
Abstract
Bioactivity-guided separation of the methanol extract of Calophyllum scriblitifolium bark led to the isolation of five new pyranocoumarins, caloforines A–E (1–5) and two new coumarins, caloforines F and G (6 and 7). Their structures were elucidated by 1D and 2D NMR spectroscopy, and their absolute configurations were investigated by a combination of CD spectroscopy and DFT calculation. Caloforines A–F (1–6) showed moderate antimalarial activity against Plasmodium falciparum 3D7 strain.
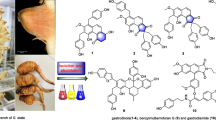
Graphical abstract

Similar content being viewed by others


Introduction
Malaria is the largest parasitic protozoan infection in humankind in which malaria infects humans through the anopheles mosquito. Malaria is not only widespread throughout the tropics but also occurs in many temperate regions. In light of this problem, scientists have turned to naturally occurring compounds obtained from plants recurrently used in traditional medicine [1]. As part of our ongoing effort to isolate and identify novel antimalarial natural products from a variety of natural sources, our laboratory reported the discovery of various types of skeletal natural products showing antimalarial activity [2,3,4,5,6,7,8].
Calophyllum, the largest genus in the Calophyllaceae family, is composed of over 200 species distributed mainly in the tropical region [9]. Plants of this genus have been reported for several ethnomedicinal uses in the traditional systems of medicine [10]. In addition, plants of this genus have been reported to contain xanthones, flavonoids, acylphloroglucinols, terpenoids and chromanones [11, 12]. The scientific study of the genus Calophyllum revealed that it is a rich source of bioactive secondary metabolites showing a wide range of biological activities. Some coumarins from C. flavoranulum showed activity against Plasmodium berghei parasite [13].
We have reported Calophyllum chromanones (calofolic acids A–F) showing dose-dependent vasorelaxation activity on isolated rat aorta, have been isolated from the tropical tree C. scriblitifolium [14]. In our search for new bioactive compounds, we investigated the MeOH extract of C. scriblitifolium which showed antimalarial activity. Bioactivity-guided separation of the extract led to the isolation of five new pyranocoumarins, caloforines A–E (1–5) and two new coumarins, caloforines F and G (6 and 7) (Fig. 1). Structure elucidation of 1–7 and the antimalarial activity of the isolated coumarins, caloforines A–F (1–6) are reported herein.

Structures of 1–7
Results and discussions
Caloforine A (1) was obtained as an optically active colorless amorphous solid, − 40 (c 1.0, MeOH) and revealed to have the molecular formula C17H20O5, requiring eight degrees of unsaturation, by HRESIMS. The IR spectrum showed an important absorption at 1717 cm−1 for the unsaturated ester carbonyl group. The UV spectrum (λmax 221, 232, 259, and 321 nm) indicated the presence of a coumarin chromophore. The 1H and 13C NMR data (Tables 1 and 3) and HSQC spectrum of 1 revealed the presence of seventeen carbon signals due to three sp3 methines, one sp3 methylene, four methyls, seven sp2 quaternary carbons, and two sp2 methines. Among them, three sp2 quaternary carbons (δC 153.5, 156.7, and 160.4) and one methyl carbon (δC 56.0) were attributed to those attached to an oxygen atom.
The gross structure of 1 was elucidated by analysis of 2D NMR data including the 1H-1H COSY, HSQC, and HMBC spectra. Analyses of the HSQC and 1H-1H COSY spectra revealed the presence of two partial structures; a (C-9 ~ C-13) and b (C-14 ~ C-15). The connections between two partial structures and coumarin skeleton were deduced mainly from the HMBC correlations of H-9, H2-14, and H3-15 (Fig. 2). In addition, the presence of a methoxy group at C-7 was deduced from the HMBC correlations of OCH3-7, H-8 and H-9 to C-7. Thus, 1 was revealed as a tetrahydropyranoyl coumarin as shown in Fig. 1, which may be derived from tiglyl moiety at C-6, with an ethyl group at C-4 and a methoxy group at C-7.

Selected 2D NMR correlations of 1
The relative stereochemistry of C-9, C-10, and C-11 was elucidated from 3 J coupling constants as in the case of those of calanolides from Calophyllum lanigerum [15]. The 3JH10-H11 constant (10.8 Hz) indicated anti relation between H-10 and H-11, whereas that of 3JH9-H10 (3.5 Hz) syn relation between H-9 and H-10.
