virulence include the outer hyaluronic acid capsule which, like the M protein, imparts antiphagocytic properties to the bacteria. Both the M protein and the hyaluronic acid capsule are attached to the cell wall and cell membrane of the bacteria. The M protein binds to complement regulatory protein factor H and fibrinogen to exert its antiphagocytic properties, but the binding to fibrinogen and kininogen also activates the complement/contact pathways leading to the release of bradykinin and the increase in vascular permeability seen in severe or invasive Streptococcal infections such as necrotizing fasciitis, sepsis, or toxic shock syndrome. A list of selected virulence factors in group A streptococcal infection is shown in Table 7.
For colonization or disease to occur, the streptococcus must be able to adhere to host tissues, otherwise, they will be removed by the normal mechanical and immunological clearance mechanisms of our immune systems. Adhesins on the surface of the bacterial interact with tissue specific ligands, and multiple adhesins can enhance virulence. The process of adhering of bacteria to pharyngeal mucosa or dermal epithelium triggers an inflammatory cascade that can lead to cytokine production and inflammatory cell chemotaxis. One of the functions of the M protein is to act as an adhesion, but there are many other Streptococcal proteins that serve the same function.
A C5a peptidase on the surface of the streptococcus can also contribute to its antiphagocytic activity by cleavage of C5a on its polymorphonuclear leukocyte binding site. By adulthood, most people will have developed antibodies to C5a, in contrast to only 15% of children less than 10 years of age.
It has been demonstrated that antibodies directed against the M protein on the surface of group A streptococcus can opsonize and lead to phagocytosis of the organism. The antibody is directed against the hypervariable region of the M protein and is type specific. Immunity is generally long lasting, and has been shown to persist for up to 32 years in many individuals, though in some people, no protective immunity develops. It has also been suggested that antibodies against the more highly conserved C-region of the M protein play a more significant role in passive immunity. For this reason, there have been a number of different approaches for the development of a vaccine (see below).
Molecular Mimicry in the Pathogenesis of Acute Rheumatic Fever
Antibodies directed against M proteins have been found to cross-react with various self proteins, and this is believed to be the primary mechanism for the development of acute rheumatic fever [43]. In the case of rheumatic heart disease, the self proteins include those found in cardiac myocardium, cardiac connective tissue, valvular tissue, and the smooth muscle cells of arteries. Specific proteins to which autoantibodies are found include fragments of the myosin heavy chain in Indian patients and creatine kinase and dihydrolipoamide dehydrogenase in a Russian population. It has also been reported that the rheumatogenic epitopes reside in the S2 region of human cardiac myosin, and that this is fairly consistent worldwide, irrespective of the emm serotype [44].
The process of molecular mimicry begins during a group A streptococcal infection, and involves the presentation of antigen in the context of major histocompatibility complex (MHC) by antigen presenting cells to CD4 T cells. Helper T cells will then activate B cells to stimulate the production of antibodies against streptococcal cell wall. However, these antibodies may also recognize self-molecules in the joints, myocardium, and other tissues, leading to inflammation, production of inflammatory cytokines, stimulation of the complement pathway, recruitment of neutrophils and macrophages, which ultimately result in the clinical manifestations seen in rheumatic fever.
The inflammatory response in rheumatic fever is a complex cascade of events that can simultaneously occur in multiple tissues. In the heart, autoantibodies to myosin or perivascular connective tissue have been demonstrated. Aschoff bodies are granulomatous nodules that can be seen on light microscopy that are characteristic of rheumatic heart disease. They are composed of fibrinous material, lymphocytes, macrophages, and plasma cells surrounding a necrotic center and represent connective tissue inflammation in the heart. The macrophages can coalescence to form giant cells or “catapillar” cells.
Aschoff bodies can be present in acute rheumatic fever in all parts of the heart, including the myocardium, endocardium, and valvular tissue, thus causing a pancarditis. A pericardial exudate can form, leading to pericarditis. Involvement of the endocardium is more serious, and the fibrinous necrosis and tissue damage that ensues can affect closure of the heart valves, a significant contributor to mortality and morbidity of the disease. In chronic rheumatic heart disease, a cycle of recurrent inflammation and fibrinous resolution leads to leaflet thickening, shortening of the tendinous cords and commissural fusion that accompanies significant valvular dysfunction and eventual heart failure and death.
Tontsch et al. used two-dimensional immunoblot analysis to identify target antigens of autoantibodies in the sera of 56 patients with rheumatic heart disease. The target antigens included creatine kinase and two mitochondrial proteins, and to a lesser extent various stress proteins, as cardiac antigens that cross-react with streptococcal-induced antibodies [45].
The Global Distribution of M Subtypes and Impact on Morbidity
There are currently over 180 emm types identified. The number is continually increasing. Smaller alterations in emm gene sequences may change virulence patterns and these isolates are further subdivided into emm subtypes (e.g., emm68.1 differs from an emm68 reference strain by a 7-codon deletion within the coding region for the mature M protein) [46]. M proteins can be divided into two classes, I and II, depending on their reactivity with antibodies against the highly conserved C-repeat portion of the M protein. Class I proteins generally do not cross-react with class II proteins. The class I proteins are opacity factor negative and the class II proteins are opacity factor positive. Most throat infections are caused by class I M protein group A streptococcal serotypes.
The predominant isolates of emm types in the USA and Canada were studied over a period of 7 years starting in 2000 and reported in 2009. The six most common isolates in 7,040 US samples were emm types 1, 12, 28, 4, 3, and 2. In Canada, the most common emm types in 1,434 isolates were 12, 1, 28, 4, 3, and 2 [47].
The parallel question regarding emm type distribution is that of rheumatogenicity. How does the global variation in emm type distribution correlate with disease incidence? Are there certain emm types that are more rheumatogenic [48]? An analysis of eight outbreaks of rheumatic resurgence in the USA between 1980 and 1990 showed that the most common emm subtypes during these outbreaks were M types 1, 3, 5, 6, and 18. Other studies have shown that M serotypes 2, 49, 35, 40, 60, and 61 were associated with pyoderma and acute glomerulonephritis, while M types 1, 3, 5, 6, 14, 18, 19, and 24 were associated with pharyngitis and rheumatic fever [49–51]. Certain chromosomal patterns of emm genes have been associated with skin and throat infections. The global variability of emm type prevalence adds another consideration for the development of vaccines, as the optimal vaccine would be one that covers most of the common emm types worldwide [52]. Table 8 lists emm type distributions in selected countries or regions, as well as emm type with high rheumatogenicity.
It has been recently shown that horizontal gene transfers can occur from group A streptococcus to Streptococcus dysgalactiae, or group G streptococcus [53]. Group G streptococcus is a normal pathogen colonizing the throat, skin, vagina, and gastrointestinal tract of humans. The impact of this genetic transfer on the virulence of a previously known colonizing bacteria is unknown, as is the clinical significance of this observation. The authors of this study, however, suggest that the demonstration of lateral transfer of genetic material between streptococcal species may affect the epidemiology of group A streptococcus in certain endemic areas, such as Northern Australia, and may impact the way we approach vaccine development for group A streptococcus.
The Role of IgA in Group A Streptococcal Infections
Mucosal immunity may play a role in the defense of group A streptococcal infections. Serum IgA levels are frequently low in children, not reaching adult levels until adolescence. Mucosal IgA levels in saliva or in the gut may reach adult levels by 5 years of age. Group A streptococcal infections are much less frequent in adults than children. Bessen and Fischetti demonstrated that anti-M protein IgA can protect against group A streptococcal infections and prevent severe illness including delayed disseminated infection and death [54]. IgA may work by preventing bacteria from adhering to mucosal surfaces and by facilitating clearance of bacteria in conjunction with mucous production and engulfment of bacteria. This may contribute to the human host defense against group A streptococcal infections.
Genetics of Acute Rheumatic Fever and Rheumatic Heart Disease—HLA and the Environment
The observation that some ethnic groups appear to be more susceptible to develop rheumatic fever, even after correction of other factors such as poor living conditions or access to care, indicates that genetics may play a role in the pathogenesis. Genetic risk factors include polymorphisms of TNF-α and mannose-binding lectin [55], as well as several alleles in the class II MHC. The most common rheumatic fever associated HLA allele is DR7 in Turkish, Egyptian, Brazilian, and Latvian patients with rheumatic heart disease, followed by HLA-DR4 in American Caucasian and Saudi Arabian patients. Some DQ alleles, when associated with HLA-DR7 may predispose to the development of multiple valvular lesions. Genetic risk factors for group A streptococcal infection are shown in Table 9.
Independent genetic risk factors for rheumatic heart disease in patients from Turkey, Brazil, and Mexico include a polymorphism in the promoter region of the TNF-α gene at the TNFA-308G/A allele. A polymorphism in the TNFA-238G/A allele has also been found to be associated with rheumatic heart disease [56, 57]. Although the function of TNF-α as a proinflammatory cytokine compels one to believe that any polymorphism in the gene for TNF-α plays a mechanistic role in the pathogenesis of rheumatic fever, it should be noted that the gene for TNF-α is in close proximity to the MHC and the two areas may be in linkage disequilibrium. Evidence for a direct role of TNF-α in the pathogenesis of rheumatic heart disease includes the observation that mononuclear cells in the myocardium and valves of patients with rheumatic heart disease predominantly secrete interferon-γ, TNF-α, and IL-10 but very low levels of IL-4 producing cells are found. Elevated levels of human soluble tumor necrosis factor receptor I and intereleukin-1 receptor antagonist (IL-1Ra) have also been detected in the serum of patients during the acute phase of rheumatic fever, when compared with age-matched health controls [58]. The authors postulate that this increase reflects an activation of the cellular immune response and may be related to coexisting increases in proinflammatory cytokines such as TNF or IL-1.
