
Abstract
Range shifts during the Pleistocene shaped the unique phylogeographical structures of many species. Pleistocene range shifts gave currently allopatric species opportunities to occur in sympatry, likely resulting in ancient introgressions between related taxa. In our study, we investigate the range shifts and introgression patterns of three Japanese Abies species (A. firma, A. homolepis, and A. veitchii) by employing an extensive survey of 43 populations. This survey includes comprehensive analysis of both mitochondrial (mtDNA) and nuclear (18 microsatellites) genomes, in combination with species distribution modeling (SDM). It is important to note that these two types of markers provide distinct and complementary information, as they have different modes of inheritance and mutation rates. Bayesian clustering analysis indicates that the three species were clearly separated, with the exception of the A. homolepis var. umbellata population, which is considered a natural hybrid between A. homolepis and A. firma. However, mtDNA haplotypes of the four northern populations of A. firma were entirely replaced by two major haplotypes of A. homolepis and A. veitchii. The results of neighbor-net, NewHybrids, STRUCTURE analyses, and SDM suggest that historical introgression between species occurred in each geographic region, with mtDNA capture being the likely mechanism. However, contrary to these findings, the ABC coalescent analysis did not support an ancient introgression. Therefore, further validation with genome-wide level data is needed to clarify this issue. Our conclusion is that climate-induced range shifts during the Pleistocene/Holocene likely played a crucial role in the observed patterns of introgression in these species.
Similar content being viewed by others


Introduction
Pleistocene range shifts shaped the unique phylogeographical structures of numerous plant species (Knowles 2001; Hofreiter and Stewart 2009). The Pleistocene is often referred to as the glacial period because cycles of glaciation have been repeated every 100,000 years during the last 1.0 Ma in this era. These repeated glaciation cycles sometimes led to unique events such as hybridization and introgression caused by shifts in the distribution of each species (McKinnon et al. 2004a; Feliner 2014). Historical introgression and hybridization are also important factors in determining current genetic variation and may accelerate speciation and diversification (Abbott et al. 2013).
The pace of range shifts varies significantly among different species. For instance, while some plants, including tree species, exhibit relatively slower range shifts under present-day conditions, evidence suggests that they were able to expand their ranges rapidly in postglacial landscapes that were previously uninhabited (Reid’s Paradox). Tree species are typically long-lived, producing flowers year after year, and have the capacity for long-distance pollen dispersal, especially in wind-pollinated species. Consequently, when the ranges of two genetically related species become close due to distribution shifts, opportunities for hybridization may increase (Whittemore and Schaal 1991; Petit et al. 1997).
In the genus Abies, mitochondrial DNA (mtDNA) is maternally inherited and exhibits a slower mutation rate compared to nuclear DNA. This characteristic provides us with a more stable genetic marker, offering insights into long-term evolutionary history and facilitating the detection of historical hybridization events. Our study also utilizes nuclear Simple Sequence Repeats (SSRs), which, due to their relatively higher mutation rate and biparental inheritance, capture a different aspect of species’ history and help in elucidating more recent evolutionary events. These two genetic markers thus provide us with complementary perspectives on the evolutionary history of species and allow a more comprehensive understanding of how events such as hybridization and introgression have shaped current genetic variation.
In Japan, there are five native Abies species: A. sachalinensis (F. Schmidt) Mast., A. veitchii Lindl., A. mariesii Mast., A. homolepis Siebold & Zucc., and A. firma Siebold & Zucc. Of these, A. sachalinensis is found only on Hokkaido Island, while the other four species are found on Honshu and nearby islands. Abies mariesii belongs to section Amabilis, which originates from North America, but the other four Abies species belong to sections Momi and Balsamea (Farjon and Rushforth 1988; Suyama et al. 2000) of Asian origin. The three Abies (A. veitchii, A. homolepis, and A. firma) on Honshu Island and nearby islands have close phylogenetic relationships based on molecular data, although their degree of relatedness varies depending on the genetic markers used (Suyama et al. 2000). These species play ecologically important roles in each ecosystem from temperate to subalpine zones in Japan. Molecular phylogenetic analysis of the genus Abies using nuclear, plastid, and mitochondrial genomes estimates the divergence age of the two most closely related species, A. firma and A. homolepis, to be approximately 20.5 million years ago in the Early Miocene (Xiang et al. 2015). However, more recent research suggests that the divergence of A. firma and A. homolepis occurred in the late Pliocene and that the divergence of A. veitchii from the other two species occurred at the border of the Miocene and Pliocene (Semerikova et al. 2018). These findings emphasize the complex evolutionary history of these species, which may include events of introgressive hybridization as suggested by the discordant tree topologies obtained from mtDNA data (Semerikova et al. 2018). The three Abies species are currently separately distributed, but sometimes, the distribution area overlaps somewhat, especially in marginal areas. Occasional hybridization between Japanese Abies species has been observed. For instance, a hybrid between A. homolepis and A. firma (Mayr 1890; Tsumura and Suyama 1998; Aizawa and Iwaizumi 2020) as well as a hybrid between A. homolepis and A. veitchii (Isoda et al. 2000) has been reported in central Japan. Mitochondrial DNA capture in Abies species has also been reported, especially in marginal populations and areas where species overlap (Tsumura and Suyama 1998). Because the natural distribution of the three Abies species during the last glacial maximum (LGM) was more southward and at lower elevations, the three species of Abies might have been much closer than in their present-day distribution. Therefore, these Abies species represent a good model to study historical hybridization and introgression between species.
