
Abstract
While hybridization and introgression can have a strong adaptive importance, it can impede divergence of species. Quercus magnoliifolia and Q. resinosa are two endemic oak species distributed across the Mexican highlands. These species diverged ecological and morphologically; however, no nuclear genetic differentiation is evident. In this study, we determined the mechanisms that shape patterns of genetic variation and establish the role of migration and hybridization in the evolutionary history of these two oak species. To do this, Bayesian approaches were used for inference on migration rates and directionality and timing of divergence between species using chloroplast microsatellites. We then integrated species distribution models to infer the geographic distribution of Q. magnoliifolia and Q. resinosa during Last Interglacial, Last Glacial Maximum, and Mid-Holocene time frames. We failed in distinguishing a unique genetic composition for each species. Chloroplast differentiation was more congruent with geography than the taxonomic status of each species. Our study revealed that after the divergence (c. 10 Mya) of these two oak species, high rates of introgression took place at the end of the Pleistocene. Furthermore, past distribution models predicted that Q. magnoliifolia and Q. resinosa have likely been in sympatry presumable since Last Glacial Maximum and Q. resinosa probably expanded geographically towards its current distribution around Mid-Holocene. This expansion was supported by testing migration models, suggesting recent establishment of Q. resinosa to the north of Trans-Mexican Volcanic Belt. We hypothesized that after Q. magnoliifolia and Q. resinosa diverged, colonization events followed by hybridization between oaks and long-distance seed dispersal occurred, explaining the present-day patterns of distribution of chloroplast diversity. We propose that divergence of species remains mainly on loci under natural selection, providing evidence on the “porous” nature of species boundaries among oaks.
Similar content being viewed by others
Introduction
Hybridization has a major role in evolution via the passage of adaptive loci, and catalyzing species radiations (Schley et al. 2022). Regardless of the adaptive importance of hybridization and introgression, differentiation of diverged or diverging lineages can be affected by the homogenizing effect of gene flow (Soltis 2013), impeding the creation of new diversity through the production of novel genotypes and phenotypes (Abbott et al. 2013). Within the genus Quercus (L.), ancient and recent hybridization has been detected among phylogenetically distinct clades but with overlapping geographic ranges (Zhou et al. 2022). Due to the evolutionary implications, hybridization in oaks has been extensively studied and serves as an important model system for studying speciation considering high levels of gene flow (González-Rodríguez and Oyama 2005; Cavender-Bares et al. 2015; Eaton et al. 2015; Hipp 2015; Sork et al. 2016; Hipp et al. 2018; Kremer and Hipp 2020; and references therein). In addition, studies on population genetics of oak species indicate that geological and climatic changes during the last glacial period have had a significant influence on the current patterns of genetic diversity and structure. European oaks, for example, were restricted to different refugia during the last glaciation, and after the glacial maximum, oak species recolonized the continent following diverse postglacial routes producing populations with reduced genetic diversity but high genetic structure (Petit et al. 1993; Dumolin et al. 1995; Petit et al. 2002; Grivet and Petit 2003; Bagnoli et al. 2016). In contrast, oak species in North America have shown higher genetic diversity and lower genetic differentiation (Grivet and Petit 2003), suggesting that gene movement among their populations has been more stable in response to past climatic and geological events (Magni et al. 2005).
Despite hybridization has been intensively studied as an important mechanism to understand the formation of new species, several authors have reported that hybridization can occur without jeopardizing species integrity (Eaton et al. 2015; Kremer and Hipp 2020). Such findings highlight the role of hybridization, not only as a source of species diversity, but also as one of the most important dispersal mechanisms facilitating colonization of oaks (Potts and Reid 1988; Whittemore and Schaal 1991; Petit et al. 1997; Petit et al. 2004; Pollock et al. 2015; McCauley et al. 2019; Kremer and Hipp 2020). To understand how hybridization may have driven the species evolutionary history as a mechanism of dispersal may seem quite a challenge, mostly because of the difficulties to test macro- and micro-evolutionary hypotheses (Petit et al. 2004).
In Mexico, oak species have shown high genetic diversity and low genetic structure, reflecting historical population processes such as considerable range stability, altitudinal displacements, and dynamic gene flow between populations (González-Rodríguez et al. 2004; Tovar-Sánchez et al. 2008; Rodríguez-Correa et al. 2015; Ramos-Ortiz et al. 2016; Rodríguez-Correa et al. 2017; McCauley et al. 2019; Albarrán-Lara et al. 2019; Peñaloza-Ramírez et al. 2020). However, within the great geological and orographic complexity of Mexico, oak trees might reflect the formation of specific distributional patterns for each biogeographic region. In particular, the geographic distribution and geological history of oak species in the Mexican highlands may represent interesting models to understand the role of hybridization as a mechanism of species diversity but also as a potential source of long-distance colonization to invade new environments.
The Mexican highlands are a biodiversity hotspot and are part of the transition zone at the northern limit of the tropics (Myers et al. 2000; Morrone 2001; Mastretta-Yanes et al. 2015). Geology and climate have been invoked as the main drivers of the distribution of the biota in Mexican highlands (Mastretta-Yanes et al. 2015; Caballero-Rodríguez et al. 2018). The complexity of the Mexican highlands had a pivotal role in the diversification and hybridization patterns of Mexican oaks (Hipp et al. 2018; Mastretta-Yanes et al. 2018). In Mexico, oaks are particularly numerous because of high rates of lineage diversification into a wide variety of habitats and climates with the most extensive radiations occurring in montane regions (Hipp et al. 2018). However, no consensus about the patterns of genetic diversity have been found in the Neotropics. In some cases, Mexican oak species have experienced more stable climate, and thus, larger population size and relative population stability (González-Rodríguez et al. 2004; Cavender-Bares et al. 2011, 2015; Ramos-Ortiz et al. 2016; Peñaloza-Ramírez et al. 2020), and in other studies, genetic patterns were explained by a combination of geological and climatic changes causing shifts in altitudinal and latitudinal distribution (Rodríguez-Correa et al. 2017; Peñaloza-Ramírez et al. 2020).
Glacial cycles have influenced the distribution of trees in mountain communities (Ramírez-Barahona and Eguiarte 2013; Mastretta-Yanes et al. 2015, 2018), providing the opportunity for species to overlap in geographic space and come into secondary contact (Petit et al. 2004; Baena-Díaz et al. 2018). Thus, for species that have the ability to intercross with phylogenetically related species, the knowledge of historical changes of distributional range is essential to understand current geographical patterns of genetic variation (Pettengill and Moeller 2012). In this sense, the integration of phylogeography, species distribution models, and demographic evidence is necessary to gain insights on the evolutionary history of the species (Hewitt 2000; Muir and Schlötterer 2005; Baena-Díaz et al. 2018; McCauley et al. 2019).
Quercus magnoliifolia Née and Q. resinosa Liebm. are two endemic white oak species distributed across the Mexican highlands, classified in different taxonomic series: Circinatae and Macrophyllae, respectively. Recent evidence showed morphological and ecological divergence between these two oak species, but low genetic differentiation was recovered at the nuclear level presumably by retention of ancestral polymorphism due to recent divergence (Albarrán-Lara et al. 2019). Notwithstanding, intermediate morphological individuals were recorded between these two species suggesting hybridization (González-Villareal 1986; Albarrán-Lara et al. 2019). These two white oak species offer a particular useful system to investigate the relevance of historical seed dispersal to the biogeography of oaks in the Mexican highlands. Thus, in this study, we integrated phylogeography, historical demography, and species distribution models to determine the mechanisms that shape patterns of historical migration and genetic diversity structure of Q. magnoliifolia and Q. resinosa based on six chloroplast microsatellite markers.