Caloforine B (2) was determined to have the molecular formula, C18H22O5 which was larger than that of 1 by a CH2 unit. The 1H and 13C NMR data are highly similar and the presence of a methoxy group (δH 3.49) at C-9, indicating the structure of 2 as 9-methoxy caloforine A. Analysis of the 2D NMR data further supported the proposed structure (Fig. 3). In particular, the HMBC correlation of the methoxymethyl to C-9 confirmed the position of the methoxy group at C-9. The same relative stereochemistry of C-9, C-10, and C-11 as 1 was elucidated from 3 J coupling constants (3JH10-H11 12.3 Hz and 3JH9-H10 2.8 Hz) as in the case of those of 1.

Selected 2D NMR correlations of 2
The molecular formulae of caloforines C (3) and D (4) were also determined to be C17H20O5 and C18H22O5, respectively, as those of 1 and 2. Furthermore, their NMR data are also highly similar to 1 and 2, respectively. However, 3 J coupling constants of the signals associated with H-9 – H-11 were different from those of 1 and 2. The 3JH10-H11 constant (1.6 Hz) indicated syn relation between H-10 and H-11, whereas that of 3JH9-H10 (1.2 Hz) syn relation between H-9 and H-10 [15]. Analysis of the 2D NMR data (Fig. 4) supported the structures of 3 and 4 to be as shown in Fig. 1. Each stable conformer of 1 and 3 was shown in Fig. 5 to clarify the relationship of 3JH-H coupling constants associated with H-9 – H-11 (syn and anti relation).

Selected 2D NMR correlations of 3, 4 and 5

Stable conformers of 1 and 3
Caloforine E (5) was revealed to have the molecular formula C18H22O5 by HRESIMS. It’s NMR data are highly similar to 3. However, the signals [(δH 2.98 (2H, m) and 1.23 (3H, t, 7.3)] for ethyl group at C-4 in 3 are not observed in 5, and a propyl signal [(δH 2.90 (2H, m), 1.65 (2H, m), and 1.05 (3H, t, 7.5)] are observed instead. The proposed structure as shown in Fig. 1 was confirmed through analysis of the 2D NMR data (Fig. 4).
Electronic circular dichroism (ECD) may provide a powerful approach to the determination of the absolute configuration of natural products [16]. The absolute configuration of caloforines A–E (1–5) was assigned by comparing the experimental CD spectra shown in Fig. 6 and the calculated CD spectra. CD calculation was performed by Turbomole 7.1 [17] using RI-TD-DFT-B3LYP/def2-SVPD level of theory on RI-DFT-B3LYP/def2-SVP optimized geometries. The experimental CD spectra show a similar CD pattern compared to calculated CD spectra (Fig. 6). Therefore, the absolute configuration of caloforines A and B (1 and 2) was proposed to be 9S, 10R, 11S and that of caloforines C–E (3–5) 9R, 10R, 11R as shown in Fig. 1.

Experimental and calculated CD spectra of 1–5
Caloforine F (6) was revealed to have the molecular formula C19H22O5 by HRESIMS. The IR spectrum showed two absorptions at 1735 and 1660 cm−1 for unsaturated ester carbonyl and unsaturated ketone groups, respectively. The UV spectrum (λmax 226, 242, and 322 nm) indicated the presence of a coumarin chromophore.
The 1H and 13C NMR data (Tables 1 and 3) and HSQC spectrum of 6 revealed the presence of nineteen carbon signals due to one ketone, one ester ketone, two methoxyls, two sp3 methylenes, three methyls, three sp2 methines, and seven sp2 quaternary carbons. Among them, three sp2 quaternary carbons (δC 155.8 156.7, and 159.4) and two methyl carbons (δC 56.3 and 63.7) were attributed to those attached to an oxygen atom.
The gross structure of 6 was elucidated by analysis of 2D NMR data including the 1H-1H COSY, HSQC, and HMBC spectra (Fig. 7). Analyses of the HSQC and 1H-1H COSY spectra revealed the presence of two partial structures; a (C-11 ~ C-12) and b (C-14 ~ C-16). The connections between the partial structure a and a coumarin skeleton were deduced mainly from the HMBC correlations of H-14 and H2-15. In addition, the presence of a methoxy group at C-5 and C-7 was deduced from the NOESY correlation between OCH3-5 and H2-14, and the HMBC correlations of OCH3-7 and H-8 to C-7. Thus, 6 was revealed as a coumarin with a propyl group at C-4, an angelyl moiety at C-6, and two methoxy groups at C-5 and C-7.