Abies firma is a representative tree species of temperate forests in Japan. However, several threats are causing concern for the health and survival of these forests, including air pollution, logging, and overgrazing by deer populations. It is also important to note that old growth forests of Abies firma have become rare. Abies homolepis is ranked as Near Threatened by the IUCN and maintains a large population on the Pacific side of the Honshu mountain range, but the population is shrinking due to logging in the peripheral forests. Abies veitchii still forms extensive mono-dominant forests, in the high mountainous areas of Honshu; however, its population size is decreasing in lower elevation areas such as Kii mountain range. Although none of these species are forestry target species, they are major tall tree species of Japan’s subalpine, cool-temperate to temperate forests. Ensuring their continued role as forest dominants is crucial to the stability of these forest ecosystems into the future. However, there is limited knowledge regarding the geographic distribution of genetic diversity and genetic structure, which are fundamental to the conservation and management of these species.
In this study, we focused on (1) clarifying the evolutionary relationship between three Japanese Abies species; (2) inferring the natural distribution area of each species and where species overlapped during LGM and other periods using species distribution modeling (SDM); and then (3) discussing how the current populations of the three Abies species were established and how to conserve these trees.
Materials and methods
Investigated species and sample collection
Abies firma has a continuous wide distribution across Honshu, Shikoku, and Kyushu Islands at altitudes between 50 and 1600 m above sea level (Liu 1971) (Fig. 1). Abies homolepis is found on the Pacific Ocean side of central Honshu and Shikoku, at elevations of 1000–1800 m (Fig. 1), generally growing in an environmental zone that is intermediate between A. firma and A. veitchii. Abies veitchii is predominantly found in central Honshu at elevations between 1200 and 2800 m. Apart from these central populations, there are isolated populations within the Kii mountain region on the Kii Peninsula and in Shikoku. Within Shikoku, two populations are known: one on Mt. Ishizuchi and another on Mt. Tsurugi. For this study, we sampled only the population from Mt. Ishizuchi (Fig. 1). Abies homolepis var. umbellata is considered a natural hybrid between A. firma and A. homolepis by their phenotype and genetic data (Mayr 1890; Aizawa and Iwaizumi 2020). The plant differs from A. homolepis by the greenish ripening cones, with exserted bract scales.

The mtDNA haplotype distribution in populations of three Abies species. a The haplotype network for the three Abies species is shown. b The mtDNA haplotypes in 22 populations of A. firma. c The mtDNA haplotypes in eight populations of A. homolepis including one population of A. homolepis var. umbellata. d The mtDNA haplotypes in 12 populations of A. veitchii. The shaded area is the natural distribution area (Hayashi 1960) of this species
Fresh needles were collected from 804 trees representing 43 populations dispersed widely across the natural distribution of these three Abies species (Table 1). The three species included 419 individuals from 22 populations of A. firma, 153 individuals from nine populations of A. homolepis (including one population of A. homolepis var. umbellata), and 232 individuals from 12 populations of A. veitchii. Among them, 27 populations were the same as in the previous study of Tsumura and Suyama (1998). In each population, the distance between sampled trees was at least 25 m. The collected materials were stored at − 30 ℃ prior to DNA extraction.
Mitochondrial DNA analysis
For mitochondrial DNA analysis, two to 12 samples per population (346 samples in total) were randomly selected (Table 1). We used the modified CTAB method to extract the total DNA from the collected samples (Tsumura et al. 1995). Five mitochondrial DNA regions (nad1; 258 bp, nad5; 274 bp, matR; 211 bp, rps3; 381 bp, and cox1; 266 bp) were amplified using primer pairs (Supplemental Table 1). PCR amplifications were carried out using a Model 9700 GeneAmp PCR system (Applied Biosystems) in reaction mixtures with a total volume of 10 μL, containing 20 mmol/L Tris–HCl (pH 8.0), 50 mmol/L KCl, 2 mmol/L MgCl2, 0.2 mmol/L of each deoxynucleoside triphosphate, 0.2 μmol/L of each primer, 5 ng template DNA, and 1.0 units of Taq polymerase (Promega). The following thermal profile was used: 3-min denaturation at 94 °C, followed by 30 cycles of 45-s denaturation at 94 °C, 30-s annealing at 55–64 °C, and 30-s extension at 72 °C, with a final extension step of 72 °C for 10 min. Amplification products were separated by electrophoresis in 2% (w/v) agarose gels run in 1 × TAE buffer. The gels were then stained with ethidium bromide and visualized under UV light. Each PCR fragment was sequenced using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and ABI Prism 3100 DNA sequencer (Applied Biosystems).