Materials and methods
Study systems
Quercus magnoliifolia is distributed from the Sierra Madre Occidental, through the Trans-Mexican Volcanic Belt to the Sierra Madre del Sur (Valencia 2004; Albarrán-Lara et al. 2010, 2019). This species inhabits tropical dry forests and pine-oak forests from 375 to 2,900 m of elevation (Valencia 2004; Albarrán-Lara et al. 2019). Quercus resinosa has a restricted geographical distribution, from the west of the Trans-Mexican Volcanic Belt, through southern parts of the Sierra Madre Occidental to the Central Plateau (Albarrán-Lara et al. 2010, 2019) in xeric scrublands and pine-oak forests from 1300 to 2500 m (Valencia 2004). Both species produce ovoid acorns annually, but their size may differ between species: Q. magnoliifolia produces solitary acorns (20-30 mm in length; 10-25 mm in diameter), meanwhile Q. resinosa produces solitary to grouped (2 to 3) acorns (15-35 mm in length; 15-30 mm in diameter) (González-Villareal 1986). Previous studies have shown that both species co-occur frequently and hybridize within the Trans-Mexican Volcanic Belt and the southern part of the Sierra Madre Occidental (González-Villareal 1986; Albarrán-Lara et al. 2010, 2019).
Sampling populations
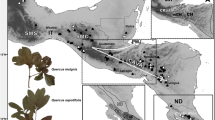
Leaf tissue from 397 trees in 61 populations of Q. magnoliifolia, Q. resinosa, and intermediate like-forms (hereafter hybrids) was collected across four Mexican biogeographic provinces (Morrone et al. 2017): Sierra Madre Occidental (SMO), Central Mexican Plateau (CP), Trans-Mexican Volcanic Belt (TMVB), and Sierra Madre del Sur (SMS) (Table 1; Fig. 1). We corroborated the taxonomic identity of Quercus species and hybrids by examining leaf shape as described in Albarrán-Lara et al. (2019). This procedure resulted in 145 individuals of Q. magnoliifolia, 121 hybrids, and 131 of Q. resinosa. Hybrid individuals were uniformly distributed in most localities, Q. resinosa were identified at the north of TMBV, whereas Q. magnoliifolia along TMBV and SMS (see Online Resource 1, Fig. S1). Population names, geographical location, biogeographic region, and morphological assignment for each individual are listed in Table S1

Sample collection localities of Quercus magnoliifolia, Quercus resinosa, and their hybrids. The geographical position of the 61 sampling points is represented by diamonds. Populations were collected across four Mexican biogeographic provinces: SMO, Sierra Madre Occidental; SMS, Sierra Madre del Sur; TMVB, Trans-Mexican Volcanic Belt; CP, Central Mexican Plateau. The numbers correspond to PopID in Table 1
DNA extraction and chloroplast microsatellites genotyping
To isolate DNA, we used cetyltrimethylammonium bromide (CTAB) protocol with an additional phenol-chloroform cleaning step (Lefort and Douglas 1999). The isolated DNA was diluted with deionized water to a final concentration of 20 ng/μL and stored at -20°C. Six molecular markers derived from chloroplast microsatellites short simple repeats (cpSSR) described in Weising and Gardner (1999) (Ccmp10) and Deguilloux et al. (2003) (μdt1, μd3, μdt4, μcd4, and μcd5) were amplified using multiplex PCR reaction by QIAGEN Multiplex PCR Kit. The thermal cycling program consisted of one cycle at 95°C for 15 min and then 35 cycles, each of 95°C for 30 s, annealing at 50°C for 1:30 min and extension at 72°C for 1 min. A final extension at 60°C for 30 min was included. All amplification reactions were carried out in 5 μl volumes including 1X Multiplex PCR Master Mix, 2 μМ each primer, deionized water, and 20 ng DNA. Multiplex PCR products were diluted 1:1 in deionized water and run in an ABI-PRISM 3100-Avant sequencer with the GeneScan-500 LIZ size standard included (Applied Biosystems, Foster City, California, USA). The software Peak Scanner v1.0 (Applied Biosystems, Foster City, California, USA) was used to determine the size of alleles.
Genetic diversity and phylogeographic structure
Genetic diversity estimates were assessed per sampled location in terms of number of haplotypes (A), number of private haplotypes (Ph), and Nei’s index of genetic diversity (Hs) estimated without bias (Nei 1973) using Haplotype Analysis software (Eliades and Eliades 2009). To identified trends in Hs with respect to latitude, longitude, and elevation, we performed Spearman’s rank correlation (rho) in R version 4.1.3 (R core Team 2022). In addition to previous parameters of genetic diversity, the number of shared haplotypes (Sh), effective number of haplotypes (Eh), and haplotypic richness (Rh) were also estimated for both species (Q. magnoliifolia and Q. resinosa) and their hybrids.
To assess the genetic differentiation, we first computed FST among species (and hybrid forms) and among sampling collection using Haplotype Analysis software. We also evaluated the effect of geographic distance on genetic structure among populations across the entire range covered by the two oak species by performing a Mantel test with 9999 permutations. We measured genetic distance as FST / (1- FST) and calculated geographic distances (km) between sampling collection using the R package “fossil” (Vavrek 2011). Then, AMOVA was run to determine the proportion of genetic variance explained by 1) grouping by species and hybrids, 2) grouping by biogeographic provinces (SMO, CP, TMVB, and SMS), and 3) grouping localities of Q. resinosa at the north of TMVB and grouping Q. magnoliifolia localities along TMBV and SMS (with no distinction of hybrids). AMOVA ran with 10,000 permutations to compute p-values using Arlequin version 3.5 (Excoffier and Lischer 2010).
The genealogical relationship between haplotypes was inferred by Haplotype network based on minimum spanning network with 1000 permutations using the R package “pegas” (Paradis 2010). Finally, we inferred genetic partition using tess3r (Caye et al. 2016) by estimates of ancestry proportions and ancestral allele frequencies. We performed 10 independent runs for each K value, which ranged from 1 to 15, and retained repetitions of K with the lowest root mean squared errors. To determine the optimum value of K, we employed a cross-validation method to identify when the curve exhibits a plateau or starts increasing again (Caye et al. 2016).
Approximate Bayesian Computation (ABC)
To test for different hypothesis of historical demography between Q. magnoliifolia and Q. resinosa, we contrasted two evolutionary scenarios using DIYABC version 2.1.0 (Cornuet et al. 2014). Evolutionary scenario A modeled a relative ancient divergence from a common ancestor (with an effective population size N1+N2) of Q. magnoliifolia and Q. resinosa at a time t2 followed by recent unidirectional introgression from Q. magnoliifolia (N2) to Q. resinosa (N1) at a time t1. Evolutionary scenario B described a simple species divergence at a time t2. As starting point, we considered t2 and t1 as the time of divergence of the two species (~8 Mya) proposed by Hipp et al. (2018) and the timing for the last glaciation event (~29,000 – 19,000 years), respectively, both translated into generational time. The later was considered as the first event of flowering and seed production, which occur between 15 and 25 years for most oak species (Johnson et al. 2002). After fine-tuning, prior distributions of demographic parameters were set as follow: Normal distribution for N1 and N2 (min = 10, max = 500, mean = 100, sd = 50; min = 100, max = 1000, mean = 200, sd = 50, respectively). Uniform for t1 with min = 1000, max = 3500; t2, min = 300,000, max = 500,000; and the unidirectional introgression rate (r2) with min = 0.01, max = 0.7. We assumed a stepwise mutation model with a mean mutation rate of 1 · 10-5 – 3 · 10-3. The summary statistics of each simulation included the mean number of alleles, mean genetic diversity, mean size variance, and FST. We computed 1,000,000 simulations for each evolutionary model and selected the 1% of the simulated data closest to the observed data to assess the posterior probabilities of scenarios based on logistic regression and direct method (Cornuet et al. 2014).