Selected 2D NMR correlations of 6 and 7
Caloforine G (7) was revealed to have the molecular formula C18H20O6 by HRESIMS. The IR spectrum showed two absorptions at 1731 and 1662 cm−1 for unsaturated ester carbonyl and unsaturated ketone groups, respectively. Furthermore, the NMR data of 6 and 7 are highly similar to each other. However, the signals associated with the angelyl moiety in 6 is not observed in 7, and sp2 methylene and an oxygen-bearing doublet methyl signals (δC 128.3, δH 5.79 and 6.18, and δC 22.2, δH 1.48 and δC 66.5, δH 4.87 for 7) are observed instead. Furthermore, an ethyl signal instead of a propyl at C-4 is also observed. Therefore, 7 should have a 3-hydroxy-2-methylenebutanoyl moiety instead of an angelyl moiety. Analysis of the 2D NMR data (Fig. 7) supported the structure of 6 and 7 to be as shown in Fig. 1. Caloforine G (7), which was considered to be racemate, showed no optical rotation.
Coumarins, which exhibited various biological properties such as antimicrobial, anti-inflammatory, enzyme inhibitory properties, and so on, also displayed potential in vitro anti-plasmodial and in vivo antimalarial activities [18, 19]. Moreover, many of coumarin derivatives. have already been used in clinical practice for the treatment of several diseases.
Caloforines A–G (1–7) were tested for the antimalarial activity against P. falciparum 3D7 strain. The result showed that most of them showed moderate in vitro antimalarial activity [the half-maximal (50%) inhibitory concentration (IC50) = 9.4 ~ 35.5 µM, respectively.] (Table 2), whereas 7 did not (> 50 µM). The activity for pyranocoumarins 1–5 might be not influenced by their stereochemistry and substituent patterns.
Experimental section
General experimental procedures
Optical rotations were measured on a JASCO DIP-1000 polarimeter. UV spectra were recorded on a Shimadzu UVmini-1240 spectrophotometer and IR spectra on a JASCO FT/IR-4100 spectrophotometer. CD spectra were recorded on a JASCO J-820 polarimeter. High-resolution ESI MS was obtained on a JMS-T100LP (JEOL). 1H and 2D NMR spectra were measured on a 400 MHz or 600 MHz spectrometer at 300 K, while 13C NMR spectra were on a 100 MHz or 150 MHz spectrometer. The residual solvent peaks were used as internal standard (δH 7.26 and δC 77.0 for CDCl3). Standard pulse sequences were used for the 2D NMR experiments.
Material
The barks of C. scriblitifolium were collected in Mersing, Malaysia in July 2013. The botanical identification was made by Mr. Sani Miran, Herbarium of Universiti Kebangsaan Malaysia. Voucher specimens (Herbarium No. SM2299) are deposited in the Herbarium of Universiti Kebangsaan Malaysia.
Extraction and isolation
The barks of C. scriblitifolium (1.0 kg) were extracted with MeOH to obtain 130.8 g of extract. A part of the MeOH extract (50 g) was successively partitioned with n-hexane, EtOAc, n-BuOH and water. The EtOAc and n-BuOH-soluble materials were combined (25.5 g), and further separated by HP-20 column chromatography (H2O/MeOH 1:0 → 4:1 → 3:2 → 1:4 → 0:1, 100% Acetone) to obtain 3 fractions (eb-1–3). The third fraction (eb-3, 1.1 g) was further separated with a silica gel column (Hexane/EtOAc 1:0 → 9:1 → 4:1 → 7:3 → 6:4, CHCl3/MeOH 0:1 → 50:1 → 30:1 → 10:1 → 0:1) to obtain 8 fractions (eb-3–1–7). Further separation of the fraction elucted by Hexane/EtOAc 1:0 → 9:1, by ODS HPLC (Nacalai tesque Cosmosil MS-II, 10 × 250 mm, 70% MeOH at 4.7 mL/min, UV detection at 254 nm) yielded caloforines A (1, 0.7 mg, 0.0007%, tR 56.4 min), B (2, 1.1 mg, 0.0011%, tR 55.6 min), C (3, 2.5 mg, 0.0008%, tR 59.7 min), D (4, 0.8 mg, 0.0008%, tR 58.6 min), E (5, 0.9 mg, 0.0011%, tR 54.8 min), F (6, 0.4 mg, 0.0004%, tR 57.9 min), and G (7, 1.0 mg, 0.0010%, tR 54.6 min).