Other genetic factors relevant to rheumatic fever and rheumatic heart disease include single nucleotide polymorphisms in the promoter region of IL-10, IL-6, and higher variable number of tandem repeats in the IL-1Ra gene. A study was conducted where 50 children with rheumatic heart disease with a mean age of 12.2 years were compared with 98 healthy unrelated controls. Higher risk genotypes for rheumatic heart disease included TNF-α-308 A/A, IL-10-1082 A/A (odds ratio, 37.4), TNF-α-308 A/A, IL-10-1082 G/G (odds ratio, 31.6), TNF-α-308 A/A, IL-1Ra A1/A1 (odds ratio, 7.23), and IL-10-1082 A/A, IL-1Ra A1/A1 (odds ratio 7.2). Genotypes that conferred low risk for the development of rheumatic heart disease included TNF-α-308 G/A, IL-10-1082 G/A (odds ratio, 0.08), TNF-α-308 G/A, IL1Ra A1/A2 (odds ratio, 0.18), IL-10-1082 G/A, IL2Ra A1/A2 (odds ratio, 0.19), and TNF-α-308 G/G, IL-10-1082 G/A (odds ratio, 0.23) [59]. Interestingly, while there appears to be a significant genetic component, the right environmental conditions might be required to create the gene-environment interaction necessary for the development of acute rheumatic fever, as it has been observed that twins do not usually both get rheumatic fever.
Role of T Cells in Rheumatic Heart Disease—T Cell Mimicry
The chronic inflammatory response in rheumatic heart disease involves T cell infiltration into the surface endothelium of the valve [60]. Immunochemical staining of the valve shows the presence of CD4+ or CD8+ cells. There is significant homology between cardiac myosin and strep M proteins. CD4+ cells infiltrating heart tissue are specific for streptococcal antigens and cross-reacting heart tissue, and are thought to be important mediators of heart valve damage [61]. The lack of IL-4 production by valvular infiltrating CD4+ cells is thought to result in a lack of regulation of the inflammatory process within the valve, and has been used to explain why the valvular tissue is targeted in many cases of rheumatic fever.
Anti-strep antibodies directed against the valvular epithelium lead to inflammation and upregulation of adhesion molecules. This leads to lymphocyte infiltration. A monoclonal antibody mAB3.B6 recognizes cardiac myosin and laminin in the valve. Laminin is homologous with Streptococcal M proteins and cardiac myosins. It is a 900kD protein molecule consisting of three chains, A, B1, and B2. The laminin sequence HTQNT is shared with cardiac myosin, and HTQNT has also been found to have homology with light meromyosin peptides. If there is a common sequence in various proteins of different regions of the heart, including valvular endothelium, valvular matrix, cardiac myosin, heavy and light meromyosin, this could explain the inflammatory pancarditis seen in rheumatic heart disease [62].
Peripheral blood mononuclear cells (PBMC) from patients with rheumatic heart disease were stimulated in vitro with opsonized group A streptococcus from patients with acute rheumatic fever and compared with PBMCs from controls. The proliferative responses to a 50- and a 54-kDa myocardial protein after stimulation increased in PBMCs from the patient group, but not the control group. It is suggested that these two myocardial proteins contain a putative antigen that is recognized by T cells from rheumatic heart disease patients. Exposure to group A streptococcus enhances the ability of the T cells to respond to this antigen [63]. Human T cell clones specific for M protein were also demonstrated to cross-react with human cardiac myosin and laminin [64].
This two-step hypothesis has been described and attempts to explain the sequence of events that lead to pathogenesis of rheumatic heart disease [65]. Firstly, antibodies are produced during the acute group A streptococcal disease against the bacterial carbohydrate antigen, N-acetyl-glucosamine, laminin and the M protein. The cross-reactive anti-group A carbohydrate antibodies initiate the disease by reacting with cardiac myosin. These antibodies also are directed against the valvular epithelium [62]. Activation of the valvular endothelium produces an inflammatory cascade beginning with the activation of vascular cell adhesion molecule-1. This allows M protein specific T cells to gain entry into the tissue, leading to the formation of Aschoff bodies consisting of macrophages and T cells beneath the endothelium. The T cells are directed against shared epitopes of cardiac myosin and M protein. This is a Th1 response that leads to scarring of the valve. Scarred tissue then becomes neovascularized, allowing disease progression, which ultimately leads to irreversible deformation of the valve, chrodae tendinae, and eventual malfunction of the heart [66].
The Role of Co-infection in the Development of Rheumatic Fever
It has been suggested that rheumatic fever does not result from group A streptococcus alone, but is potentially a result of a co-pathogen, such as a virus. Another suggestion was that a co-pathogen contributed synergistically to the pathogenesis along with group A streptococcus. This has not been substantiated. Olgunturk studied the possibility of hepatitis B, C, rubella, herpes simplex virus one, and Epstein–Barr virus (EBV) as a possible co-pathogen involved in the development of rheumatic fever and found that only EBV DNA analysis was positive in all acute rheumatic fever patients. The authors concluded that there was no evidence to support the synergism theory for the pathogenesis of acute rheumatic fever [67].
The Pathogenesis of Sydenham’s Chorea
Sydenham’s chorea results from cross-reactivity between the surface of neuronal cells and group A carbohydrate epitopes N-acetyl-beta-d-flucosamine and lysoganglioside. An antibody designated as mAb24 has greatest avidity for ganglioside, and induces increased calcium calmodulin dependent (CaM) protein kinase II levels in a neuroblastoma cell line [68]. The effect of this antibody can be elicited in Lewis rats after direct injection via the intrathecal route, suggesting that the antibody can cross the blood–brain barrier and induce antibody mediated cell signaling leading to the clinical signs of Sydenham’s chorea. In vitro, the antibodies can induce tritiated dopamine release. The movement disorder of Sydenham’s can be induced by dopamine release in the caudate putamen area of the brain. Elevated serum anti-CaM kinase II antibodies are not found during the convalescent phase of the disease. Other neuronal protein targets of cross-reacting antibodies in Sydenham’s chorea include tubulin [69] and a number of basal ganglia antigens of unknown function [70]. Interestingly, that antibodies found in Sydenham’s chorea do not show the same pattern as that seen in pediatric autoimmune neuropsychiatric disorders associated with group A streptococcus (PANDAS) [71]. In fact, in one study there was no difference in anti-basal ganglia antibodies in 15 children with a diagnosis of PANDAS when compared with 15 healthy controls [72]. An upregulation of Th2 cytokines in the serum and cerebrospinal fluid has been reported in patients with Sydenham’s chorea [73].
It is now clear that the mechanism of rheumatic fever is a complex cascade of inter-related factors and processes, including molecular mimicry, involvement of T cells, monocytes [74] and phagocytic cells, leading to the production of inflammatory cytokines and damage to heart and brain tissue. The pathogenesis of Sydenham’s chorea or PANDAS involves a different antibody pattern than that of carditis, and potentially a different pathogenic mechanism as well. Post-streptococcal glomerulonephritis may also be a severe sequelae of group A streptococcal pharyngitis, but the pathogenesis may yet have different facets; in this case, the involvement of immune complexes. A schematic of the possible pathogenic mechanisms involved in the autoimmune effects of group A streptococcal infection is shown in Fig. 2.

Pathogenesis of acute rheumatic fever and rheumatic heart disease
Global Strategies to Eliminate Rheumatic Fever in the Twenty-First Century
Development of a Vaccine for Group A Streptococcus
The development of a vaccine to prevent group A streptococcus infections has for the most part targeted the M protein. Early studies in the 1960s with a type 3 M protein vaccine was unfortunately unsuccessful, as there was a higher incidence of acute rheumatic fever in those vaccinated than was typically observed in the general population. The numbers in this study were small. However, one of the biggest concerns with vaccine development for group A streptococcus is that there is always the risk that coverage of certain M proteins may itself lead to the development of autoantibodies that cross-react with heart tissue. On the other hand, leaving out many of the M proteins associated with virulence limits the effectiveness of the vaccine. Investigators trying to develop the ideal vaccine have struggled with this dilemma for decades while trying to find the optimal balance between effectiveness and safety.
Many of the early vaccine trials have focused on developing antibodies to the hypervariable N-terminal region of the M protein because these antibodies have been found to be able to opsonize and ultimately lead to phagocytosis of the organism. But these antibodies are generally type specific, and since there are over 180 M subtypes identified so far, one of the difficulties in developing an effective vaccination program is finding a solution to ensure widespread coverage of multiple M protein strains. A 26-valent vaccine directed against the M protein serotypes commonly found in pharyngitis, invasive GAS disease, and rheumatogenic GAS disease was tested in healthy adult volunteers, and no induction of cross-reacting antibodies directed against self tissues was detected [75].