Past changes in species distribution
To gain insights about the more recent evolutionary history of the two oak species, we reconstructed species distribution models during the Mid-Holocene (MH; ~ 6,000 years BP), Last Glacial Maximum (LGM; ~ 21,000 years BP), and Last Interglacial (LIG; ~ 120,000 years BP). To do this, a database of 917 and 390 occurrence records was compiled from herbarium collections (MEXU, IEB, XAL) and GBIF data to Q. magnoliifolia and Q. resinosa, respectively. To diminish the spatial autocorrelation effects of aggregation in the modeling procedure (e.g., Zorrilla-Azcué et al. 2021), all records except one within 10 km2 were deleted, leaving a database with 450 and 220 occurrence records for Q. magnoliifolia and Q. resinosa, respectively, that was used in further analysis. Nineteen bioclimatic variables for the locations of each sampled population were obtained from the WorldClim Global v1.0 database (Hijmans et al. 2005) with a 0.8° resolution. Bioclimatic variables with high correlations (> 0.8) were eliminated to avoid collinearity, resulting in nine variables that were used in the subsequent analysis: annual mean temperature, mean diurnal range of temperature, temperature seasonality, mean temperature of the warmest quarter, mean temperature of the coldest quarter, annual precipitation, precipitation seasonality, precipitation in the wettest quarter, and precipitation in the driest quarter.
To model the ecological niche, we used a random partition method to split the available data into training and test sets. Fifteen randomized subsets containing 60% of the records (270 occurrence records for Q. magnoliifolia and 130 for Q. resinosa) were run as independent analyses using MaxEnt 3.3.3 (Phillips et al. 2006; Phillips and Dudik 2008). We obtained a model for each subset (considered replicates) using a default convergence threshold with a maximum number of 1000 iterations. Then, the obtained models were binarized using the probabilities at which 90% of the input occurrence points were included in each prediction as a threshold (Zhu et al. 2016). Model validation was performed using the kappa index (Cohen 1960). We selected three models with the highest kappa index (> 0.86 for Q. magnoliifolia and > 0.82 for Q. resinosa) as the best subset of models that were subsequently used to project the distribution to past climate scenarios. The overlapping area of the three best models was considered the current distribution.
The best subsets of current niche models were projected into the reconstructed climatic conditions during the Mid-Holocene, Last Glacial Maximum, and Last Interglacial periods. For the Mid-Holocene and Last Glacial Maximum periods, we used three general circulation models: the Community Climate System Model (CCSM), Model for Interdisciplinary Research on Climate (MIROC), and Max Planck Institute for Meteorology Earth System Model (MPI-ESM) (Cooper et al. 2016; Zhu et al. 2016); and for the Last Interglacial period, only the CCSM was used (Otto-Bliesner et al. 2008). Before projection into the past, we identified whether non-analogous climatic conditions were present between the past scenarios with respect to our current model (climate anomaly analysis proposed by Silva et al. (2020)); temperature seasonality and precipitation in the driest quarter variables were identified to have anomalous behavior in the Last Interglacial period, and both variables were therefore excluded in this projection. The intersection area of the replicates with the highest kappa index was considered the distribution range of the species in each time horizon. The maps of each period of each species were intersected to determine a probable area of sympatry between both species. The number of pixels (0.08°) was counted for each horizon for each species, for MH and LGM only the area where the three models coincide was quantified.
Migration models
Along with the species distribution modelling, we tested migration models that describe different hypotheses of ancient populations’ connectivity. We implemented the Bayesian inference strategy from Migrate-n version 3.7.2 (Beerli and Felsenstein 2001; Beerli 2006; Beerli and Palczewski 2010) to estimate historical population size (Θ = Neμ) and migration rates (Nm = ΘM/1). This method can be used to compare marginal likelihoods and Bayes Factor (BF) to test the best fit of the data. Here, we compared three migration models: The full migration model (model 1) considering all populations exchange migrants at individual rates (Fig. S2); model 2 described the areas of Q. magnoliifolia based on distribution models during the LGM, including the prediction of recent colonization of Q. resinosa towards the north (pop 2), and Q. magnoliifolia to the east of TMVB (pop 3) (Fig. S2); and model 3 describing a similar migration model suggested by Q. castanea Née in Mexico (Peñaloza-Ramírez et al. 2020), in which pop 3 was historically connected with southern populations (SMS; pop 4) and only received migrants recently from the west of TMVB (pop 1; Fig. S2). We first ran migrate-n using the complete dataset; however, migration parameters did not converge after one million steps and using a variety of running schemes, probably because many private haplotypes prevented chain convergence. Thus, we constructed a data subset including only haplotypes present in three or more localities. In this way, we conserved putative ancestral haplotypes that could have had enough time to disperse. This data subset left us with 229 individuals, and 36 sampling localities (available at https://doi.org/10.5281/zenodo.7641499).
Based on this subset, we ran migrate-n again and fitted prior distributions using the full migration model. Uniform prior distribution was as follow: Θmin= 0; Θmax=5; delta = 0.05; and Mmin= 0; Mmax= 60; delta = 6. Each model implemented the Brownian motion model with initial values computed using FST. We performed three replicates with a static heading scheme using four short chains and a single long chain. We recorded 1,000,000 steps with sample increment of 10; sampling 30,000,000 of parameter values and discarding 100,000 trees per chain. We evaluated chain convergence examining the posterior distribution of histograms of each parameter looking for a smooth curve with a single peak. Finally, to determine the best fit model, we used the marginal likelihoods by thermodynamic integration with Bezier approximation to calculate BF and posterior probabilities (Beerli and Palczewski 2010).
Results
Genetic diversity
We obtained a total of 103 haplotypes (A) with a large fraction of private ones (Table S2), which accounted for 71 % (Ph = 73) of the total number of haplotypes identified through 61 localities; 12 of them in high frequencies (≥ 0.5). Private haplotypes were distributed in 38 localities, 15 at the north of TMVB, and 23 along the TMVB and SMS (Fig. S3). We also observed nine sampling localities completely composed of private haplotypes (Table S2; Fig. S3). The rest of the haplotypes were restricted to two or three localities. Notwithstanding, we identified nine haplotypes with a wider geographical distribution (hap-7, hap-9, hap-10, hap-12, hap-18, hap-38, hap-75, hap-56, hap-44; Fig. S3). For instance, hap-9 occurred in most of the geographic distribution of both species, whereas hap-12 was restricted to the current distributional range of Q. resinosa.
Genetic diversity (Hs) was different among all populations (Table S3), ranging from no genetic diversity (Hs = 0) to very high (Hs = 0.9), with no particular geographic trend in latitude, longitude or elevation (rho = -0.1, p = 0.4; rho = 0, p = 0.9; rho = -0.07, p = 0.5, respectively); even when we tested trends based only on localities that resembled a linear distribution on vertical (rho = 0.16, p = 0.5) and horizontal axis (rho = 0.06, p = 0.8). Quercus magnoliifolia, hybrid individuals, and Q. resinosa shared ten haplotypes with a similar contribution among them (Fig. 2). Hybrids resulted with higher values of genetic diversity except for the number of private haplotypes (Table 2).

Minimum spanning network inferred between Quercus magnoliifolia, Quercus resinosa and their hybrids. Circle sizes proportional to the haplotype frequencies; the area within circles proportional to the number of observed individuals of Q. magnoliifolia (white), hybrids (gray), and Q. resinosa (black). Mutational steps are symbolized by dashes. Numbers represent the haplotype IDs shared among all taxa
Genetic and phylogeographic structure
Genetic differentiation between species and hybrids, and among populations revealed low values of FST which ranged from 0.002 (Q. magnoliifolia vs. hybrids) to 0.004 (Q. magnoliifolia vs. Q. resinosa), and from 0 to 0.012 among populations. We did not detect that geographic distance had an influence on genetic structure (r = 0.05, p = 0.14). The AMOVA results showed no genetic differentiation when grouping by species and hybrids (FCT = 0.013, p = 0.098) or following biogeographic provinces (FCT = 0.032, p = 0.077) (Table 3). However, when we took into account the geographic distribution of Q. resinosa at the north and Q. magnoliifolia along the TMVB and SMS, AMOVA suggested low but significant genetic differentiation (FCT = 0.04; p = 0.03) (Table 3).