Caloforine A (1): white amorphous solid; − 40 (c 1.0, MeOH); IR (film) υmax 3447, 2971, 2931, 1717, 1604 cm−1; UV (MeOH) λmax 221 (ε 44,354), 232 (17,146), 259 (7782), 321 (16,538) nm; CD (MeOH) λmax 215 (Δε -14.3) nm; 1H and 13C NMR data, see Tables 1 and 3; ESIMS m/z 327 (M + H)+. HRESIMS m/z 305.1387 [calcd for C17H21O5 (M + H)+: 305.1389].
Caloforine B (2): white amorphous solid; − 63 (c 1.0, MeOH); IR (film) υmax 3465, 2954, 1731, 1699, 1605 cm−1; UV (MeOH) λmax 212 (ε 34,121), 236 (12,434), 259 (5819), 324 (12,561) nm; CD (MeOH) λmax 213 (Δε -12.33) nm; 1H and 13C NMR data, see Tables 1 and 3; ESIMS m/z 319 (M + H)+. HRESIMS m/z 319.1552 [calcd for C18H23O5 (M + H)+: 319.1545].
Caloforine C (3): white amorphous solid; + 30 (c 1.0, MeOH); IR (film) υmax 3447, 2966, 2924,1727, 1604 cm−1; UV (MeOH) λmax 207 (ε 9587), 232 (8404), 259 (4676), 321 (6834) nm; CD (MeOH) λmax 213 (Δε 3.77) nm; 1H and 13C NMR data, see Tables 1 and 3; ESIMS m/z 305 (M + H)+. HRESIMS m/z 305.1392 [calcd for C17H21O5 (M + H)+: 305.1389].
Caloforine D (4): white amorphous solid; + 16 (c 1.0, MeOH); IR (film) υmax 3465, 2964, 1731, 1604 cm−1; UV (MeOH) λmax 232 (ε 27,280), 259 (12,003), 323 (5286), 343 (7570) nm; CD (MeOH) λmax 216 (Δε 5.62) nm; 1H and 13C NMR data, see Tables 1 and 3; ESIMS m/z 319 (M + H)+. HRESIMS m/z 319.1545 [calcd for C18H23O5 (M + H)+: 319.1545].
Caloforine E (5): white amorphous solid; + 20 (c 1.0, MeOH); IR (film) υmax 2925, 1731, 1657, 1604 cm−1; UV (MeOH) λmax 206 (ε 21,415), 233 (8696), 259 (4263), 323 (6188) nm; CD (MeOH) λmax 214 (Δε 4.14) nm; 1H and 13C NMR data, see Tables 1 and 3; ESIMS m/z 319 (M + H)+. HRESIMS m/z 319.1544 [calcd for C18H23O5 (M + H)+: 319.1545].
Caloforine F (6): white amorphous solid; IR (film) υmax 2924, 1735, 1660 cm−1; UV (MeOH) λmax 206 (ε 24,031), 226 (11,585), 242 (8474), 322 (6984) nm; 1H and 13C NMR data, see Tables 1 and 3; ESIMS m/z 331 (M + H)+. HRESIMS m/z 331.1548 [calcd for C19H23O5 (M + H)+: 331.1545].
Caloforine G (7): white amorphous solid; IR (film) υmax 2961, 1731, 1662, 1602 cm−1; UV (MeOH) λmax 205 (ε 29,797), 223 (15,842), 320 (5019) nm; 1H and 13C NMR data, see Tables 1 and 3; ESIMS m/z 333 (M + H)+. HRESIMS m/z 333.1350 [calcd for C18H21O6 (M + H)+: 333.1338].
CD calculation
The conformations were obtained using Monte Carlo analysis with MMFF94 force field and charges on Macromodel 9.1. CD calculations were performed in Turbomole 7.1 using RI-TD-DFT-B3LYP/def2-SVPD level of theory on RI-DFT-B3LYP/def2-SVP optimized geometries [17].