All identified sequences were aligned using Clustal-W (Thompson et al. 1994) and verified by visual inspection using MEGA version 5.0 (Tamura et al. 2011). A matrix of combined sequences was constructed for 346 individuals examined.
Multilocus mtDNA haplotypes were defined by assembling single-locus mtDNA genotypes with DnaSP software (Rozas et al. 2003). The number of haplotypes and their respective mtDNA diversity were then calculated for each population. We calculated nucleotide diversity (π), Tajima’s D (Tajima 1989) using DnaSP. Evolutionary relationships between haplotypes were constructed with minimum-spanning trees estimated from mtDNA sequences using software NETWORK ver.4.2 (Fluxus Engineering, https://www.fluxus-engineering.com/sharenet.htm).
Microsatellite analysis
Genotypes of all collected sample trees were determined using 21 polymorphic microsatellite loci (SSR: Simple Sequence Repeat, Supplemental Table 2): Abfi13, Abfi18 (Saito et al. 2005), As08, As09, As13 (Lian et al. 2007), NFF7 and NFH15 (Hansen et al. 2005), c66, c4656, c14606, c16096, c32446, c49, c7912, c13359, c14033, c14394, c11017, c17556, c28104, and c32410 (Uchiyama et al. 2013). Polymerase chain reaction (PCR) was carried out using a Multiplex PCR Kit (QIAGEN) as followed the protocol of the kit. The fragment analysis of PCR products was conducted using a 3100-Avant Genetic Analyzer (Applied Biosystems), and microsatellite genotypes were scored using Genotyper version 3.7 software (Applied Biosystems). Three loci (Abfi13, NFF7, and c14606) suspected of having null alleles based on MICROCHECKER v2.2.3 (Van Oosterhout et al. 2004) were excluded from further analysis.
To assess genetic diversity within populations, we calculated observed heterozygosity (HO), expected heterozygosity (HE), and fixation index (FIS) to investigate deviation from Hardy–Weinberg equilibrium using FSTAT 2.9.3.2. (Goudet 2001) and allelic richness (AR), private allelic richness (private AR) using HP-RARE software (Kalinowski 2005). A hierarchical analysis of molecular variance (AMOVA) was performed using GenAlex var. 6.5 (Peakall and Smouse 2012) to describe the distribution of genetic variation within populations, between populations within species and between species. We also performed a phylogenetic network analysis of the populations of three Abies species using the neighbor-net approach (Bryant and Moulton 2004). SplitsTree4 (Huson and Bryant 2006) was used to construct the phylogenetic network using the pairwise FST values between populations to understand the evolutionary relationships between populations of the three species. We employed the F-model with a Bayesian clustering approach proposed by Pritchard et al. (2000) to detect population structure and determine the most appropriate number of subpopulations (K) for interpreting the data, without prior information about the number of locations at which the populations were sampled. We performed runs with a burn-in of 300,000, followed by 1,000,000 iterations. The number of K we adopted was where the log likelihood of the data, ln P(X|K) (Pritchard et al. 2000), or ΔK, the rate of change of ln P(X|K) between successive K values (Evanno et al. 2005), was maximal. The Clumpak (Kopelman et al. 2015) and Structure Harvester (Earl and Vonholdt 2012) servers were used to draw the resulting bar plots and calculate the number of K. In order to identify potential F1 or recently introgressed individuals using 18 microsatellite loci, a Bayesian clustering method implemented in NewHybrids software v.1.1 (Anderson and Thompson 2002) was used. We employed the standard 6 genotype categories (Parent1, Parent2, F1, F2, BC1, and BC2). We ran five independent analyses (each using a different random starting value) with 500,000 MCMC after a period of 500,000 burn-in cycles using the Jeffreys-like prior. We assessed convergence by visually reviewing the P(z) values from the various runs, which we averaged over the five runs.
We looked for any recent bottlenecks in population size using a standard differences test (Cornuet and Luikart 1996) and Wilcoxon’s signed rank test in the BOTTLENECK software (Piry et al. 1999) under the assumptions for both the infinite allele mutation model (IAM) and the two-phase model (TPM), under which 70% of the mutations are assumed to occur under the stepwise mutation model and 30% under the infinite allele model.