The distribution of haplotype frequencies confirmed the extensive haplotype sharing between Q. magnoliifolia or Q. resinosa and putative hybrids (Fig. 2). In most cases, haplotypes were separated by one mutational step. The haplotype network revealed a single ancestral haplotype (hap-9) from which all others derived. Interestingly, this ancestral and widely distributed haplotype (Fig. 2), was closely related with hap-12. Note that in several hap-12 derived haplotypes, the proportion of Q. resinosa individuals increased (Fig. 2), suggesting some chloroplast differentiation between Q. magnoliifolia and Q. resinosa (irrespective of hybrids). In addition, geographic distribution of hap-12 and its position within the haplotype network (Fig. 2) suggested colonization events that could take place to the north.
Regarding TESS analysis, cross-validation suggested an optimum genetic partition at K = 8; however, the error increased from K= 4 to K = 8 (Fig. 3A). Thus, we found K = 3 as a reasonable number of genetic subdivisions (Fig. 3B, C). The pattern of genetic differentiation obtained from K = 3 yielded similar results as from AMOVA and the haplotype network: high genetic admixture between species and hybrids, but with an apparent differentiation between the geographic distribution of Q. resinosa and Q. magnoliifolia (Fig. 3B, C).

Clustering results for 397 individual trees from 61 localities. a) Cross-validation score from TESS3 plotted against the number of ancestral populations (K). b) Bar plot of the probability of assignment to K = 3 for each individual (vertical bars). Probability is proportional to the color length. c) Represents the geographical distribution for the same three genetic clusters (K = 3) suggested by TESS3. The proportion of colors in each pie charts reflects the proportion of ancestry to each genetic cluster. Biogeographic provinces: SMO, Sierra Madre Occidental; SMS, Sierra Madre del Sur; TMVB, Trans-Mexican Volcanic Belt; CP, Central Mexican Plateau
Approximate Bayesian computation
Evolutionary scenarios tested in DIYABC (Fig. 4) showed a clear statistical support for evolutionary scenario A, which modeled recent introgression and ancient divergence from a common ancestor. Data from the prior distribution and the observed data set were within the posterior predictive distribution, suggesting that scenario A explained better the observed data (Fig. S4). Assuming a generational time of 25 years (Johnson et al. 2002), ABC analysis suggested that Q. resinosa and Q. magnoliifolia diverged back to c. 10 Mya followed by recent introgression with a mean rate of 0.67 at c. 35,000 years ago (Table 4).

Approximate Bayesian Computation (ABC) analysis. Schematic representation of models tested. Scenario A (left) represents species divergence followed by unidirectional introgression (r2) from Q. magnoliifolia (N2) to Q. resinosa (N1). Scenario B (right) represents divergence between Quercus species from a common ancestor with population size N1+N2
Past changes in species distribution
The kappa values of the three best replicates for Q. magnoliifolia ranged between 0.86 and 0.89, while for Q. resinosa ranged between 0.82 and 0.87, indicating agreement for the three models contrasting the current distribution of both species. The most important climatic variables were the seasonality of temperature and precipitation for Q. magnoliifolia, while for Q. resinosa were seasonality of temperature and mean temperature in warmest quarter. Past distribution models for Q. resinosa suggested a more patchy and restricted geographical distribution than observed today (Fig. 5); whereas for Q. magnoliifolia, models predicted a more stable geographic distribution over time. During the LIG, Q. magnoliifolia and Q. resinosa were likely in sympatry; however, the predicted area of contact becomes more extensive during LGM, in which a large fraction of the distribution range of Q. resinosa lied within that of Q. magnoliifolia. Towards the Mid-Holocene relevant changes in geographic distribution were predicted for Q. resinosa, which migrated northward to reach its current distribution. This occurred parallel with the expansion of sympatric areas from LGM to Mid-Holocene (Fig. 5). Regarding Q. magnoliifolia, past distribution models suggested that this species had a wider geographic distribution since LIG; and likely experienced shorter geographic expansion eastward since LGM to Mid-Holocene (Fig. 5).

Species distribution models during the Mid-Holocene (6,000 years BP), Last Glacial Maximum (~ 21,000 years BP), and Last Interglacial (~ 120,000 years BP) for Quercus magnoliifolia (left), area of sympatry (middle), and Quercus resinosa (right). The intersection area of the replicates with the highest kappa index was projected as the distribution range of the species in each time horizon. Asterisks indicate the surface (km2) where the three models (MIROC, CCSM, and MPI) coincide for Last Glacial Maximum and Mid-Holocene
Migration models
The comparison of migration models showed strong support for model 2 (posterior probability = 0.9999; Table 5). The best-fitting model described the recent establishment of Q. resinosa at the north of the TMVB and to the east of some Q. magnoliifolia populations (Fig. S2). Those populations were likely linked by unidirectional gene flow from the west of TMVB. Estimated average Nm values ranged between 1.67 and 22.9 individuals per generation with the highest gene flow occurring northward for Q. resinosa populations (Fig. S2).
Discussion
We investigated the patterns of genetic variation of two Mexican oaks, Q. magnoliifolia and Q. resinosa. We did not distinguish a unique genetic composition for each species; however, we found a chloroplast differentiation that was congruent with geography than the taxonomic status of each species or their previously described hybrids (Albarrán-Lara et al. 2019). Our study revealed that after divergence (c. 10 Mya), high rates of introgression took place at the end of the Pleistocene (c. 35,000 years). Furthermore, past distribution models suggested that Q. magnoliifolia and Q. resinosa were largely in sympatry during the LGM, and during the Mid-Holocene, Q. resinosa likely expanded towards its current distribution. This was also supported by the migration models tested which suggested a recent establishment of Q. resinosa in the north of TMVB. In the light of this evidence, we propose that the pattern of genetic variation between both oak species reflect an important role of not only introgression but also migration by means of historical seed dispersal events.
Introgression and migration events explain the present-day distribution of chloroplast diversity
Oak clades have a long evolutionary history in which their ancestors originated during the Paleocene (Zhou et al. 2022). In fact, fossil evidence indicated that Quercus had a long presence during this epoch in the Northern Hemisphere, forming a subtropical flora with temperate elements (Barrón et al. 2017). In North America, fossil records suggest that Quercus arrived in the middle Eocene (ca. 44 Ma) (Barrón et al. 2017). Later, as the climate cooled during the Eocene-Oligocene transition, oaks migrated southward and underwent a parallel diversification before and after colonizing Mexico around 14–20 Mya (Hipp et al. 2018). Since then, oaks have colonized a wide variety of environments from xeric scrublands to temperate forests of Mexico.
Within Quercus, colonization of new territories could be facilitated by interspecific gene exchanges and seed-dispersers (Petit et al. 2004; Kremer and Hipp 2020; Zhou et al. 2022). Evidence involving hybridization and long-distance dispersal in range expansions were earlier documented for European or North American oaks (Whittemore and Schaal 1991; Petit et al. 1997). Hybridization and its potential for facilitating range expansion has been observed in Mexican oaks as well with the expansion of the subsection Racemiflorae at its northern edge (McCauley et al. 2019). In the case of Q. robur L. – Q. petrea (Matt.) Liebl. complex within Europe, Petit et al. (2004) established a model of continuous cycles of long-distance seed dispersal for the establishment of new populations of Q. robur, followed by interspecific pollen flow of Q. petrea into the newly formed populations. Repetitive introgression events and selection in favor to this successional species allowed the emergence of Q. petrea types into the new population bordered by Q. robur. Introgression-driven migration was an active mechanism during post-glacial colonization events in these oak species (Kremer and Hipp 2020).