Parasite strain culture
P. falciparum laboratory strain 3D7 was obtained from Prof. Masatsugu Kimura (Osaka City University, Osaka, Japan). For the assessment of antimalarial activity of the compounds in vitro, the parasites were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 0.5 g/L L-glutamine, 5.96 g/L HEPES, 2 g/L sodium bicarbonate (NaHCO3), 50 mg/L hypoxanthine, 10 mg/L gentamicin, 10% heat-inactivated human serum, and red blood cells (RBCs) at a 3% hematocrit in an atmosphere of 5% CO2, 5% O2, and 90% N2 at 37 °C as previously described [20]. Ring-form parasites were collected using the sorbitol synchronization technique [21]. Briefly, the cultured parasites were collected by centrifugation at 840 g for 5 min at room temperature, suspended in a fivefold volume of 5% D-sorbitol (Nacalai Tesque, Kyoto, Japan) for 10 min at room temperature, and then they were washed twice with RPMI 1640 medium to remove the d-sorbitol. The utilization of blood samples of healthy Japanese volunteers for the parasite culture was approved by the institutional review committee of the Research Institute for Microbial Diseases (RIMD), Osaka University (approval number: 22–3).
Antimalarial activity
Ring-form-synchronized parasites were cultured with compounds 1–7 at sequentially decreasing concentrations (50, 15, 5, 1.5, 0.5, and 0.15 µM) for 48 h for the flow cytometric analysis using an automated hematology analyzer, XN-30. The XN-30 analyzer was equipped with a prototype algorithm for cultured falciparum parasites (prototype; software version: 01–03, (build 16)) and used specific reagents (CELLPACK DCL, SULFOLYSER, Lysercell M, and Fluorocell M) (Sysmex, Kobe, Japan) [22, 23]. Approximately 100 µL of the culture suspension diluted with 100 µL phosphate-buffered saline was added to a BD Microtainer MAP Microtube for Automated Process K2 EDTA 1.0 mg tube (Becton Dickinson and Co., Franklin Lakes, NJ, USA) and loaded onto the XN-30 analyzer with an auto-sampler as described in the instrument manual (Sysmex). The parasitemia (MI-RBC%) was automatically reported [22]. Then 0.5% DMSO alone or containing 5 µM artemisinin was used as the negative and positive controls, respectively. The growth inhibition (GI) rate was calculated from the MI-RBC% according to the following equation:
The IC50 was calculated from GI (%) using GraphPad Prism version 5.0 (GraphPad Prism Software, San Diego, CA, USA) [24].
References
Wiesner J, Ortmann R, Jomaa H, Schlitzer M (2003) New antimalarial drugs. Angew Chem Int Ed 42:5274–5293
Tang Y, Nugroho AE, Hirasawa Y, Tougan T, Horii T, Hadi AHA, Morita H (2019) Leucophyllinines A and B, bisindole alkaloids from Leuconotis eugeniifolia. J Nat Med 73:533–540
Amelia P, Nugroho AE, Hirasawa Y, Kaneda T, Tougan T, Horii T, Morita H (2019) Indole alkaloids from Tabernaemontana macrocarpa Jack. J Nat Med 73:820–825
Nugroho AE, Hirasawa Y, Kaneda T, Shirota O, Matsuno M, Mizukami H, Morita H (2021) Triterpenoids from Walsura trichostemon. J Nat Med 75:415–422
Nugroho AE, Ono Y, Jin E, Hirasawa Y, Kaneda T, Rahman A, Kusumawati I, Tougan T, Horii T, Zaini NC, Morita H (2021) Bisindole alkaloids from Voacanga grandifolia leaves. J Nat Med 75:408–414
Amelia P, Nugroho AE, Hirasawa Y, Kaneda T, Tougan T, Horii T, Morita H (2021) Two new bisindole alkaloids from Tabernaemontana macrocarpa Jack. J Nat Med 75:633–642
Hirasawa Y, Yasuda R, Minami W, Hirata M, Nugroho AE, Tougan T, Uchiyama N, Hakamatsuka T, Horii T, Morita H (2021) Divaricamine A, a new anti-malarial trimeric monoterpenoid indole alkaloid from Tabernaemontana divaricata. Tetrahedron Lett 83:153423
Nugroho AE, Okabe M, Hirasawa Y, Wong CP, Kaneda T, Tougan T, Horii T, Morita H (2021) A novel trimeric triterpene from Chisocheton ceramicus Miq. Natural Product commun. https://doi.org/10.1177/1934578X211053202inpress