Species distribution modeling
We predicted the possible distribution of the three Abies species at the LGM (ca. 21 ka) and under current climate conditions using species distribution models, assessing the effects of historical range shifts on the population distributions. The generalized additive model (GAM; Elith et al. 2006) was used to develop species distribution models for the species using the “mgcv” package in R 2.15.0 (R Development Core Team 2012). The following four climatic variables that are important for the distribution of conifers in Japan (Tanaka et al. 2012; Tsuyama et al. 2014, 2015) have been extracted from the WorldClim database (http://www.worldclim.org) at a spatial resolution of 30 s: mean daily minimum temperature of the coldest month (MTCM) measures extreme cold; mean temperature of the warmest quarter (MTWQ) measures effective heat quantity for plant growth; precipitation of the warmest quarter (PRWQ) provides a measure of water supply during the growing season; precipitation of the coldest quarter (PRCQ) is a water supply measure during winter, mainly supplied as snow in mountainous areas. We verified that the variance inflation factor (VIF) values for all explanatory variables were below ten, indicating that these variables did not have significant multicollinearity. We used presence/absence data for Abies species as a response variable and the four climatic variables as explanatory variables. We extracted the current natural distribution records for the species from the Phytosociological Relevé Database (PRDB, Tanaka 2012), which provided the presence/absence data. Duplicate presence/absence records in each grid cell were removed to decrease the effect of spatial autocorrelation. After the thinning, we obtained 1187 presences and 11,144 absences for A. firma, 356 presences and 11,975 absences for A. homolepis, and 211 presences and 12,120 absences for A. veitchii. Model performance was evaluated by calculating the area under the curve (AUC) (Thuiller et al. 2003) based on predicted presence/absence and the validation data bootstrapped from all the training data with 100 repetitions. Relative importance of variables (IOV) values were calculated based on Akaike weight for all the models developed using all combinations of explanatory variables (Burnham and Anderson 2002) in the “MuMIn” package in R. The potential habitats under LGM climate conditions were predicted using the Model for Interdisciplinary Research on Climate (MIROC) version 3.2_hires (K-1 Model Developers 2004) and the Community Climate System Model version 4 (CCSM4, Gent et al. 2011), both global circulation models from the WorldClim dataset with a spatial resolution of 2.5 min. Additionally, the potential habitats were projected for the Mid-Holocene (MH) and Last Interglacial (LIG) climate conditions to assess temporal changes. CCSM4 was used for MH, and CCSM2 was employed for LIG (see Otto-Bliesner et al. 2006 for further details). These data were from the WorldClim dataset with a spatial resolution of 30 s.
Results
Mitochondrial DNA polymorphism in the three Abies species
We found eight nucleotide substitutions and one insertion/deletion in five regions of mtDNA, which total length of the sequences was 1390 bp (Supplemental Table 1): the nad1 region had two nucleotide substitutions and one insertion/deletion, nad5 and rps3 had two substitutions each and the other regions, matR and coxI had only one substitution. A total of 12 haplotypes were detected.
The haplotype diversity was higher in A. firma and A. homolepis including the putative hybrid species A. homolepis var. umbellata but very low in A. veitchii (Table 2), and the nucleotide diversity was also higher in the former two species but very low in A. veitchii. The result of Tajima’s D test was that A. firma had the highest value, 1.1990 and A. veitchii the lowest, − 1.2132, but the difference was not statistically significant.
The haplotypes present were similar in A. firma and A. homolepis (Table 2 and Supplemental Table 4). Of the seven haplotypes detected in A. firma and A. homolepis, six were common to the two species. Unique haplotypes were also detected in each species, namely, haplotypes VIII (fYKS) and XII (fMTE) in A. firma, V (hYTG) in A. homolepis, IX (uMTM) in A. homolepis var. umbellata, and X (vIKW, vONT, vKSK) and XI (vNIK) in A. veitchii.
Genetic variation between species was low, GPT = 0.0761 and was also very low within populations, HP/HT = 0.0485; most of the variation was between populations of species, GST = 0.8754 (Table 3).
Mitochondrial DNA haplotype distribution in the three Abies species
Abies firma has seven haplotypes, of which the haplotype I was the most common accounting for 43.8% of individuals in this species, followed by haplotype II (17.7%), III (15.3%) and IV (10.3%) (Supplemental Table 4). The proportion of the remaining four haplotypes was less than 5%. Haplotype I was dominant in the western part of Japan, haplotype II was present on the Pacific Ocean side of central Japan, haplotype III was found on the Sea of Japan side of western Japan, and haplotype IV appeared in most northern area of the species’ natural distribution (Fig. 1). The minor haplotypes were distributed mainly on the Sea of Japan side of western Japan. The two most northern populations shared the same haplotype as the dominant haplotype of A. veitchii. Three populations—fTKB, fKYS, and fDND—in central Japan shared the same haplotype as the dominant haplotype of A. homolepis.
Abies homolepis has six haplotypes, of which haplotype II was dominant, accounting for 55.7% of individuals in this species, followed by haplotype I, which is the dominant type of A. firma. The remaining five haplotypes were present in less than 10% of individuals. The major haplotype, II, was found in central Japan, the main natural distribution area of this species (Fig. 1). The second major haplotype, I, was only found in the two isolated western populations—hODG and hISZ—and this is the major haplotype of A. firma.
Abies veitchii has only three haplotypes, of which haplotype IV accounted for 88.2% of individuals; the other two haplotypes were found in five populations (Fig. 1). Population vISZ has a unique haplotype V for this species, although it is also present in population hISZ of A. homolepis and the fHNZ and fONZ populations of A. firma.
SSR genetic diversity within the three Abies species
In A. firma, the allelic richness (Ar) in the populations ranged from 1.640 (fHNZ) to 1.920 (fDND), with an average of 1.751. The private allelic richness (private Ar) was 0.010 (fHNZ, fYKA, and fHTK) to 0.060 (fMTE, fDND), with an average of 0.030 using 16 polymorphic loci (Supplementary Fig. 2, Table 1). The expected heterozygosity (He) was 0.211 (fOKB) to 0.318 (fDND), with an average of 0.258.