In the light of our results and in the absence of nuclear differentiation previously reported by Albarrán-Lara et al. (2019), we propose that after Q. magnoliifolia and Q. resinosa diverged (~10 Mya) it is likely that repetitive cycles of long-distance seed dispersal and interspecific pollen flow may have played a role in the establishment of Q. resinosa in its current geographical distribution, and thus influence the present-day distribution of chloroplast diversity. According to many proxy reconstructions, temperatures during the LIG were warmer and wetter than present (reviewed by Suh et al. 2020), whereas during the last glacial period, dry and cool conditions prevailed in the tropics (Braconnot et al. 2007), suggesting that climatic conditions could favor the population persistence of both Quercus species during LGM, as was shown by our results (Fig. 5). As climate changed towards the Holocene, warmer and wetter conditions prevailed (Metcalfe et al. 2000; Harrison et al. 2003). Due to wide preferences in temperature and annual precipitation regimens of Q. magnoliifolia (Albarrán-Lara et al. 2019), unidirectional introgression events at the end of the Pleistocene detected in this study could involve genetic variation that allowed Q. resinosa successfully overcome Holocene changes.
A similar scenario was hypothesized in a genome-wide sequencing study, suggesting that introgressed temperature tolerance alleles in co-occurring Quercus species were involved in the expansion of recipient species to higher latitudes (Leroy et al. 2020). If this is correct, Q. resinosa expanded northward parallel to sympatric areas (Fig. 5) by means of repetitive cycles of long-distance seed dispersal and interspecific pollen flow with Q. magnoliifolia, allowing Q. resinosa to reach its current geographical distribution and explain the low chloroplast differentiation. Although it has been suggested that Quercus species have lower seed dispersal potential, it has been reported that long distance dispersal also occurs mainly by bird species such as Cyanocitta, Aphelocoma, Corvus and Pica species (Steele and Smallwood 2002). Additionality, recent phylogenetic analysis has suggested close relationship with rodent (Sciuridae) and bird seed-dispersers in different periods of time: earlier following the split of Quercus ancestors, and after, during the Miocene (Zhou et al. 2022). This highlights that seed dispersers may be more important in determining the distribution of genetic variation in Mexican Quercus than previously thought. On the other hand, these repetitive cycles of hybridization and long-distance dispersal offer a plausible explanation for the wider geographical distribution of hybrids found in this study (Fig. S1), especially for those localities outside of the current area of sympatry proposed in a previous study (Albarrán-Lara et al. 2019).
Finally, past distribution models predicted that Q. magnoliifolia and Q. resinosa have been largely in sympatry since the LGM, providing the opportunity for genetic exchange, as is commonly found in Quercus species that co-occur in mixed stands (González-Rodríguez and Oyama 2005; Cavender-Bares et al. 2015; Hipp 2015; Sork et al. 2016; Hipp et al. 2018). Although, Q. magnoliifolia and Q. resinosa today occur in different environments, they showed some degree of niche overlap in which a proportion of the niche of Q. resinosa was included into that of Q. magnoliifolia (Albarrán-Lara et al. 2019). In this sense, several studies have shown that climatic niche similarity promotes interspecific gene flow in diverse organisms (Arteaga et al. 2011; Andújar et al. 2014; Ortego et al. 2014; Wu et al. 2015), suggesting that ancient hybridization could be facilitated by the niche-overlap between oak species. From these hybrid zones, colonization events could also take place at great distances, invading new environments and differentiating in their current ecological niches (McCauley et al. 2019). The dispersal-migration processes are congruent with the model of invasion for Quercus species described by Petit et al. (2004).
Genetic diversity patterns
There is no consensus about the patterns of genetic diversity in oaks worldwide. However, within the Neotropics, high numbers of haplotypes and within-population diversity is usually observed. For instance, populations of Q. magnoliifolia and Q. resinosa were characterized, in general, by comparable cpDNA diversity (Hs = 0.5–0.9) to other Quercus distributed in Mexico, such as Q. castanea, Q. insignis, Q. sapotifolia, Q. affinis–Q. laurina, Q. crassifolia–Q. crassipes complex and oaks of the subsection Racemiflorae (Tovar-Sánchez et al. 2008; Ramos-Ortiz et al. 2016; Rodríguez-Correa et al. 2017; McCauley et al. 2019; Peñaloza-Ramírez et al. 2020). This level of chloroplast diversity attests for hybridization events as important drivers of high levels of genetic diversity observed, but also that species had persisted long enough to accumulate variation. Hybridization has prevented other factors being detectable, such as isolation by distance or associations between genetic diversity with latitude and altitude that track shifts in species distribution even in the absence of predictive distribution models.
The geographic distribution of hap-12 at the north of TMVB and their derived position from ancestral hap-9 (Fig. S3; Fig. 2) suggested colonization events that were supported by the migration model, indicating a recent establishment of Q. resinosa at the north of TMVB. This highlights the relevance of TMVB as a bridge between biogeographic provinces directly influencing the genetic variation of co-distributed taxa (Tovar-Sánchez et al. 2008; Bryson et al. 2018). Results from migration models indicated that Q. resinosa dispersed from the western part of TMBV northward into the Sierra Madre Occidental and Central Mexican Plateau, probably very recently in geological time. The derived condition of hap-12 and its geographic distribution were also consistent with this colonization route. The TMVB was previously proposed as well as an important hotspot for oak species diversity, as well as important zone for hybridization and introgression processes (Tovar-Sánchez et al. 2008; Bryson et al. 2018; Albarrán-Lara et al. 2019), allowing the contact between previously isolated oak species in other biogeographic regions, and thus, acquired new haplotypes by secondary contact. Interestingly, the pattern of shared haplotypes among Q. magnoliifolia-Q. resinosa and hybrids further supported, as explain in the previous section, that hybridization may have occurred more than once, since both ancestral haplotypes and relatively more recent haplotypes (Fig. 2) were shared between species.
Finally, our results together with previously reported nuclear variation (Albarrán-Lara et al. 2019) suggest that Q. magnoliifolia and Q. resinosa do not represent isolated gene pools. This contrasts with other Quercus species from other sections inhabiting Mexico, in which despite frequent hybridization remain genetically, morphological, and ecologically distinct (e.g., Tovar-Sánchez et al. 2008; Rodríguez-Correa et al. 2017). Notwithstanding, a similar pattern was observed previously between Q. minima (Sarg.) Small and Q. geminata Small in southeastern United Stated (Cavender-Bares et al. 2015). Some authors argue that species boundaries are semipermeable with permeability varying as a function of genome region (Harrison and Larson 2014). Thus, hybridizing taxa often remain distinct for only part of the genome. Barton and Hewitt (1985) suggested that sequences on a chromosome become homogenized over time by gene flow, except in small regions subject to divergent directional selection. Later, Wu (2001) proposed that for recently diverged species few genes contribute to reproductive isolation, whereas gene exchange can occur in other parts of the genome. In this way, chloroplast loci studied here and those previously assessed with nuclear loci, probably exchanged between Q. magnolifoliia and Q. resinosa, leading to large genetic admixture between them. Thus, it is likely that divergence between these Quercus species remains only in loci under natural selection. Scanning a large proportion of the genome to detect genome-wide patterns of divergence during reproductive isolation may improve our understanding about how Quercus species evolve in the face of gene flow (Nosil and Feder 2012).