Stevens PF (1980) A revision of the old world species of Calophyllum (Guttiferae). J Arnold Arbor 61:117
Gupta S, Gupta P (2020) The genus Calophyllum: review of ethnomedicinal uses, phytochemistry and pharmacology. Bioact Nat Prod Drug Discov. https://doi.org/10.1007/978-981-15-1394-7_5
Alarcón AB, Cuesta-Rubio O, Pérez JC, Piccinelli AL, Rastrelli L (2008) Constituents of the cuban endemic species Calophyllum pinetorum. J Nat Prod 71:1283–1286
Cao S, Low K-N, Glover RP, Crasta SC, Ng S, Buss AD, Butler MS (2006) Sundaicumones A and B, polyprenylated acylphloroglucinol derivatives from Calophyllum sundaicum with weak activity against the glucocorticoid receptor. J Nat Prod 69:707–709
Abbas J, Syafruddin D (2014) Antiplasmodial evaluation of one compound from Calophyllum flavoranulum. Indo J Chem 14:185–191
Nugroho AE, Sasaki T, Kaneda T, Hadi AHA, Morita H (2017) Calofolic acids A-F, chromanones from the bark of Calophyllum scriblitifolium with vasorelaxation activity. Bioorg Med Chem Lett 27:2124–2128
Kashman Y, Gustafson KR, Fuller RW, Cardellina JH II, McMahon JB, Currens MJ, Buckheit RW Jr, Hughes SH, Cragg GM, Boyd MR (1992) The calanolides, a novel HIV-inhibitory class of coumarin derivatives from the tropical rainforest tree, Calophyllum lanigerum. J Med Chem 35:2735–2743
Nugroho AE, Morita H (2019) Computationally-assisted discovery and structure elucidation of natural products. J Nat Med 73:687–695
TURBOMOLE V7.1 (2009) A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1987–2007
Cebrián-Torrejón G, Spelman K, Leblanc K, Muñoz-Durango K, Gutiérrez ST, Ferreira ME, de Arias AR, Figadère B, Maciuk AFA, Grellier P, Cech NB, Poupon E (2011) The antiplasmodium effects of a traditional South American remedy: Zanthoxylum chiloperone var. angustifolium against chloroquine resistant and chloroquine sensitive strains of Plasmodium falciparum. Braz J Pharmacog 21:652–661
Subeki S, Matsuura H, Takahashi K, Yamasaki M, Yamato O, Maede Y, Katakura K, Kobayashi S, Trimurningsih T, Chairul C, Yoshihara T (2005) Anti-babesial and anti-plasmodial compounds from Phyllanthus niruri. J Nat Prod 68:537–539
Trager W, Jensen JB (1976) Human malaria parasites in continuous culture. Science 193(4254):673–675. https://doi.org/10.1126/science.781840
Lambros C, Vanderberg JP (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420
Tougan T, Suzuki Y, Itagaki S, Izuka M, Toya Y, Uchihashi K, Horii T (2018) An automated haematology analyzer XN-30 distinguishes developmental stages of falciparum malaria parasite cultured in vitro. Malar J 17:59. https://doi.org/10.1186/s12936-018-2208-6
Toya Y, Tougan T, Horii T, Uchihashi K (2021) Lysercell M enhances the detection of stage-specific Plasmodium-infected red blood cells in the automated hematology analyzer XN-31 prototype. Parasitol Int 80:102206. https://doi.org/10.1016/j.parint.2020.102206
Tougan T, Toya Y, Uchihashi K, Horii T (2019) Application of the automated haematology analyzer XN-30 for discovery and development of anti-malarial drugs. Malar J 18:8. https://doi.org/10.1186/s12936-019-2642-0
Acknowledgements
We thank Prof. Masatsugu Kimura (Osaka City University, Osaka, Japan) for the kind gift of the 3D7 strain, Mr. Yuji Toya and Dr. Kinya Uchihashi (Sysmex) for the setting of the XN-30 analyzer and Ms. Toshie Ishisaka and Ms. Sawako Itagaki for their technical assistance. This work was partly supported by JSPS KAKENHI (JP 19K07152 to MH) and (JP 16K08309 to AEN), Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ogasawara, A., Noguchi, R., Shigi, T. et al. Caloforines A–G, coumarins from the bark of Calophyllum scriblitifolium. J Nat Med 76, 645–653 (2022). https://doi.org/10.1007/s11418-022-01613-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-022-01613-6