In A. homolepis, the allelic richness (Ar) in the populations ranged from 1.750 (hYTG, hCUS) to 1.840 (hNIK, hISZ), with an average of 1.794. The private allelic richness (private Ar) was 0.050 (hCUS) to 0.120 (hFUJ), with an average of 0.091 using 15 polymorphic loci (Supplementary Fig. 3, Table 1). The expected heterozygosity (He) was 0.239 (hYTG) to 0.272 (hMKN), with an average of 0.257.
In A. veitchii, the allelic richness (Ar) in the populations ranged from 1.560 (vAZM) to 1.900 (vDIB and vNIK), with an average of 1.791. The private allelic richness (private Ar) was 0.030 (vMSN) to 0.080 (vONT), with an average of 0.054 using 18 polymorphic loci (Supplementary Fig. 4, Table 1). The expected heterozygosity (He) was 0.202 (vAZM) to 0.293 (vNIK), with an average of 0.265.
No significant deviations from HWE were detected in any of the populations in three species. (FIS, P < 0.05).
Genetic structure within and between the three Abies species in nuclear DNA
Genetic differentiation between species was FST = 0.252 and GST = 0.257. The result of the hierarchical analysis of molecular variation (AMOVA) showed that 39.44% (P < 0.01) of the variation was between species, 5.55% (P < 0.01) was between populations within species, and 55.01% (P < 0.01) was within populations of species (Supplemental Table 5).
Bayesian clustering using 43 populations of the three Abies species demonstrated that the model with K = 3 provided satisfactory explanations of the observed data, as judged by the delta K value (Fig. 2). Clusters I, II, and III of the STRUCTURE analysis were A. firma, A. homolepis, and A. veitchii, respectively, and their F values were 0.1730, 0.1433, and 0.2030, respectively. The neighbor-net tree constructed based on the pairwise FST values between 43 populations of the three species showed that the species were clearly separated (Fig. 3). Both results of STRUCTURE and the neighbor-net tree indicated that population uMTM of A. homolepis var. umbellata was between A. firma and A. homolepis.

Results of STRUCTURE analysis of three Abies species using SSR data. a STRUCTURE results for 42 populations of the three Abies species, including A. homolepis var. umbellata, which is of hybrid origin. The estimated optimal number of clusters is three based on ln P(X/K) and ΔK. b Results of the STRUCTURE analysis for A. firma, where the estimated optimal number of clusters is two based on ln P(X/K) and four based on ΔK. c Results of the STRUCTURE analysis for A. homolepis, where the estimated optimal number of clusters is four based on ln P(X/K) and ΔK. d Results of the STRUCTURE analysis for A. veitchii, where the estimated optimal number of clusters is seven based on ln P(X/K) and ΔK

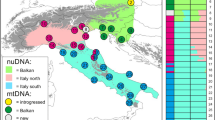
A population network based on pair-wise FST values between populations of the three Abies species constructed by SplitsTree4 (Huson and Bryant 2006), utilizing nuclear SSR data. The size of pie charts is proportional to population genetic diversity (He: heterozygosity). Color of pie chart indicates the mtDNA haplotype in each population
Genetic differentiation between populations in A. firma was low (FST = 0.051) but isolation by distance was observed (R2 = 0.264, P < 0.001, Supplemental Fig. 1). The genetic relationship between 21 populations in the neighbor-net tree for A. firma roughly corresponded to their geographical locations (Fig. 3). In STRUCTURE analysis, K = 2 and 4 were most appropriate when assessed on the basis of ln P(X|K) and ΔK, respectively. Cluster II (F = 0.1581) was dominant in western Japan, and frequencies changed gradually along the natural distribution (Fig. 2). The Wilcoxon test indicated that population fHTK experienced a bottleneck (P < 0.05).
Genetic differentiation between populations in A. homolepis was also low (FST = 0.044), and no apparent isolation by distance was observed. STRUCTURE analysis indicated K = 4 to be the most appropriate value on the basis of both ln P(X|K) and ΔK (Fig. 2). Unique clusters were detected in three populations (hCUS, hODG, and hISZ) in western Japan. There was no evidence of bottlenecks in any population.
Genetic differentiation between populations in A. veitchii was a little higher (FST = 0.071) than for the other two species. Isolation by distance was clearly observed (R2 = 0.5607, P < 0.001), and the genetic relationship between populations in the neighbor-net tree roughly reflected their geographical locations, in which two peripheral populations—vAZM and vISZ—had relatively long branches, while populations around Chubu district were close to each other. Structure analysis indicated K = 7 to be the most appropriate number based on both ln P(X|K) and ΔK (Fig. 2). First, two southern populations (vMSM and vISZ) were separated at K = 2, then a cluster represented by the northern population (vAZM) was detected at K = 3. At the optimal K = 7, these three populations were identified as distinct and unique clusters. There was no evidence of bottlenecks in any population.