An alternative is to direct the antibodies against the more highly conserved C-terminal region of the M protein in order to increase the likelihood of broader coverage. However, the highly conserved C-repeat section of the M protein molecule possesses a coiled coil dimeric structure that cross-reacts with a mammalian tropomyosin and parts of the myosin molecule which also has a similar coiled coil protein structure. Prospective vaccines developed that react with highly conserved C regions of a recombinant M6 protein displayed low level cross-reactivity with the denatured form of at least one of the following mammalian proteins: laminin, light meromyosin, heavy meromyosin, myosin, and cardiac tropomyosin [76].
Pandey in Australia has recently reported on the development of a vaccine that targets the B cell epitope on the conserved region of the M protein called J8 [77]. J8 is itself non-immunogenic, so it was haptenized with diphtheria toxoid (DT). This conjugated vaccine, along with alum as an adjuvant, was found to be protective in mice. High J8 titers correlated with survival rates. It was demonstrated that passive transfer of anti-J8 antibodies afforded protection but was not long lasting. On the other hand, evidence of an active protective immunity was presented which showed that depletion of CD4+ cells but not CD8+ cells resulted in a failure to sustain long lasting immunity in mice immunized with the J8-DT vaccine.
Vaccines can also be developed to target other cell surface molecules that may play a role in the virulence of group A streptococcus. Carbohydrate moieties conjugated to tetanus toxoid have been studied in mice and have led to lower death rates. Passive transfer of anti-carbohydrate antibodies provides passive protection. “Naturally occurring” levels of anti-carbohydrate antibodies increases with age [78, 79], and a study of health Mexican children demonstrated that titers of group A streptococcal carbohydrate antibodies correlates with group A streptococcal colonization. Studies on cross-reactivity of the antibodies with human heart tissue have been inconsistent, but the more recent studies failed to demonstrate any cross-reactivity, leading investigators to consider group A streptococcal carbohydrates, which are highly conserved and circumvent the problem of type-specificity, as a viable candidate for vaccine research.
Adhesins (Sfb1, LTA, and FBP54))
Adhesins are an important part of the pathogenesis of rheumatic fever and are involved in attachment of group A streptococcus to host tissues. Adhesions are often the first step leading to the inflammatory process which involves release of cytokines and amplification of the inflammatory response. Sfb1 or fibronectin binding protein 1 is a streptococcal adhesion that has been a target of vaccine research [80]. In mice, intranasal immunization using Sfb1 by itself or coupled with cholera toxin B as an adjuvant led to the production of anti-Sfb1 IgG and IgA. Following immunization, mice were protected from almost certain mortality when challenged with group A streptococcus compared with mice that were not immunized. Sfb1 is only present in about seventy percent of group A streptococcal strains. Strains that express Sfb1 have an increased adherence to human epithelial cells, and may be associated with a higher rate of upper respiratory infections [81].
Another adhesion, lipoteichoic acid, has also been under investigation as a vaccine candidate. Immunization of mice led to increased serum IgG and pharyngeal IgA to lipoteichoic acid, and transfer of pharyngeal washings from immunized mice led to passive protection from adherence of group A streptococcus [82]. Other promising adhesion candidates include fibronectin binding protein 54 (FBP54), which is not present in all group A streptococcal strains. Immunization with FPB54 improves survival in mice who are subsequently infected by group A streptococcus [83].
C5a Peptidase
C5a peptidase from streptococcus is a large surface protein of about 130 kDa molecular weight. It is a serine peptidase and is also an important factor defining virulence in groups A, B, C, and G streptococci. This molecule functions as a cleaving agent of chemokine C5a at its lymphocyte binding site, inhibiting recruitment of phagocytes, and facilitates nasopharygeal lymphoreticular tissue colonization by streptococci. While no human vaccine studies have been done with this antigen, animal studies have shown that intranasal vaccination with C5a peptidase induces opsonizing antibody production, improves streptococcal clearance from oral mucosa [84] and protects against multiple M types [85]. Adults tend to have higher natural titers of Ig A and IgG to C5a peptidase in their saliva than do children [86]. Because this protein is common to multiple groups of streptococci, a vaccine based on this molecule could potentially protect against multiple types of infections that result from the various streptococcal species.
Cysteine Proteases and Pyogenic Exotoxins
The pyogenic exotoxin, streptococcal pyogenic exotoxin B (SPEB), is a cysteine protease that is expressed in most strains. Deletion mutations of the SPEB gene have been associated with less pathogenicity than the wild type strain. Because of its potential role as a virulence factor, SPEB has been the candidate for vaccine development. Both active and passive immunization using SPEB as an immunogen resulted in increased survival time after infection in a mouse model of group A streptococcal infection [87]. A peptide derived from streptolysin S toxin has also been used to produce antibodies in rabbits against the toxin, that act by enhancing phagocytosis in synergy with anti-M protein antibodies [88].
Streptococcal Pili
The streptococcal pili are surface appendages anchored to the cell wall that facilitate epithelial cell adherence, including keratinocytes, pharyngeal cells and skin. Because there is little variation in pili between group A streptococcus serotypes, this has been approached as a potential target for a vaccine. In fact, streptocococcal pili vaccines have resulted in the induction of an immune response in animal models.
Whole Cell Vaccines
Attempts to develop a whole cell vaccine have been studied in a limited fashion. The concern would be persistent cross-reactivity with cardiac antigens and high rates of serious complications. One strategy would be to eliminate certain M proteins from the surface of the streptococcus. Several M-negative mutant strains have been constructed and used in animal trials. There was significant protection from subsequent challenge by the wild type bacteria [89, 90]. Early attempts at a vaccine targeted heat-killed group A streptococcus or partially purified group A streptococcus [91]. A selection of potential vaccine candidates in the treatment of group A streptococcal infection is shown in Table 10.
Novel Vaccine Development Strategies in the Pursuit of a Group A Streptococcal Vaccine
Peptide Vaccines
Use of peptides from the variable terminal of the M protein would make sense if one wanted to customize vaccines for a specific geographic location, but because the M proteins are so diverse, this would create a logistical nightmare because of the large variety of vaccines that would need to be produced. Specific peptides portions derived from the conserved regions of M proteins have been studied as vaccine candidates. Peptide 145 was not a successful candidate because T cells specific for p145, a minimally conserved epitope from C terminus of the M protein, react with human heart tissue. Antibodies to p145 opsonize multiple GAS strains and T cells specific for p145 occur naturally in humans without evidence of cardiac disease, but it is possible that p145 can induce an immune response when administered as a vaccine, and can potentially lead to unwanted cardiac side effects. Other chimeric peptides, J2, J7, J8, and J14 have also been studied as potential vaccine candidates, as they react with human sera from endemic group A streptococcal populations [92]. Assembly of these peptides into polymeric structures as a novel method of conjugation has been promising [93], but problems with the methods of synthesis and batch reproducibility have hindered ongoing research into this methodology [94].
Lipid Core Peptide Technology
Lipid core technology has been utilized in construction of a J8 conjugate vaccine. Lipid core peptide vaccines utilize a lipid moiety as an adjuvant to increase peptide immunogenicity [95]. An alum conjugated J8 vaccine has been shown to induce antibody production and confer protection in a mouse model of group A streptococcus [96].
DNA Vaccines
DNA vaccines have shown promise in the development of vaccines for a variety of bacterial infections [97]. DNA is often used as an adjuvant in order to induce innate immune responses. For example, using unmethylated CpG conjugated with the vaccine candidate can bind to Toll-like receptor 9 and generate specific innate and acquired immune responses to the target protein. However, a DNA vaccine for Streptococcus has not been synthesized at this time.
Nanoparticle Vaccines
There is an increasing role of nanoparticles in the development of new vaccine technology. Nanoscale synthetic polymers as carriers do not induce self-recognizing antibodies, unlike other carriers, such as keyhole-limpet hemocyanin, which can lead to antibodies against the carriers [98]. An intranasal vaccine based on this delivery system involved conjugation of a peptide derived from the C-terminal highly conserved region of the M protein, and the resultant polyacrylate dendritic polymer induced IgG antibodies against the peptide, and in vitro opsonization of group A streptococcus [98].
Mucosal Delivery
The argument behind the development of mucosal vaccination stems from the pathologic mechanism for certain infections, in which the infectious agent gains entry by attachment to mucosal surfaces, as in the case of group A streptococcus. Early blockage of group A streptococcal colonization would be the optimal method to prevent infection. The role of IgA has also been considered in the development of mucosal vaccines to group A streptococcus. Many of the experiments on group A streptococcal vaccine development have been done using intranasal administration of vaccine to mice [99]. Results have shown that induction of specific IgA production in these mice is possible, along with IgG production in serum. Some of these studies have already been discussed above.