Conclusions
To study the evolutionary history of Q. magnoliifolia and Q. resinosa, we assessed the genetic variation of chloroplast molecular markers implementing different approaches. Although we did not recognize a gene pool for each species, we found evidence that suggested an evolutionary history involving colonization by hybridization and long-distance seed dispersal between these two oak species. It is likely that by means of these mechanisms both Q. magnoliifolia and Q. resinosa show their current population genetic structure and distribution preventing strong signals of genetic divergence. In the face of such complex phylogeographic pattern, we assume that divergence between species remains mainly on loci under natural selection, providing evidence about the “porous” nature of species boundaries among oaks.
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJ, Bierne N et al (2013) Hybridization and speciation. J Evol Biol 26:229–246. https://doi.org/10.1111/j.1420-9101.2012.02599.x
Albarrán-Lara AL, Mendoza-Cuenca L, Valencia-Avalos S, González-Rodríguez A, Oyama K (2010) Leaf fluctuating asymmetry increases with hybridization and introgression between Quercus magnoliifolia and Quercus resinosa (Fagaceae) through an altitudinal gradient in Mexico. Int J Plant Sci 171:310–322. https://doi.org/10.1086/650317
Albarrán-Lara AL, Petit RJ, Kremer A, Caron H, Peñaloza-Ramírez JM, Gugger PF, Dávila-Aranda P, Oyama K (2019) Low genetic differentiation between two morphologically and ecologically distinct giant-leaved Mexican oaks. Plant Syst Evol 305:89–101. https://doi.org/10.1007/s00606-018-1554-8
Andújar C, Arribas P, Ruiz C, Serrano J, Gómez-Zurita J (2014) Integration of conflict into integrative taxonomy: Fitting hybridization in species delimitation of Mesocarabus (Coleoptera: Carabidae). Mol Ecol 23:4344–4361. https://doi.org/10.1111/mec.12793
Arteaga MC, McCormack JE, Eguiarte LE, Medellín RA (2011) Genetic admixture in multidimensional environmental space: Asymmetrical niche similarity promotes gene flow in armadillos (Dasypus novemcinctus). Evolution 65:2470–2480. https://doi.org/10.1111/j.1558-5646.2011.01329.x
Baena-Díaz F, Ramírez-Barahona S, Ornelas JF (2018) Hybridization and differential introgression associated with environmental shifts in a mistletoe species complex. Sci Rep 8(1):5591. https://doi.org/10.1038/s41598-018-23707-6
Bagnoli F, Tsuda Y, Fineschi S, Bruschi P, Magri D, Zhelev P, Paule L, Simeone MC, González-Martínez SC, Vendramin GG (2016) Combining molecular and fossil data to infer demographic history of Quercus cerris: insights on European eastern glacial refugia. J Biogeogr 43:679–690. https://doi.org/10.1111/jbi.12673
Barrón E, Averyanova A, Kvaček Z, Momohara A, Pigg KB, Popova S, Postigo-Mijarra JM, Tiffney BH, Utescher T, Zhou ZK (2017) The fossil history of Quercus. In: Gil-Pelegrín E, Sancho-Napik D, Peguero-Pina JJ (eds) Oaks physiological ecology. Springer Cham, pp 39–105. https://doi.org/10.1007/978-3-319-69099-5_3
Barton NH, Hewitt GM (1985) Analysis of hybrid zones. Annu Rev Ecol Syst 16:113–148. https://www.jstor.org/stable/2097045. Accessed 14 Sept 2023
Beerli P (2006) Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinform 22:341–345. https://doi.org/10.1093/bioinformatics/bti803
Beerli P, Felsenstein J (2001) Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc Natl Acad Sci U S A 98:4563–4568
Beerli P, Palczewski M (2010) Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185:313–326. https://doi.org/10.1534/genetics.109.112532
Braconnot P, Otto-Bliesner B, Harrison S, Joussaume S, Peterchmitt J, Abe-Ouchi A, Crucifix M, Driesschaert E, Fichefet T, Hewitt CD, Kageyama M, Kitoh A, Lainé A, Loutre M-F, Marti O, Merkel U, Ramstein G, Valdes P, Weber SL et al (2007) Results of PMIP2 coupled simulations of the Mid-Holocene and Last Glacial Maximum-Part 1: experiments and large-scale features. Clim Past 3:261–277. https://doi.org/10.5194/cp-3-261-2007
Bryson RW, Zarza E, Grummer JA, Parra-Olea G, Flores-Villela O, Klicka J, McCormack JE (2018) Phylogenomic insights into the diversification of salamanders in the Isthmura bellii group across the Mexican highlands. Mol Phylogenet Evol 125:78–84. https://doi.org/10.1016/j.ympev.2018.03.024
Caballero-Rodríguez D, Correa-Metrio A, Lozano-García S, Sosa-Nájera S, Ortega B, Sanchez-Dzib Y, Aguirre-Navarro K, Sandoval-Montaño A (2018) Late-Quaternary spatiotemporal dynamics of vegetation in Central Mexico. Rev Palaeobot Palynol 250:44–52. https://doi.org/10.1016/j.revpalbo.2017.12.004
Cavender-Bares J, Gonzalez-Rodriguez A, Pahlich A, Koehler K, Deacon N (2011) Phylogeography and climatic niche evolution in live oaks (Quercus series Virentes) from the tropics to the temperate zone. J Biogeogr 38:962–981. https://doi.org/10.1111/j.1365-2699.2010.02451.x
Cavender-Bares J, González-Rodríguez A, Eaton DAR, Hipp AAL, Beulke A, Manos PS (2015) Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Mol Ecol 24:3668–3687. https://doi.org/10.1111/mec.13269
Caye K, Deist TM, Martins H, Michel O, François O (2016) TESS3: Fast inference of spatial population structure and genome scans for selection. Mol Ecol Resour 16:540–548. https://doi.org/10.1111/1755-0998.12471
Cohen J (1960) A coefficient of agreement for nominal scales. Educ Psychol Meas 20:37–46. https://doi.org/10.1177/001316446002000104
Cooper DM, Dugmore AJ, Gittings BM, Scharf AK, Wilting A, Kitchener AC (2016) Predicted Pleistocene-Holocene range shifts of the tiger (Panthera tigris). Divers Distrib 22:1199–1211. https://doi.org/10.1111/ddi.12484
Cornuet JM, Pudlo P, Veyssier J, Dehne-Garcia A, Gautier M, Leblois R, Marin JM, Estoup A (2014) DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism DNA sequence and microsatellite data. Bioinform 30:1187–1189. https://doi.org/10.1093/bioinformatics/btt763
Deguilloux MF, Dumolin-Lapegue S, Gielly L, Grivet D, Petit RJ (2003) A set of primers for the amplification of chloroplast microsatellites in Quercus. Mol Ecol Notes 3:24–27. https://doi.org/10.1046/j.1471-8286.2003.00339.x
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256. https://doi.org/10.1007/BF00220937
Eaton DAR, Hipp AAL, González-Rodríguez A, Cavender-Bares J (2015) Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69:2587–2601. https://doi.org/10.1111/evo.12758
Eliades N-G, Eliades DG (2009) Haplotype Analysis: software for analysis of haplotype data. Forest genetics and forest tree breeding. Georg-August University Goettingen, Germany. http://www.uni-goettingen.de/de/67064.html. Accessed 14 Sept 2023
Excoffier L, Lischer HEL (2010) Arlequin suite version 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
González-Rodríguez A, Bain JF, Golden JL, Oyama K (2004) Chloroplast DNA variation in the Quercus affinis-Q. laurina complex in Mexico: Geographical structure and associations with nuclear and morphological variation. Mol Ecol 13:3467–3476. https://doi.org/10.1111/j.1365-294X.2004.02344.x