The results of the NewHybrids analysis indicated that most of the individuals of A. homolepis var. umbellata were derived from hybrids between A. firma and A. homolepis (F2 or later) (Fig. 4a). This is consistent with the results of the STRUCTURE analysis. F2 hybrids for this combination were detected in populations at the edge of the distribution (fKMG) and in an isolated population in Shikoku (fISZ) as shown in Table 1. A small number of individuals classified as F2 were also detected within each species, providing evidence of hybridization between these species. For the A. firma × A. veitchii combination, hybrids were only detected in A. veitchii populations (vKSK, vODR, vDIB, vFUJ, and vMIS). The location vKSK showed evidence of hybridization with both A. firma and A. homolepis. However, only two individuals were detected as F1, both in A. veitchii (specifically in A. homolepis × A. veitchii crosses), each from different locations (vKSK, vODR) (Fig. 4b). This information is provided in Table 1, where we have highlighted the populations where hybrids were detected.

Hybridization plot obtained using the software NewHybrids between a A. firma and A. homolepis; b A. homolepis and A. veitchii; c A. firma and A. veitchii. F1 = first-generation hybrid, F2 = offspring of two F1 parents, BC1 = back-cross of F1 hybrid to parent species 1, BC2 = back-cross of F1 hybrid to parent species 2
Species distribution modeling
The mean and standard deviation of the AUC in the GAM for A. firma, A. homolepis, and A. veitchii were 0.84 ± 0.005, 0.96 ± 0.002, and 0.97 ± 0.002, respectively, which indicates reasonable prediction accuracy (Swets 1988).
IOV values showed that all the explanatory variables had a value of almost 1.00 for A. firma and A. homolepis. As for A. veitchii, MTCM was associated with lower IOV values (0.46) than other variables (PRWQ, PRCQ, and MTWQ, 1.00). Presence/absence data showed that A. firma grows in areas with warmer winter and summer (median and range of MTCM for 95% of the presence records were − 3.1 and − 9.0 to − 1.2 ℃; MTWQ: 22.2 and 18.3 to 25.1 ℃) than the other two species (A. homolepis, MTCQ: − 9.7 and − 13.7 to − 4.4 ℃, MTWQ: 17.7 and 13.5 to 20.8 ℃; A. veitchii, MTCQ: − 12.8 and − 15.3 to − 6.6 ℃, MTWQ: 14.4 and 11.5 and 18.4 ℃). A. veitchii grows in areas with wetter summers (PRWQ: 875 and 680 to 1001 mm) than the other two species (A. homolepis, 800 and 589 to 1016 mm; A. firma, 744 and 442 to 1342 mm). The growth conditions associated with winter precipitation for the three species were similar, although A. firma is present under a wider range of conditions than the other two species (A. firma, 228 and 122 to 488 mm; A. homolepis, 208 and 146 to 266 mm; A. veitchii, 220 and 167 and 268 mm).
Predicted potential habitat (PH) for the three Abies species under the current climate was mostly consistent with actual distributions, except some areas including northern Tohoku and southern coastal areas of Hokkaido for A. firma, northeastern areas of Tohoku for A. homolepis and A. veitchii (Fig. 5). In these areas, the species are not present despite climatic conditions being suitable for them (empty habitat, Tsuyama et al. 2014). Under the MH, LGM, and LIG climates, it was predicted that the PH for A. firma would have retreated southwards and occupied lower elevation areas (Fig. 5). It was predicted that the PH for A. homolepis would have moved to lower elevation areas and expanded to southern areas under the LGM climate. It was predicted that the PH for A. veitchii under the LGM climate would have expanded to lower elevation areas, although locations would have been mostly similar to the current PH range. The northern range limits of A. firma and A. veithcii were almost consistent with current actual distributions.

The overlap of potential habitat between two species, a A. firma and A. homolepis, b A. homolepis and A. veitchii, and c A. firma and A. veitchii, under climatic conditions of four periods: the first row shows under the current, the second under the MH, the third under the LGM, and the bottom under the LIG. The abbreviation PH in the legend shows potential habitat
Discussion
Distribution shift and overlap in the range of Abies species between the LGM and interglacial period
The natural distribution of the three Abies species retreated southward and to lower elevations during the LGM because of the cooler temperatures and lower precipitation compared with the present. The distribution of these Abies species overlapped not only during the LGM but continues to do so (Fig. 5). The area of overlap was much larger, and individuals would have been present at a higher density during the LGM, especially A. firma with A. homolepis, and A. homolepis with A. veitchii. However, there was little overlap between A. firma and A. veitchii predicted by this model. During the LGM, the climate was cooler than at present, and thus, flowering phenology between species would have been much closer, meaning that hybridization between species would have been more likely to occur, especially in marginal populations. In Quercus species, hybridization frequently occurs in marginal populations (Matsumoto et al. 2009; Nagamitsu et al. 2020). Abies firma and A. homolepis sometimes form hybrids in the contact zone around 1000–1100 m elevation, which is the upper distribution limit of A. firma in the mountain regions of Honshu (Aizawa and Iwaizumi 2020).