Prevention of Group A Streptococcal Infections and their Sequelae with Antibiotics
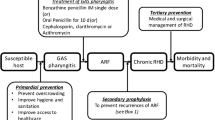
Primary Prevention
At the present time, prevention of an initial attack of rheumatic fever after a group A streptococcal infection (primary prevention) and prevention of recurrences (secondary prevention) are accomplished with the use of antibiotics. The American Heart Association recommendations for antibiotic selection and dosaging are shown in Fig. 3 [100]. In this scheme, the recommendation is to use penicillin V or amoxicillin orally as a first choice, followed by benzathine penicillin G [101]. In penicillin allergic individuals, the choices are azithromycin, clindamycin, a cephalosporin, and clarithromycin. Note that erythromycin is no longer recommended in most protocols. There may be some variation between recommendations from various parts of the world, based on the health care delivery systems of different regions. For example, in India, where there is a higher risk of rheumatic fever than in Western countries, the recommendation is to use benzathine penicillin G as a first choice, followed by oral penicillin V, then azithromycin and then cephalexin. This recommendation is likely made as a result of concerns related to compliance or loss of follow-up [102].

Treatment algorithm and antibiotic dosing for group A streptococcal pharyngitis [100]
The use of Amoxicillin in primary prevention of rheumatic fever was compared with penicillin V in 152 patients between 4–18 years of age. There was no significant difference in clinical or bacteriological response between amoxicillin 750 mg by mouth once daily for 10 days and penicillin V 250 mg by mouth three times a day for 10 days [103]. There was also no difference in clinical or bacteriological response between Amoxicillin 50 mg kg−1 day−1 once daily up to 750 mg/day for 10 days and pencicillin V 250 mg by mouth three times a day in 157 patients over 3 years of age [104]. Once daily amoxicillin was not found to be inferior to twice daily amoxicillin with regard to bacteriologic failure rate or adverse effects [105]. Similar findings were reported when comparing once daily amoxicillin with twice daily penicillin V [106].
A meta-analysis done on the use of antibiotics in the primary prevention of rheumatic fever [107] showed that these studies were poorly designed but that the consensus was that rheumatic fever can be prevented by the use of antibiotics. The overall protective effect was 70%, with a CI of 95%, and a relative risk of 0.32.
Cephalosporins may induce more rapid eradication of GAS and in some cases more effective, but may not provide any greater benefit in preventing rheumatic fever.
Secondary Prevention
Secondary prevention involves the use of prophylactic antibiotics for years, which brings up the subject of antibiotic resistance. In fact, it has been suggested that the resurgence of rheumatic fever in some parts of the developed world may be a result of the development of antibiotic resistance.
A Cochrane database analysis of randomized and quasi-randomized trials comparing the use of penicillin with control, oral versus parenteral penicillin, and various intramuscular schedules of treatment as secondary prevention of recurrent rheumatic fever revealed several interesting findings. Firstly, it was no surprise that streptococcal infections and the risk of rheumatic fever recurrence was significantly reduced in penicillin treated patients compared with controls, though there were some studies that did not show any difference. Secondly, intramuscular penicillin seemed to be superior to oral penicillin in the prevention of recurrent attacks of rheumatic fever, and thirdly, though it seemed that there were fewer instances of rheumatic fever recurrences in a group of patients receiving two-weekly versus four-weekly intramuscular penicillin, this did not reach statistical significance. The authors of this Cochrane database analysis concluded that intramuscular penicillin is the drug of choice for secondary prevention of rheumatic fever and that injections should be given every 2 weeks [108].
It has been demonstrated that secondary prevention of recurrent rheumatic fever reduces the burden of disease in many developing countries, including Cambodia and Mozambique. Echocardiographic screening is an important tool to detect the disease, as awareness and recognition is often suboptimal in these regions. Significant cardiac and neurologic manifestations of acute rheumatic fever may be reduced by cost-efficient secondary prophylaxis with antibiotics [1].
In some areas, secondary prevention alone has proven to be insufficient in reducing the overall burden of disease in terms of prevalence. Investigators in New Zealand, where a highly successful program of secondary prevention, but none for primary prevention is in place, cite the combined primary and secondary comprehensive prevention program in Cuba [109], as a effective approach to reduction of overall disease burden [110].
Improved Access to Care and Patient and Healthcare Provider Education
Successful prevention of rheumatic fever and rheumatic heart disease by early administration of antibiotics requires awareness and recognition of the potential consequences of group A streptococcus. Enhanced surveillance programs in Queensland, Australia have led to an improvement in notification rates of acute rheumatic fever, thus impacting on the success of secondary prevention programs [111].
However, access to care can overcome the risk of developing rheumatic fever stemming from poor living conditions and other environmental factors. In 1960s, access to community primary health care clinics in Baltimore led to a 60% decrease in RF [112].
Other public health measures include the creation of registries for acute rheumatic fever and rheumatic heart disease, improvement in the awareness of these diseases and proper training of nurses and triage personnel to recognize signs of the disease. Public health measures needed to improve the outcome of this group of diseases are shown in Fig. 4.

Public health measures needed to combat the global human burden of group A streptococcal disease
The Economic Burden of Strep A Pharyngitis
It is estimated that in the USA, about 7.3 million pediatric outpatient encounters are attributable to a sore throat [113]. Most of these are viral, but anywhere between 1/6 to 1/3 may be due to group A streptococcus. In the USA, up to 1,300 deaths can be attributed to group A streptococcus annually [114, 115]. The economic costs of morbidity and treatment of group A streptococcus are high, even in a developed country such as the USA. It is even higher in underdeveloped countries or in the developing world, or in areas where rheumatic fever may be endemic, as treatment of the long-term sequelae of group A streptococcus are significantly higher than the costs of prevention.
Direct medical costs include physician office visits, emergency department or urgent care visits, diagnostic costs, either by culture or rapid diagnostic strep tests, cost of medications, follow-up visits, and most importantly, the costs of medical and surgical treatment of rheumatic heart disease. Other direct costs may include those that can be associated with treatment of other short and long-term sequelae of rheumatic fever, including Sydenham’s chorea, PANDAS, post-streptococcal polyreactive arthritis, and the treatment of other sequelae of group A streptococcal pharyngitis, such as post-streptococcal glomerulonephritis.
Indirect costs include losses in work or school productivity, personal sick time costs, child care costs, costs of transportation to and from physician’s offices or emergency rooms, and again, the costs of care services related to the disability related consequences of rheumatic fever and rheumatic heart disease. All in all, the direct, or medical-related cost per group A streptococcal pharyngitis was estimated to be approximately $118 per case, and indirect cost was estimated to be $87. The total societal cost per year among children in the USA is extrapolated to be between $224 and $539 million. For each case of group A streptococcal pharyngitis, a child would miss a mean of 1.9 days of school. Significant loss of work days was also observed in a number of parents, including some in which a second parent also missed work [116]. A study in Brazil that was published in 2001 estimated the annual cost of rheumatic fever to be approximately $51 million [117]. A study in 2007 from India demonstrated that the most cost-effective prevention measure for rheumatic fever was primary prevention [118].
Treatment of Rheumatic Heart Disease and Sydenham’s Chorea
Prevention is clearly the most important factor in the treatment of group A streptococcal sequelae. Treatment of established rheumatic heart disease varies according to the degree of severity of the heart damage, primarily valvular disease. Patients should be closely monitored for the development of valve failure and congestive heart failure, whether they have prosthetic valves or not. The 10-year mortality of patients undergoing mechanical valve replacement is high. Valve repair is generally the preferred surgery over valve replacement, as there is lower surgical risk and better preservation of left ventricular function [119–121]. In addition, there is a lesser need for anticoagulant therapy in the former group, and outcomes are better.
Surgical treatment of symptomatic mitral valve stenosis include percutaneous balloon mitral valvuloplasty and surgical valvuloplasty [122–124]. The 10-year re-stenosis rate is 35%. Unfortunately, these procedures should be performed by surgeons and at a center in which there is sufficient experience to optimize good outcomes, and this is not always available in developing countries, leading to a significantly higher cost of treatment, as eluded to in the previous section. Moreover, because awareness and recognition of the disease is often delayed in developed countries, surgical intervention is often not conducted until patients are older, which is also associated with higher morbidity.
Other considerations in the treatment of rheumatic heart disease include dental prophylaxis for subacute bacterial endocarditis, and ongoing secondary prophyaxis of group A streptococcal infections to prevent recurrences of rheumatic fever. In severe cases of rheumatic heart disease that have undergone valve replacement, anticoagulation becomes necessary as well. Ongoing monitoring should include regular visits to a cardiologists and periodic echocardiography, the frequency of which varies as a function of disease severity.
Summary
Group A streptococcal infections of the upper respiratory tract and skin are still some of the most common infectious diseases of the current era. Invasive group A streptococcal disease is responsible for many deaths throughout the world, especially in developing countries or underdeveloped areas of the world. The treatment of group A streptococcal infections of the pharynx is the primary prevention of rheumatic fever. This has reduced the incidence of rheumatic fever dramatically in the developed world, where antibiotics are readily available and access to care is prompt and unrestricted. Secondary prevention with long-term antibiotic prophylaxis has reduced the recurrence rate of rheumatic fever and its most serious component, rheumatic heart disease. However, there has been a recent resurgence in rheumatic fever in developed countries, which has been postulated to be related to the emergence of resistant strains of group A streptococcus. This, and the fact that incidence of rheumatic fever in the developing world is still high, has propelled other approaches to management of the problem, such as development of a vaccine.