González-Rodríguez A, Oyama K (2005) Leaf morphometric variation in Quercus affinis and Q. laurina (Fagaceae), two hybridizing Mexican red oaks. Bot J Linn Soc 147:427–435. https://doi.org/10.1111/j.1095-8339.2004.00394.x
González-Villareal LM (1986) Contribución al conocimiento del género Quercus (Fagaceae) en el estado de Jalisco. In: Colección de la flora de Jalisco. Instituto de Botánica, Universidad de Guadalajara, Jalisco, México, pp 130–136 http://hdl.handle.net/20.500.12104/49531
Grivet D, Petit RJ (2003) Chloroplast DNA phylogeography of the hornbeam in Europe: evidence for a bottleneck at the outset of postglacial colonization. Conserv Genet 4:47–56. https://doi.org/10.1023/A:1021804009832
Harrison SP, Kutzbach JE, Liu Z, Bartlein PJ, Otto-Bliesner B, Muhs D, Prentice IC, Thompson RS (2003) Mid-Holocene climates of the Americas: a dynamical response to changed seasonality. Clim Dyn 20:663–688. https://doi.org/10.1007/s00382-002-0300-6
Harrison RG, Larson EL (2014) Hybridization, introgression, and the nature of species boundaries. J Hered 105(S1):795–809. https://doi.org/10.1093/jhered/esu033
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913. https://doi.org/10.1038/35016000
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high-resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. https://doi.org/10.1002/joc.1276
Hipp AL (2015) Should hybridization make us skeptical of the oak phylogeny? Int Oaks 26:9–18. https://www.researchgate.net/publication/278683227. Accessed 14 Sept 2023
Hipp AL, Manos PS, González-Rodríguez A, Hahn M, Kaproth M, McVay JD, Valencia-Ávalos S, Cavender-Bares J (2018) Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. New Phytol 217:439–452. https://doi.org/10.1111/nph.14773
Johnson PS, Shirley SR, Rogers R (2002) The ecology and silviculture of oaks. CABI, Boston, MA, 503. https://lccn.loc.gov./2018043937. Accessed 14 Sept 2023
Kremer A, Hipp A (2020) Oaks: an evolutionary success story. New Phytol 226:987–1011. https://doi.org/10.1111/nph.16274
Lefort F, Douglas GC (1999) An efficient micro-method of DNA isolation from mature leaves of four hardwood tree species Acer, Fraxinus, Prunus and Quercus. Ann For Sci 56:259–263. https://doi.org/10.1051/forest:19990308
Leroy T, Louvet J, Lalanne C, Le Provost G, Labadie K, Aury J-M, Delzon S, Plomion C, Kremer A (2020) Adaptive introgression as a driver of local adaptation to climate in European white oaks. New Phytol 226:1171–1182. https://doi.org/10.1111/nph.16095
Magni CR, Ducousso A, Caron H, Petit RJ, Kremer A (2005) Chloroplast DNA variation of Quercus rubra L. in North America and comparison with other Fagaceae. Mol Ecol 14:513–524. https://doi.org/10.1111/j.1365-294X.2005.02400.x
Mastretta-Yanes A, Moreno-Letelier A, Piñero D, Jorgensen TH, Emerson BC (2015) Biodiversity in the Mexican highlands and the interaction of geology, geography and climate within the Trans-Mexican Volcanic Belt. J Biogeogr 42:1586–1600. https://doi.org/10.1111/jbi.12546
Mastretta-Yanes A, Xue AT, Moreno-Letelier A, Jorgensen TH, Alvarez N, Piñero D, Emerson BC (2018) Long-term in situ persistence of biodiversity in tropical sky islands revealed by landscape genomics. Mol Ecol 27:432–448. https://doi.org/10.1111/mec.14461
McCauley RA, Cortés-Palomec AC, Oyama K (2019) Species diversification in a lineage of Mexican red oak (Quercus section Lobatae subsection Racemiflorae) – the interplay between distance, habitat, and hybridization. Tree Genet Genomes 15:27. https://doi.org/10.1007/s11295-019-1333-x
Metcalfe SE, O’Hara SL, Caballero M, Davies SJ (2000) Records of late Pleistocene-Holocene climatic change in Mexico - a review. Quat Sci Rev 19:699–721. https://doi.org/10.1016/S0277-3791(99)00022-0
Morrone JJ (2001) Homology, biogeography and areas of endemism. Divers Distrib 7:297–300. https://doi.org/10.1046/j.1366-9516.2001.00116.x
Morrone JJ, Escalante T, Rodríguez-Tapia G (2017) Mexican biogeographic provinces: Map and shapefiles. Zootaxa 4277:277–279. https://doi.org/10.11646/zootaxa.4277.2.8
Muir G, Schlötterer C (2005) Evidence for shared ancestral polymorphism rather than recurrent gene flow at microsatellite loci differentiating two hybridizing oaks (Quercus spp.). Mol Ecol 14:549–561. https://doi.org/10.1111/j.1365-294X.2004.02418.x
Myers N, Mittermeier RA, Mittermeier CG, da Fonseca GAB, Kent J (2000) Biodiversity hotspots for conservation priorities. Nature 403:853–858. https://doi.org/10.1038/35002501
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70:3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Nosil P, Feder JL (2012) Genomic divergence during speciation: Causes and consequences. Philos Trans R Soc B: Biol Sci 367:332–342. https://doi.org/10.1098/rstb.2011.0263
Ortego J, Gugger PF, Riordan EC, Sork VL (2014) Influence of climatic niche suitability and geographical overlap on hybridization patterns among southern Californian oaks. J Biogeogr 41:1895–1908. https://doi.org/10.1111/jbi.12334
Otto-Bliesner BL, Marshall SJ, Overpeck JT, Miller GH, Hu A, CAPE Last Interglacial Project members (2008) Simulating Arctic climate warmth and icefield retreat in the last interglaciation. Science 311:1751–1753. https://doi.org/10.1126/science.1120808
Paradis E (2010) Pegas: an R package for population genetics with an integrated-modular approach. Bioinform 26:419–420. https://doi.org/10.1093/bioinformatics/btp696
Peñaloza-Ramírez JM, Rodríguez-Correa H, González-Rodríguez A, Rocha-Ramírez V, Oyama K (2020) High genetic diversity and stable Pleistocene distributional ranges in the widespread Mexican red oak Quercus castanea Née (1801) (Fagaceae). Ecol Evol 10:4204–4219. https://doi.org/10.1002/ece3.6189
Petit RJ, Kremer A, Wagner DB (1993) Geographic structure of chloroplast DNA polymorphisms in European oaks. Theor Appl Genet 87:122–128. https://doi.org/10.1007/BF00223755
Petit RJ, Pineau E, Demesure B, Ducouso A, Kremer A (1997) Chloroplast DNA footprints of postglacial recolonization by oaks. Proc Natl Acad Sci U S A 94:9996–10001. https://doi.org/10.1073/pnas.94.18.9996
Petit RJ, Brewer S, Bordács S, Burg K, Cheddadi R, Coart E, Cottrell J, Csaikl UM, van Dam B, Deans JD, Espinel S, Fineschi S, Finkeldey R, Glaz I, Goicoechea PG, Jensen JS, König AO, Lowe AJ, Madsen SF et al (2002) Identification of refugia and postglacial colonization routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manag 156:49–74. https://doi.org/10.1016/S0378-1127(01)00634-X
Petit RJ, Bodénès C, Ducousso A, Roussel G, Kremer A (2004) Hybridization as a mechanism of invasion in oaks. New Phytol 161:151–164. https://doi.org/10.1046/j.1469-8137.2003.00944.x
Pettengill JB, Moeller DA (2012) Phylogeography of speciation: Allopatric divergence and secondary contact between outcrossing and selfing Clarkia. Mol Ecol 21:4578–4592. https://doi.org/10.1111/j.1365-294X.2012.05715.x
Phillips SJ, Dudik M (2008) Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31:161–175. https://doi.org/10.1111/j.0906-7590.2008.5203.x
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259. https://doi.org/10.1016/j.ecolmodel.2005.03.026