Mitochondrial DNA polymorphisms and their relationship between species
Some haplotypes are shared between species because of ancient introgression or shared polymorphism before speciation (incomplete lineage sorting). Examples of such processes can be found in Tasmanian eucalypts (McKinnon et al. 2004b). Bidirectional organelle capture has occurred between A. firma and A. homolepis, between A. firma and A. veitchii, and between A. homolepis and A. veitchii. The direction of organelle capture was from the species with the major distribution to the one with a marginal distribution. Mitochondrial DNA is generally inherited maternally in Pinaceae (Neale and Sederoff 1988; Mogensen 1996), and it is likely that hybridization between species occurred during the glacial period when the distributions of pairs of species were much closer. Thereafter, during the interglacial, hybrids are likely to have formed repeatedly with the dominant species for many generations, so that only mitochondrial DNA remained from the original species. Organelle capture in plants is a common phenomenon associated with introgression between species (Rieseberg and Soltis 1991). In the case of A. firma, the three most northern populations have the main mtDNA haplotype of A. veitchii, and the southern three populations have the main mtDNA haplotype of A. homolepis. However, the distribution overlaps little between A. firma and A. veitchii according to the SDM results, and thus, mtDNA haplotype IV in A. firma in the northern populations may have originated through crossing with a hybrid between A. veitchii and A. homolepis. Additionally, extinct Abies populations could have left their mitochondrial haplotypes in extant species. For instance, A. veitchii was found in the lowland Kanto region during the LGM (Momohara et al. 2016), where A. firma now harbors a mitotype most common in A. veitchii. The mtDNA haplotype VI is found in populations of A. firma in the Chugoku district and populations of A. homolepis and A. veitchii on Shikoku Island; they are genetically very close to the main mtDNA haplotype IV of A. veitchii. However, this haplotype is found only in western Japan, like haplotypes III and VIII, and thus, probably the same process of mtDNA acquisition may have occurred.
In addition to our studies on Japanese Abies species, other research has also reported similar findings. For example, Semerikova and Semerikov (2014) examined mitochondrial DNA (mtDNA) variation and reticulate evolution of the Abies genus, employing three regions of mtDNA, and found a correlation between species’ geographical distribution and assignment to a particular mtDNA haplotype group. These findings suggest that past migrations and species hybridizations have led to the capture of alien mtDNA, thereby resulting in incongruence between mtDNA and chloroplast DNA (cpDNA) phylogenies. Another study by Semerikova et al. (2011) on Abies species from the Russian Far East provided evidence of post-glacial history and introgression in Abies. They demonstrated that coastal Abies populations suffered less from climatic fluctuations during Pleistocene glacial periods than their more continental counterparts and that Sakhalin was a major area of introgression. Shao and Xiang (2015) combined morphological characters and mtDNA and cpDNA sequence data to infer species delimitation and phylogeography of the Abies chensiensis complex. They found that cpDNA sequence variation aligned with the morphological results, while mtDNA genealogy was more strongly related to geographical distribution. These studies collectively indicate that the patterns of shared mtDNA haplotypes and complex genetic interactions observed in Japanese Abies species are not unique but are common across the genus. This pattern may result from similar historical and geographical factors such as past climatic events, species migration, and hybridization, which influence the genetic diversity and distribution of Abies species.
On the other hand, it is critical to note that introgression is not the only mechanism that could explain the observed pattern of shared mtDNA haplotypes. It is quite plausible that ancient polymorphisms also played a significant role. In this scenario, shared haplotypes could have been maintained in these species from a common ancestor, rather than being transferred between species through hybridization events. Importantly, the potential underestimation of past Abies species distributions by SDMs might further underscore the role of ancient polymorphisms. This underestimation is suggested by the fossil record, such as the findings that A. veitchii was found in the lowland Kanto region during the Last Glacial Maximum (LGM), where A. firma now harbors a mitotype most common in A. veitchii (Momohara et al. 2016). While SDMs provide valuable insights into the potential distribution of species, they are reliant on current and historical climate data and may not fully capture the complex demographic history and geographical distributions of past populations. Thus, both introgression and ancient polymorphisms should be considered as contributing factors to the patterns of shared mtDNA haplotypes in Japanese Abies species.
Nuclear DNA diversity within and between populations and genetic structure in the three Abies species
The observed heterozygosity (HO) and expected heterozygosity (HE) values for the three Abies species in this study are lower compared to other gymnosperms in Japan such as Pinus densiflora (HE: 0.863; Iwaizumi et al. 2013), Larix kaempferi (HE: 0.713; San Jose-Maldia 2010), and Cryptomeria japonica (HE: 0.628; Kimura et al. 2014).
A similar level of genetic diversity is seen in another Abies species, Abies sachalinensis (HE: 0.398; Kitamura et al. 2020), although its distribution is limited to Hokkaido. In contrast, species such as Pinus pumila (HS: 0.225; Tani et al. 1996) and Pinus parviflora (HS: 0.259; Tani et al. 2003) show even lower levels of genetic diversity. However, these examples, derived from allozyme data, represent species with patchy distributions. Therefore, the relatively low genetic diversity found in the three Abies species in our study compared to other conifers in Japan suggests that genetic drift may have had a significant impact on these Abies species.