Although this process has been ongoing since the 1960s, there is no vaccine. One of the problems is that there are a large number of M subtypes, and that most of the vaccines are developed against the hypervariable N terminal of the M protein, and are type specific. Broad coverage is thus not achievable. Vaccines developed against the hypervariable M region have also been shown to potentially cause rheumatic heart disease. Vaccines have been developed against other proteins on the surface of the bacterium, which provides some advantages but may be accompanied by new problems, and none of these have entered clinical trials at this time. Eventually, it would seem inevitable that a vaccine will be developed, but the effectiveness of this vaccine in eradicating the disease and preventing rheumatic fever is yet unknown. In the future, a better understanding of the innate and acquired immunity of rheumatic fever may also lead to the development of novel biologics that may be able to prevent this very debilitating and lethal complication of group A streptococcal infection.
In view of the absence of a vaccine, prevention remains the primary control measure. “An ounce of prevention is worth a pound of cure” applies to rheumatic fever and rheumatic heart disease throughout the globe. The pre-requisite for prevention among both patients and care givers are awareness and early recognition [125]. Public education regarding the potential consequences of a seemingly treatable and otherwise innocuous illness, pharyngitis [126], can help increase awareness and early treatment of the disease, providing for improved outcomes, decreased morbidity and mortality and a higher quality of life. Since rheumatic fever is the highest in indigenous populations in the Pacific Islands, New Zealand, and Australia, cultural sensitivities must be appreciated [24].
The issue of environmental factors and autoimmunity continues to attract significant attention and indeed was the focus of the 7th Congress of Autoimmunity in Slovenia [127–139]. Interestingly, the role of bacterial agents in triggering autoimmunity was only a minor subject in the overall theme and special issue published from this Congress [140]. On the other hand, it is clear that microbiology and the role of bacterial agents in triggering autoimmunity is a very important subject [141–143]. It is interesting to point out that had the latency time for rheumatic fever and strep infection been measured in years, rather than in days to weeks, we would still be unaware of the etiological association. Our understanding of the complex and interactive roles of epigenetics and the environment in the development of autoimmune diseases is far from complete [144–151]. Yet, this knowledge is critically important as we attempt to develop new therapies for diseases [152–154] such as rheumatic fever and rheumatic heart disease.
References
The Lancet Neurology (2010) Neurological burden of acute rheumatic fever and rheumatic heart disease: the case for action. Lancet Neurol 9:447
Rullan E, Sigal LH (2001) Rheumatic fever. Curr Rheumatol Rep 3:445–452
Lee JL, Naguwa SM, Cheema GS, Gershwin ME (2009) Acute rheumatic fever and its consequences: a persistent threat to developing nations in the 21st century. Autoimmun Rev 9:117–123
Bland EF (1960) Declining severity of rheumatic fever. A comparative study of the past four decades. N Engl J Med 262:597–599
Pastore S, De Cunto A, Benettoni A, Berton E, Taddio A, Lepore L (2010) The resurgence of rheumatic fever in a developed country area: the role of echocardiography. Rheumatology (Oxford) 50(2):396–400
Veasy LG, Tani LY, Hill HR (1994) Persistence of acute rheumatic fever in the intermountain area of the United States. J Pediatr 124:9–16
Veasy LG, Wiedmeier SE, Orsmond GS et al (1987) Resurgence of acute rheumatic fever in the intermountain area of the United States. N Engl J Med 316:421–427
Mortimer EA Jr, Rammelkamp CH Jr (1956) Prophylaxis of rheumatic fever. Circulation 14:1144–1152
Tibazarwa KB, Volmink JA, Mayosi BM (2008) Incidence of acute rheumatic fever in the world: a systematic review of population-based studies. Heart 94:1534–1540
Carapetis JR, Steer AC, Mulholland EK, Weber M (2005) The global burden of group A streptococcal diseases. Lancet Infect Dis 5:685–694
Carapetis J, Steer A (2010) Prevention of rheumatic fever. Pediatr Infect Dis J 29:91–92, author reply 2
Hoffmann S, Henrichsen J, Schmidt K (1988) Incidence and diagnosis of acute rheumatic fever in Denmark, 1980 and 1983. A retrospective analysis of the fulfillment of the revised Jones criteria in hospitalized patients. Acta Med Scand 224:587–594
Lennon D (2009) In: Feigen R, Cherry JD, Demmier-Harrison GJ, Kaplan SL (eds) Feigen and Cherry’s Textbook of Pediatric Infectious Diseases, 6th edn. Saunders, Philadelphia, pp 419–434
Martin JM, Barbadora KA (2006) Continued high caseload of rheumatic fever in western Pennsylvania: possible rheumatogenic emm types of streptococcus pyogenes. J Pediatr 149:58–63
Wald ER, Dashefsky B, Feidt C, Chiponis D, Byers C (1987) Acute rheumatic fever in western Pennsylvania and the tristate area. Pediatrics 80:371–374
Chun LT, Reddy DV, Yim GK, Yamamoto LG (1992) Acute rheumatic fever in Hawaii: 1966 to 1988. Hawaii Med J 51:206–211
Centers for Disease Control (1988) Acute rheumatic fever among Army trainees—Fort Leonard Wood, Missouri, 1987–1988. MMWR Morb Mortal Wkly Rep 37:519–522
Leads from the MMWR (1988) Acute rheumatic fever at a Navy training center—San Diego, California. JAMA 259:1782–1787
Pastore S, De Cunto A, Benettoni A, Berton E, Taddio A, Lepore L (2011) The resurgence of rheumatic fever in a developed country area: the role of echocardiography. Rheumatology (Oxford) 50:396–400
Olgunturk R, Aydin GB, Tunaoglu FS, Akalin N (1999) Rheumatic heart disease prevalence among schoolchildren in Ankara, Turkey. Turk J Pediatr 41:201–206
Vinker S, Zohar E, Hoffman R, Elhayany A (2011) Incidence and clinical manifestations of rheumatic fever: a 6 year community-based survey. Isr Med Assoc J 12:78–81
Thakur JS, Negi PC, Ahluwalia SK, Vaidya NK (1996) Epidemiological survey of rheumatic heart disease among school children in the Shimla Hills of northern India: prevalence and risk factors. J Epidemiol Community Health 50:62–67
Chockalingam A, Gnanavelu G, Elangovan S, Chockalingam V (2003) Current profile of acute rheumatic fever and valvulitis in southern India. J Heart Valve Dis 12:573–576
White H, Walsh W, Brown A et al (2011) Rheumatic heart disease in indigenous populations. Heart Lung Circ 19:273–281
Carapetis JR, Currie BJ, Mathews JD (2000) Cumulative incidence of rheumatic fever in an endemic region: a guide to the susceptibility of the population? Epidemiol Infect 124:239–244
Parnaby MG, Carapetis JR (2010) Rheumatic fever in Indigenous Australian children. J Paediatr Child Health 46:527–533
Seckeler MD, Barton LL, Brownstein R (2010) The persistent challenge of rheumatic fever in the Northern Mariana Islands. Int J Infect Dis 14:e226–e2269
Kohler LA, Alik T, Kaplan EL, Anderson FL (2010) A pilot study for the primary prevention of rheumatic fever in Kosrae, Federated States of Micronesia. Pac Health Dialog 16:99–108
Steer AC, Jenney AJ, Oppedisano F et al (2008) High burden of invasive beta-haemolytic streptococcal infections in Fiji. Epidemiol Infect 136:621–627
Longo-Mbenza B, Bayekula M, Ngiyulu R et al (1998) Survey of rheumatic heart disease in school children of Kinshasa town. Int J Cardiol 63:287–294
Bergmark R, Bergmark B, Blander J, Fataki M, Janabi M (2010) Burden of disease and barriers to the diagnosis and treatment of group a beta-hemolytic streptococcal pharyngitis for the prevention of rheumatic heart disease in Dar Es Salaam, Tanzania. Pediatr Infect Dis J 29:1135–1137
Abdel-Moula AM, Sherif AA, Sallam SA, Mandil AM, Kassem AS, Zaher SR (1998) Prevalence of rheumatic heart disease among school children in Alexandria, Egypt: a prospective epidemiological study. J Egypt Public Health Assoc 73:233–254
Massell BF (1962) Factors in the pathogenesis of rheumatic fever recurrences. J Maine Med Assoc 53:88–93
Ozdemir O, Oguz D, Atmaca E, Sanli C, Yildirim A, Olgunturk R (2010) Cardiac troponin T in children with acute rheumatic carditis. Pediatr Cardiol 32:55–58