Pollock LJ, Bayly MJ, Vesk PA (2015) The roles of ecological and evolutionary processes in plant community assembly: The environment, hybridization, and introgression influence co-occurrence of Eucalyptus. Am Nat 185:784–796. https://doi.org/10.1086/680983
Potts BM, Reid JB (1988) Hybridization as a dispersal mechanism. Evolution 42:1245–1255. http://www.jstor.org/stable/2409008. Accessed 14 Sept 2023
R Core Team (2022) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org/. Accessed 14 Sept 2023
Ramírez-Barahona S, Eguiarte LE (2013) The role of glacial cycles in promoting genetic diversity in the Neotropics: The case of cloud forests during the Last Glacial Maximum. Ecol Evol 3:725–738. https://doi.org/10.1002/ece3.483
Ramos-Ortiz S, Oyama K, Rodríguez-Correa H, González-Rodríguez A (2016) Geographic structure of genetic and phenotypic variation in the hybrid zone between Quercus affinis and Q. laurina in Mexico. Plant Species Biol 31:219–232. https://doi.org/10.1111/1442-1984.12109
Rodríguez-Correa H, Oyama K, MacGregor-Fors I, González-Rodríguez A (2015) How are oaks distributed in the Neotropics? A perspective from species turnover, areas of endemism, and climatic niches. Int J Plant Sci 176:222–231. https://doi.org/10.1086/679904
Rodríguez-Correa H, Oyama K, Quesada M, Fuchs EJ, Quezada M, Ferrufino L, Valencia-Ávalos S, Cascante-Marín A, González-Rodríguez A (2017) Complex phylogeographic patterns indicate Central American origin of two widespread Mesoamerican Quercus (Fagaceae) species. Tree Genet Genomes 13:1–14. https://doi.org/10.1007/s11295-017-1147-7
Schley RJ, Twyford AD, Pennington RT (2022) Hybridization: a ‘double-edged sword’ for Neotropical plant diversity. Bot J Linn 199:331–356. https://doi.org/10.1093/botlinnean/boab070
Silva AP, Mukherjee S, Ramakrishnan U, Fernandes C, Björklund M (2020) Closely related species show species-specific environmental responses and different spatial conservation needs: Proinailurus cats in the Indian subcontinent. Sci Rep 10:18705. https://doi.org/10.1038/s41598-020-74684-8
Soltis PS (2013) Hybridization, speciation and novelty. J Evol Biol 26:291–293. https://doi.org/10.1111/jeb.12095
Sork VL, Riordan E, Gugger PF, Fitz-Gibbon S, Wei X, Ortego J (2016) Phylogeny and introgression of California scrub white oaks (Quercus section Quercus). Int Oaks 27:61–74. https://www.researchgate.net/publication/308632217. Accessed 14 Sept 2023
Steele MA, Smallwood PD (2002) Acorn dispersal by birds and mammals. In: McShea WJ, Healey WM (eds) Oak forest ecosystems: ecology and management for wildlife. CABI, Baltimore, pp 182–195
Suh YJ, Diefendorf AF, Freimuth EJ, Hyun S (2020) Last interglacial (MIS 5e) and Holocene paleohydrology and paleovegetation of midcontinental North America from Gulf of Mexico sediments. Quat Sci Rev 227:106066. https://doi.org/10.1016/j.quascirev.2019.106066
Tovar-Sánchez E, Mussali-Galante P, Esteban-Jiménez R, Piñero D, Arias DM, Dorado O, Oyama K (2008) Chloroplast DNA polymorphism reveals geographic structure and introgression in the Quercus crassifolia x Quercus crassipes hybrid complex in Mexico. Botany 86:228–239. https://doi.org/10.1139/B07-128
Valencia AS (2004) Diversidad del género Quercus (Fagaceae) en México. Bol Soc Bot Méx 75:33–53 https://doi.org/10.17129/botsci.1692
Vavrek MJ (2011) Fossil: palaeoecological and palaeogeographical analysis tools. Palaeontol Electron 14:16. https://www.uv.es/pe/2011_1/238/238.pdf. Accessed 14 Sept 2023
Weising KR, Gardner C (1999) A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome 42:9–19. https://doi.org/10.1139/g98-104
Whittemore AT, Schaal BA (1991) Interspecific gene flow in sympatric oaks. Proc Natl Acad Sci U S A 88:2540–2544. https://doi.org/10.1073/pnas.88.6.2540
Wu C-I (2001) The genic view of the process of speciation. J Evol Biol 14:851–865. https://doi.org/10.1046/j.1420-9101.2001.00335.x
Wu Z, Ding Z, Yu D, Xu X (2015) Influence of niche similarity on hybridization between Myriophyllum sibiricum and M. spicatum. J Evol Biol 28:1465–1475. https://doi.org/10.1111/jeb.12667
Zhu GP, Ye Z, Du J, Zhang DL, Zhen YH, Zheng CG, Zhao L, Li M, Bu WJ (2016) Range wide molecular data and niche modeling revealed the Pleistocene history of a global invader (Halyomorpha halys). Sci Rep 6:23192. https://doi.org/10.1038/srep23192
Zhou BF, Yuan S, Crowl AA, Liang YY, Shi Y, Chen XY, An QQ et al (2022) Phylogenomic analyses highlight innovation and introgression in the continental radiations of Fagaceae across the Northern Hemisphere. Nat Commun 13:1320. https://doi.org/10.1038/s41467-022-28917-1
Zorrilla-Azcué S, González-Rodríguez A, Oyama K, González MA, Rodríguez-Correa H (2021) The DNA history of a lonely oak: Quercus humboldtii phylogeography in the Colombian Andes. Ecol Evol 11:6814–6828. https://doi.org/10.1002/ece3.7529
Acknowledgements
The authors thank to three anonymous reviewers that improved the final version of this manuscript. We also thank Víctor Rocha Ramírez for technical support, and the use of Chihuil, the bioinformatics server at UNAM (Unidad Mazatlán) for statistical analysis support.
Funding
This study was supported by the graduate program Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM) and by CONACYT doctoral scholarship (188873) and UC MEXUS-CONACYT postdoctoral fellowship (1010/680/2012; 1010/375/2013) to ALAL. This research was supported by DGAPA-PAPIIT, UNAM (grants IN209108, IN229803, and IV201015) to KO, CONACYT-SEMARNAT (grant 2004-C01-97), CONACYT (grants 23728 and 241036) to KO, CONACYT-ECOS-NORD (grant M03-A01) to KO, and CONACYT (Ciencia de Frontera) (grant 263962) to HRC.
Author information
Authors and Affiliations
Contributions
AAL, PDA, and KO designed the study and supervised the whole project. AAL and JMPR performed the field and molecular work. AAL, MOZ, CTM, JMPR, IMD-C, HRC, and KO analyzed the data and interpret the results. The manuscript was written and revised by all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Data Archiving Statement
The datasets generated and analyzed during the current study will be public available in the open access repository of Zenodo [https://doi.org/10.5281/zenodo.7641524, https://doi.org/10.5281/zenodo.7641499].
Additional information
Communicated by J. Chen
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Fig. 1
(PDF 4.23 mb)
Supplementary Fig. 2
(PDF 613 kb)
Supplementary Fig. 3
(PDF 2.44 mb)
Supplementary Fig. 4
(PDF 164 kb)
Supplementary Table 1
(XLSX 28.4 kb)
Supplementary Table 2
(XLSX 12.7 kb)
Supplementary Table 3
(XLSX 14.4 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Albarrán-Lara, A., Ochoa-Zavala, M., Torres-Miranda, A. et al. Long-distance dispersal drives the genetic variation and historical demography of Quercus magnoliifolia and Quercus resinosa (Fagaceae) in the Mexican highlands. Tree Genetics & Genomes 19, 45 (2023). https://doi.org/10.1007/s11295-023-01619-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-023-01619-5