Szekely P, Farmer MB (1964) Rheumatic fever and rheumatic heart disease; natural history and preventive aspects. Public Health 78:78–84
Bisno AL, Gerber MA, Gwaltney JM Jr (1997) Kaplan EL, Schwartz RH. Diagnosis and management of group A streptococcal pharyngitis: a practice guideline. Infectious Diseases Society of America. Clin Infect Dis 25:574–583
Forward KR, Haldane D, Webster D, Mills C, Brine C, Aylward D (2006) A comparison between the Strep A Rapid Test Device and conventional culture for the diagnosis of streptococcal pharyngitis. Can J Infect Dis Med Microbiol 17:221–223
Sheeler RD, Houston MS, Radke S, Dale JC, Adamson SC (2002) Accuracy of rapid strep testing in patients who have had recent streptococcal pharyngitis. J Am Board Fam Pract 15:261–265
Gieseker KE, Roe MH, MacKenzie T, Todd JK (2003) Evaluating the American Academy of Pediatrics diagnostic standard for Streptococcus pyogenes pharyngitis: backup culture versus repeat rapid antigen testing. Pediatrics 111:e666–e670
Rogo TO, Schwartz RH, Ascher DP (2011) Comparison of the Inverness Medical Acceava Strep A Test With the Genzyme OSOM and Quidel QuickVue Strep A Tests. Clin Pediatr (Phila) 50(4):294–296
Gerber MA, Shulman ST (2004) Rapid diagnosis of pharyngitis caused by group A streptococci. Clin Microbiol Rev 17:571–580, table of contents
McDonald MI, Towers RJ, Fagan P, Carapetis JR, Currie BJ (2007) Molecular typing of Streptococcus pyogenes from remote Aboriginal communities where rheumatic fever is common and pyoderma is the predominant streptococcal infection. Epidemiol Infect 135:1398–1405
Zabriskie JB (1967) Mimetic relationships between group A streptococci and mammalian tissues. Adv Immunol 7:147–188
Ellis NM, Kurahara DK, Vohra H et al (2010) Priming the immune system for heart disease: a perspective on group A streptococci. J Infect Dis 202:1059–1067
Tontsch D, Pankuweit S, Maisch B (2000) Autoantibodies in the sera of patients with rheumatic heart disease: characterization of myocardial antigens by two-dimensional immunoblotting and N-terminal sequence analysis. Clin Exp Immunol 121:270–274
Beall B, Gherardi G, Lovgren M, Facklam RR, Forwick BA, Tyrrell GJ (2000) emm and sof gene sequence variation in relation to serological typing of opacity-factor-positive group A streptococci. Microbiology 146(Pt 5):1195–1209
Shulman ST, Tanz RR, Dale JB et al (2009) Seven-year surveillance of north american pediatric group a streptococcal pharyngitis isolates. Clin Infect Dis 49:78–84
Stollerman GH (1991) Rheumatogenic streptococci and autoimmunity. Clin Immunol Immunopathol 61:131–142
Stollerman GH (1997) Rheumatic fever. Lancet 349:935–942
Bisno A (1995) Non-suppurative poststreptococcal sequelae: rheumatic fever and glomerulonephritis. In: Mandell GB JE, Dolin R (eds) Principles and practice of infectious diseases. Churchill Livingstone, New York, pp 1799–1810
Bisno A (1995) Streptococcus Pyogenes. In: Mandell GB JE, Dolin R (eds) Principles and practice of infectious diseases. Churchill Livingstone, New York, pp 1786–1799
Cohen-Poradosu R, Kasper DL (2007) Group A streptococcus epidemiology and vaccine implications. Clin Infect Dis 45:863–865
Davies MR, Tran TN, McMillan DJ, Sriprakash KS (2006) Evidence for increased horizontal gene transfers among streptococci from group A streptococcal endemic regions. Int Congr Ser 1289:188–192
Bessen D, Fischetti VA (1988) Passive acquired mucosal immunity to group A streptococci by secretory immunoglobulin A. J Exp Med 167:1945–1950
Guilherme L, Kalil J (2007) Rheumatic fever: from innate to acquired immune response. Ann NY Acad Sci 1107:426–433
Hernandez-Pacheco G, Flores-Dominguez C, Rodriguez-Perez JM et al (2003) Tumor necrosis factor-alpha promoter polymorphisms in Mexican patients with rheumatic heart disease. J Autoimmun 21:59–63
Ramasawmy R, Fae KC, Spina G et al (2007) Association of polymorphisms within the promoter region of the tumor necrosis factor-alpha with clinical outcomes of rheumatic fever. Mol Immunol 44:1873–1878
Kutukculer N, Karaca NE, Sozeri BY et al (2008) Human soluble tumor necrosis factor receptor I (sTNF-RI) and interleukin-I receptor antagonist (IL-I Ra) in different stages of acute rheumatic fever. Anadolu Kardiyol Derg 8:139–142
Settin A, Abdel-Hady H, El-Baz R, Saber I (2007) Gene polymorphisms of TNF-alpha(−308), IL-10(−1082), IL-6(−174), and IL-1Ra(VNTR) related to susceptibility and severity of rheumatic heart disease. Pediatr Cardiol 28:363–371
Guilherme L, Kalil J, Cunningham M (2006) Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity 39:31–39
Guilherme L, Fae KC, Oshiro SE, Tanaka AC, Pomerantzeff PM, Kalil J (2005) Rheumatic fever: how S. pyogenes-primed peripheral T cells trigger heart valve lesions. Ann NY Acad Sci 1051:132–140
Galvin JE, Hemric ME, Ward K, Cunningham MW (2000) Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest 106:217–224
El-Demellawy M, El-Ridi R, Guirguis NI, Abdel Alim M, Kotby A, Kotb M (1997) Preferential recognition of human myocardial antigens by T lymphocytes from rheumatic heart disease patients. Infect Immun 65:2197–2205
Ellis NM, Li Y, Hildebrand W, Fischetti VA, Cunningham MW (2005) T cell mimicry and epitope specificity of cross-reactive T cell clones from rheumatic heart disease. J Immunol 175:5448–5456
Roberts S, Kosanke S, Terrence Dunn S, Jankelow D, Duran CM, Cunningham MW (2001) Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. J Infect Dis 183:507–511
Cunningham MW (2003) Autoimmunity and molecular mimicry in the pathogenesis of post-streptococcal heart disease. Front Biosci 8:s533–s543
Olgunturk R, Okur I, Cirak MY et al (2011) The role of viral agents in aetiopathogenesis of acute rheumatic fever. Clin Rheumatol 30:15–20
Kirvan CA, Swedo SE, Heuser JS, Cunningham MW (2003) Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med 9:914–920
Kirvan CA, Cox CJ, Swedo SE, Cunningham MW (2007) Tubulin is a neuronal target of autoantibodies in Sydenham’s chorea. J Immunol 178:7412–7421
Singer HS, Loiselle CR, Lee O, Garvey MA, Grus FH (2003) Anti-basal ganglia antibody abnormalities in Sydenham chorea. J Neuroimmunol 136:154–161
Kirvan CA, Swedo SE, Kurahara D, Cunningham MW (2006) Streptococcal mimicry and antibody-mediated cell signaling in the pathogenesis of Sydenham’s chorea. Autoimmunity 39:21–29
Singer HS, Loiselle CR, Lee O, Minzer K, Swedo S, Grus FH (2004) Anti-basal ganglia antibodies in PANDAS. Mov Disord 19:406–415
Church AJ, Dale RC, Cardoso F et al (2003) CSF and serum immune parameters in Sydenham’s chorea: evidence of an autoimmune syndrome? J Neuroimmunol 136:149–153
Torres KC, Dutra WO, de Rezende VB, Cardoso F, Gollob KJ, Teixeira AL (2010) Monocyte dysfunction in Sydenham’s chorea patients. Hum Immunol 71:351–354
McNeil SA, Halperin SA, Langley JM et al (2005) Safety and immunogenicity of 26-valent group a streptococcus vaccine in healthy adult volunteers. Clin Infect Dis 41:1114–1122
Vashishtha A, Fischetti VA (1993) Surface-exposed conserved region of the streptococcal M protein induces antibodies cross-reactive with denatured forms of myosin. J Immunol 150:4693–4701
Pandey M, Batzloff MR, Good MF (2009) Mechanism of protection induced by group A streptococcus vaccine candidate J8-DT: contribution of B and T-cells towards protection. PLoS ONE 4:e5147
Salvadori LG, Blake MS, McCarty M, Tai JY, Zabriskie JB (1995) Group A streptococcus-liposome ELISA antibody titers to group A polysaccharide and opsonophagocytic capabilities of the antibodies. J Infect Dis 171:593–600
Sabharwal H, Michon F, Nelson D et al (2006) Group A streptococcus (GAS) carbohydrate as an immunogen for protection against GAS infection. J Infect Dis 193:129–135
Olive C, Schulze K, Sun HK et al (2007) Enhanced protection against Streptococcus pyogenes infection by intranasal vaccination with a dual antigen component M protein/SfbI lipid core peptide vaccine formulation. Vaccine 25:1789–1797
Valentin-Weigand P, Talay SR, Kaufhold A, Timmis KN, Chhatwal GS (1994) The fibronectin binding domain of the Sfb protein adhesin of Streptococcus pyogenes occurs in many group A streptococci and does not cross-react with heart myosin. Microb Pathog 17:111–120
Yokoyama Y, Harabuchi Y (2002) Intranasal immunization with lipoteichoic acid and cholera toxin evokes specific pharyngeal IgA and systemic IgG responses and inhibits streptococcal adherence to pharyngeal epithelial cells in mice. Int J Pediatr Otorhinolaryngol 63:235–241
Kawabata S, Kunitomo E, Terao Y et al (2001) Systemic and mucosal immunizations with fibronectin-binding protein FBP54 induce protective immune responses against Streptococcus pyogenes challenge in mice. Infect Immun 69:924–930
Ji Y, Carlson B, Kondagunta A, Cleary PP (1997) Intranasal immunization with C5a peptidase prevents nasopharyngeal colonization of mice by the group A streptococcus. Infect Immun 65:2080–2087
Cleary PP, Matsuka YV, Huynh T, Lam H, Olmsted SB (2004) Immunization with C5a peptidase from either group A or B streptococci enhances clearance of group A streptococci from intranasally infected mice. Vaccine 22:4332–4341
Shet A, Kaplan EL, Johnson DR, Cleary PP (2003) Immune response to group A streptococcal C5a peptidase in children: implications for vaccine development. J Infect Dis 188:809–817
Kapur V, Maffei JT, Greer RS, Li LL, Adams GJ, Musser JM (1994) Vaccination with streptococcal extracellular cysteine protease (interleukin-1 beta convertase) protects mice against challenge with heterologous group A streptococci. Microb Pathog 16:443–450
Dale JB, Chiang EY, Hasty DL, Courtney HS (2002) Antibodies against a synthetic peptide of SagA neutralize the cytolytic activity of streptolysin S from group A streptococci. Infect Immun 70:2166–2170
Chappell JD, Stuart JG (1993) Demonstration of protection in mice from a lethal challenge of three M serotypes of Streptococcus pyogenes using an M-negative vaccine. Vaccine 11:643–648
Stjernquist-Desatnik A, Kurl DN, Schalen C, Christensen P (1990) Protective effect of heterologous gram-positive vaccine against lethal upper respiratory tract infection with type M50 group A streptococci in mice. Vaccine 8:150–152
Steer AC, Batzloff MR, Mulholland K, Carapetis JR (2009) Group A streptococcal vaccines: facts versus fantasy. Curr Opin Infect Dis 22:544–552
Brandt ER, Hayman WA, Currie B et al (1996) Opsonic human antibodies from an endemic population specific for a conserved epitope on the M protein of group A streptococci. Immunology 89:331–337
Jackson DC, O’Brien-Simpson N, Ede NJ, Brown LE (1997) Free radical induced polymerization of synthetic peptides into polymeric immunogens. Vaccine 15:1697–1705
Olive C, Toth I, Jackson D (2001) Technological advances in antigen delivery and synthetic peptide vaccine developmental strategies. Mini Rev Med Chem 1:429–438
Olive C, Batzloff M, Horvath A et al (2003) Potential of lipid core peptide technology as a novel self-adjuvanting vaccine delivery system for multiple different synthetic peptide immunogens. Infect Immun 71:2373–2383
Batzloff MR, Hayman WA, Davies MR et al (2003) Protection against group A streptococcus by immunization with J8-diphtheria toxoid: contribution of J8- and diphtheria toxoid-specific antibodies to protection. J Infect Dis 187:1598–1608
Krieg AM (2002) CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol 20:709–760
Zaman M, Skwarczynski M, Malcolm JM et al (2011) Self-adjuvanting polyacrylic nanoparticulate delivery system for group A streptococcus (GAS) vaccine. Nanomedicine 7(2):168–173
Olive C, Sun HK, Ho MF et al (2006) Intranasal administration is an effective mucosal vaccine delivery route for self-adjuvanting lipid core peptides targeting the group A streptococcal M protein. J Infect Dis 194:316–324
Gerber MA, Baltimore RS, Eaton CB et al (2009) Prevention of rheumatic fever and diagnosis and treatment of acute streptococcal pharyngitis: a scientific statement from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young, the Interdisciplinary Council on Functional Genomics and Translational Biology, and the Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Academy of Pediatrics. Circulation 119:1541–1551
Baltimore RS (2010) Re-evaluation of antibiotic treatment of streptococcal pharyngitis. Curr Opin Pediatr 22:77–82
Mayosi BM (2006) Protocols for antibiotic use in primary and secondary prevention of rheumatic fever. S Afr Med J 96:240
Feder HM Jr, Gerber MA, Randolph MF, Stelmach PS, Kaplan EL (1999) Once-daily therapy for streptococcal pharyngitis with amoxicillin. Pediatrics 103:47–51
Shvartzman P, Tabenkin H, Rosentzwaig A, Dolginov F (1993) Treatment of streptococcal pharyngitis with amoxycillin once a day. BMJ 306:1170–1172
Clegg HW, Ryan AG, Dallas SD et al (2006) Treatment of streptococcal pharyngitis with once-daily compared with twice-daily amoxicillin: a noninferiority trial. Pediatr Infect Dis J 25:761–767
Lennon DR, Farrell E, Martin DR, Stewart JM (2008) Once-daily amoxicillin versus twice-daily penicillin V in group A beta-haemolytic streptococcal pharyngitis. Arch Dis Child 93:474–478
Robertson KA, Volmink JA, Mayosi BM (2005) Antibiotics for the primary prevention of acute rheumatic fever: a meta-analysis. BMC Cardiovasc Disord 5:11
Manyemba J, Mayosi BM (2002) Penicillin for secondary prevention of rheumatic fever. Cochrane Database Syst Rev (3):CD002227
Nordet P, Lopez R, Duenas A, Sarmiento L (2008) Prevention and control of rheumatic fever and rheumatic heart disease: the Cuban experience (1986–1996–2002). Cardiovasc J Afr 19:135–140
Kerdemelidis M, Lennon DR, Arroll B, Peat B, Jarman J (2010) The primary prevention of rheumatic fever. J Paediatr Child Health 46:534–548
Hanna JN, Clark MF (2011) Acute rheumatic fever in Indigenous people in North Queensland: some good news at last? Med J Aust 192:581–584
Gordis L (1973) Effectiveness of comprehensive-care programs in preventing rheumatic fever. N Engl J Med 289:331–335
Linder JA, Bates DW, Lee GM, Finkelstein JA (2005) Antibiotic treatment of children with sore throat. JAMA 294:2315–2322
O’Brien KL, Beall B, Barrett NL et al (2002) Epidemiology of invasive group a streptococcus disease in the United States, 1995–1999. Clin Infect Dis 35:268–276
Hoge CW, Schwartz B, Talkington DF, Breiman RF, MacNeill EM, Englender SJ (1993) The changing epidemiology of invasive group A streptococcal infections and the emergence of streptococcal toxic shock-like syndrome. A retrospective population-based study. JAMA 269:384–389
Pfoh E, Wessels MR, Goldmann D, Lee GM (2008) Burden and economic cost of group A streptococcal pharyngitis. Pediatrics 121:229–234
Terreri MT, Ferraz MB, Goldenberg J, Len C, Hilario MO (2001) Resource utilization and cost of rheumatic fever. J Rheumatol 28:1394–1397
Soudarssanane MB, Karthigeyan M, Mahalakshmy T et al (2007) Rheumatic fever and rheumatic heart disease: primary prevention is the cost effective option. Indian J Pediatr 74:567–570
Yau TM, El-Ghoneimi YA, Armstrong S, Ivanov J, David TE (2000) Mitral valve repair and replacement for rheumatic disease. J Thorac Cardiovasc Surg 119:53–60
Enriquez-Sarano M, Akins CW, Vahanian A (2009) Mitral regurgitation. Lancet 373:1382–1394
Thourani VH, Weintraub WS, Guyton RA et al (2003) Outcomes and long-term survival for patients undergoing mitral valve repair versus replacement: effect of age and concomitant coronary artery bypass grafting. Circulation 108:298–304
Hernandez R, Banuelos C, Alfonso F et al (1999) Long-term clinical and echocardiographic follow-up after percutaneous mitral valvuloplasty with the Inoue balloon. Circulation 99:1580–1586
Iung B, Garbarz E, Michaud P et al (1999) Late results of percutaneous mitral commissurotomy in a series of 1024 patients. Analysis of late clinical deterioration: frequency, anatomic findings, and predictive factors. Circulation 99:3272–3278
Iung B, Vahanian A (1999) Mid-term results of percutaneous mitral balloon valvotomy. Eur Heart J 20:478–479
Steer AC, Kado J, Colquhoun S, Noonan S, Babitu T (2006) Awareness of rheumatic heart disease. Lancet 367:2118
Steer A, Colquhoun S, Noonan S, Kado J, Viale S, Carapetis J (2006) Control of rheumatic heart disease in the Pacific region. Pac Health Dialog 13:49–55
Ehrenfeld M (2010) Geoepidemiology: the environment and spondyloarthropathies. Autoimmun Rev 9:A325–A329
Hemminki K, Li X, Sundquist J, Sundquist K (2010) The epidemiology of Graves’ disease: evidence of a genetic and an environmental contribution. J Autoimmun 34:J307–J313
Invernizzi P (2009) Geoepidemiology of autoimmune liver diseases. J Autoimmun 34:J300–J306
Arnson Y, Shoenfeld Y, Amital H (2010) Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun 34:J258–J265
Berkun Y, Padeh S (2010) Environmental factors and the geoepidemiology of juvenile idiopathic arthritis. Autoimmun Rev 9:A319–A324
Chang C (2010) The immune effects of naturally occurring and synthetic nanoparticles. J Autoimmun 34:J234–J246
Chen M, Kallenberg CG (2010) The environment, geoepidemiology and ANCA-associated vasculitides. Autoimmun Rev 9:A293–A298
Deane S, Teuber SS, Gershwin ME (2010) The geoepidemiology of immune thrombocytopenic purpura. Autoimmun Rev 9:A342–A349
Meyer A, Levy Y (2010) Chapter 33: Geoepidemiology of myasthenia gravis. Autoimmun Rev 9:A383–A386
Meyer N, Misery L (2010) Geoepidemiologic considerations of auto-immune pemphigus. Autoimmun Rev 9:A379–A382
Mavragani CP, Moutsopoulos HM (2010) The geoepidemiology of Sjogren’s syndrome. Autoimmun Rev 9:A305–A310
Ranque B, Mouthon L (2010) Geoepidemiology of systemic sclerosis. Autoimmun Rev 9:A311–A318
Tobon GJ, Youinou P, Saraux A (2010) The environment, geo-epidemiology, and autoimmune disease: rheumatoid arthritis. Autoimmun Rev 9:A288–A292
Lleo A, Invernizzi P, Gao B, Podda M, Gershwin ME (2010) Definition of human autoimmunity—autoantibodies versus autoimmune disease. Autoimmun Rev 9:A259–A266
Round JL, O’Connell RM, Mazmanian SK (2010) Coordination of tolerogenic immune responses by the commensal microbiota. J Autoimmun 34:J220–J225
Doria A, Sarzi-Puttini P, Shoenfeld Y (2008) Infections, rheumatism and autoimmunity: the conflicting relationship between humans and their environment. Autoimmun Rev 8:1–4
Pordeus V, Szyper-Kravitz M, Levy RA, Vaz NM, Shoenfeld Y (2008) Infections and autoimmunity: a panorama. Clin Rev Allergy Immunol 34:283–299
Bizzaro N, Tozzoli R, Shoenfeld Y (2007) Are we at a stage to predict autoimmune rheumatic diseases? Arthritis Rheum 56:1736–1744
Borchers AT, Naguwa SM, Shoenfeld Y, Gershwin ME (2010) The geoepidemiology of systemic lupus erythematosus. Autoimmun Rev 9:A277–A287
Borchers AT, Uibo R, Gershwin ME (2010) The geoepidemiology of type 1 diabetes. Autoimmun Rev 9:A355–A365
Brooks WH, Le Dantec C, Pers JO, Youinou P, Renaudineau Y (2010) Epigenetics and autoimmunity. J Autoimmun 34:J207–J219
Chandran V, Raychaudhuri SP (2010) Geoepidemiology and environmental factors of psoriasis and psoriatic arthritis. J Autoimmun 34:J314–J321
Mackay IR (2005) The etiopathogenesis of autoimmunity. Semin Liver Dis 25:239–250
Youinou P, Pers JO, Gershwin ME, Shoenfeld Y (2010) Geo-epidemiology and autoimmunity. J Autoimmun 34:J163–J167
Shoenfeld Y, Selmi C, Zimlichman E, Gershwin ME (2008) The autoimmunologist: geoepidemiology, a new center of gravity, and prime time for autoimmunity. J Autoimmun 31:325–330
Hershko AY, Naparstek Y (2006) Autoimmunity in the era of genomics and proteomics. Autoimmun Rev 5:230–233
Shapira Y, Agmon-Levin N, Shoenfeld Y (2010) Defining and analyzing geoepidemiology and human autoimmunity. J Autoimmun 34:J168–J177
Leung PS, Dhirapong A, Wu PY, Tao MH (2010) Gene therapy in autoimmune diseases: challenges and opportunities. Autoimmun Rev 9:170–174
Oli K, Porteous J (1999) Prevalence of rheumatic heart disease among school children in Addis Ababa. East Afr Med J 76:601–605
Periwal KL, Gupta BK, Panwar RB, Khatri PC, Raja S, Gupta R (2006) Prevalence of rheumatic heart disease in school children in Bikaner: an echocardiographic study. J Assoc Physicians India 54:279–282
Oli K, Tekle-Haimanot R, Forsgren L, Ekstedt J (1992) Rheumatic heart disease prevalence among schoolchildren of an Ethiopian rural town. Cardiology 80:152–155
Steer AC, Kado J, Jenney AW et al (2009) Acute rheumatic fever and rheumatic heart disease in Fiji: prospective surveillance, 2005–2007. Med J Aust 190:133–135
Sadiq M, Islam K, Abid R et al (2009) Prevalence of rheumatic heart disease in school children of urban Lahore. Heart 95:353–357
Jaine R, Baker M, Venugopal K (2008) Epidemiology of acute rheumatic fever in New Zealand 1996–2005. J Paediatr Child Health 44:564–571
Wilson N (2010) Rheumatic heart disease in indigenous populations—New Zealand experience. Heart Lung Circ 19:282–288
Paar JA, Berrios NM, Rose JD et al (2010) Prevalence of rheumatic heart disease in children and young adults in Nicaragua. Am J Cardiol 105:1809–1814
Rizvi SF, Khan MA, Kundi A, Marsh DR, Samad A, Pasha O (2004) Status of rheumatic heart disease in rural Pakistan. Heart 90:394–399
al-Sekait MA, al-Sweliem AA, Tahir M (1990) Rheumatic heart disease in schoolchildren in western district, Saudi Arabia. J R Soc Health 110:15–16, 9
Regmi PR, Pandey MR (1997) Prevalence of rheumatic fever and rheumatic heart disease in school children of Kathmandu city. Indian Heart J 49:518–520
El-Hagrassy N, El-Chennawi F, Zaki Mel S, Fawzy H, Zaki A, Joseph N (2011) HLA class I and class II HLA DRB profiles in Egyptian children with rheumatic valvular disease. Pediatr Cardiol 31:650–656
Naito S, Kitajima K, Arakawa K (1983) HLA and rheumatic heart disease in Japanese. Am Heart J 106:1164–1167
Anastasiou-Nana MI, Anderson JL, Carlquist JF, Nanas JN (1986) HLA-DR typing and lymphocyte subset evaluation in rheumatic heart disease: a search for immune response factors. Am Heart J 112:992–997
Ayoub EM, Barrett DJ, Maclaren NK, Krischer JP (1986) Association of class II human histocompatibility leukocyte antigens with rheumatic fever. J Clin Invest 77:2019–2026
Rajapakse CN, Halim K, Al-Orainey I, Al-Nozha M, Al-Aska AK (1987) A genetic marker for rheumatic heart disease. Br Heart J 58:659–662
Guilherme L, Weidebach W, Kiss MH, Snitcowsky R, Kalil J (1991) Association of human leukocyte class II antigens with rheumatic fever or rheumatic heart disease in a Brazilian population. Circulation 83:1995–1998
Guedez Y, Kotby A, El-Demellawy M et al (1999) HLA class II associations with rheumatic heart disease are more evident and consistent among clinically homogeneous patients. Circulation 99:2784–2790
Stanevicha V, Eglite J, Sochnevs A, Gardovska D, Zavadska D, Shantere R (2003) HLA class II associations with rheumatic heart disease among clinically homogeneous patients in children in Latvia. Arthritis Res Ther 5:R340–R346
Ozkan M, Carin M, Sonmez G, Senocak M, Ozdemir M, Yakut C (1993) HLA antigens in Turkish race with rheumatic heart disease [see comment]. Circulation 87:1974–1978
Hernandez-Pacheco G, Aguilar-Garcia J, Flores-Dominguez C et al (2003) MHC class II alleles in Mexican patients with rheumatic heart disease. Int J Cardiol 92:49–54
Pruksakorn S, Currie B, Brandt E et al (1994) Identification of T cell autoepitopes that cross-react with the C-terminal segment of the M protein of group A streptococci. Int Immunol 6:1235–1244
Stoute JA, Slaoui M, Heppner DG et al (1997) A preliminary evaluation of a recombinant circumsporozoite protein vaccine against Plasmodium falciparum malaria. RTS, S Malaria Vaccine Evaluation Group. N Engl J Med 336:86–91
Brandt ER, Sriprakash KS, Hobb RI et al (2000) New multi-determinant strategy for a group A streptococcal vaccine designed for the Australian Aboriginal population. Nat Med 6:455–459
Talay SR, Zock A, Rohde M et al (2000) Co-operative binding of human fibronectin to Sfbl protein triggers streptococcal invasion into respiratory epithelial cells. Cell Microbiol 2:521–535
Guzman CA, Talay SR, Molinari G, Medina E, Chhatwal GS (1999) Protective immune response against Streptococcus pyogenes in mice after intranasal vaccination with the fibronectin-binding protein SfbI. J Infect Dis 179:901–906
Medina E, Talay SR, Chhatwal GS, Guzman CA (1998) Fibronectin-binding protein I of Streptococcus pyogenes is a promising adjuvant for antigens delivered by mucosal route. Eur J Immunol 28:1069–1077
Margarit I, Rinaudo CD, Galeotti CL et al (2009) Preventing bacterial infections with pilus-based vaccines: the group B streptococcus paradigm. J Infect Dis 199:108–115
Falugi F, Zingaretti C, Pinto V et al (2008) Sequence variation in group A streptococcus pili and association of pilus backbone types with lancefield T serotypes. J Infect Dis 198:1834–1841
Acknowledgement
The author would like to thank Mrs. Aile Reneau for her assistance with illustrations and artwork.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Chang, C. Cutting Edge Issues in Rheumatic Fever. Clinic Rev Allerg Immunol 42, 213–237 (2012). https://doi.org/10.1007/s12016-011-8271-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-011-8271-1