Generally, genetic diversity is higher in central populations and gradually decreases in the marginal populations. In our data, there is no clear pattern of genetic diversity; this may be influenced by historical hybridization and past introgression between species. Populations with mtDNA haplotypes of other species tend to exhibit higher genetic diversity; this is especially the case for the fKYS and fDND populations in A. firma and the uMTM population in A. homolepis var. umbellata. A similar result has been reported in a study of the population genetics of Pinus pumila (Tani et al. 1998), which showed a clear geographical cline in genetic diversity, although only two populations had significantly higher genetic diversity compared to the other populations. DNA analysis revealed that these populations comprised hybrids between P. pumila and P. parvifolia (Senjo et al. 1999; Watano et al. 2004). However, in the current study, even in populations with the mtDNA haplotype of the other species, marginal populations showed relatively low genetic diversity, for example, hODG and hISZ in A. homolepis and vISZ in A. veitchii.
Genetic differentiation in the nuclear genome between populations was relatively limited, despite their discontinuous natural distribution, even in populations of A. veitchii (FST = 0.069) and A. homolepis (FST = 0.043) that are relatively isolated in the higher mountains. Abies firma is found at relatively lower elevations, but its distribution area is much wider and probably dispersed population distribution than the other two Abies species, even so its genetic differentiation was also relatively low, FST = 0.045. The altitudinal distribution of A. homolepis is similar to that of Fagus crenata and Picea jezoensis (including three varieties) in central Japan, and the genetic differentiation between populations of these species is also low, 0.027 and 0.101, respectively (Aizawa et al. 2009; Hiraoka and Tomaru 2009). In the case of Pinus pumila and A. mariesii, which are found in the much higher alpine zone in central Japan, the genetic differentiation between populations is also much higher, GST = 0.170 and 0.144, respectively (Tani et al. 1996; Suyama et al. 1997). Generally, the genetic differentiation value is related to pollinators, the mating system, the distribution pattern, population history, and other factors. These species, including Abies species, are allogamous and wind pollinated, and thus, genetic differentiation is relatively low, regardless of their different altitudinal distribution.
A gentle geographical cline was detected in A. firma and A. veitchii but there is no clear cline in A. homolepis according to the nuclear DNA markers. Abies firma has a wider and lower altitudinal distribution area and thus gene flow between populations may be frequent, resulting in this gentle geographical cline. Abies veitchii is found at the highest elevations of the three Abies species, which means that each population was isolated in higher mountains during the interglacial periods; thus, a relatively clear geographical cline was established, especially in peripheral populations. Abies homolepis has an intermediate altitudinal distribution among the three Abies species, and thus, isolation and gene flow between populations have also been intermediate; its range is not as broad as A. firma, and thus, a clear geographical cline is not present in current populations.
The STRUCTURE results revealed that the three species are clearly separated, but some individuals seem share some of their genome (Fig. 2), and the populations with an mtDNA haplotype of another species have relatively higher genetic diversity. The neighbor-net showed that populations with a hybrid origin in A. firma are more closely related to populations of A. homolepis (Fig. 3). NewHybrids results showed that there are some hybrids not only F1 but also F2 and the later generations (Fig. 4). These results suggest that historical hybridization occurred during glacial periods when species distributions were much closer.
For the A. firma × A. veitchii combination, hybrids were only detected in A. veitchii populations. This suggests that there might be directional preferences for hybridization. Moreover, the location vKSK showed evidence of hybridization with both A. firma and A. homolepis, suggesting that it has favorable conditions for hybridization. However, only two individuals were detected as F1, both in A. veitchii, specifically in the A. homolepis × A. veitchii combination. This scarcity of F1s suggests that the frequency of recent hybridization events is low, or that F1 hybrids have low survival rates.
Organelle capture (organelle transfer) through introgression is a frequent phenomenon in tree species, both angiosperms and gymnosperms (Whittemore and Schaal 1991; Tsumura and Suyama 1998; Senjo et al. 1999; King and Ferris 2000; Palmé et al. 2004). In this study, we were able to confirm that the occurrence of organelle capture through introgression in the three Japanese Abies species was closely related to the distribution of these species during the LGM. In the higher latitude and/or altitude populations, especially the peripheral populations, organelle capture was likely when the species had close or overlapping distributions.
The results of network analysis, STRUCTURE analysis, NewHybrids analysis, and SDM supported the past introgression events. To gain a deeper understanding of these past events, we suggest that testing various demographic models could be a valuable approach. Furthermore, to clarify what underlies the patterns of genetic sharing between the three Abies species, we recognize the necessity for further genome-wide analysis using next-generation sequencing (NGS). Both genome-wide NGS analysis and demographic models such as those incorporating Approximate Bayesian Computation (ABC) would provide crucial insights to better distinguish between ancient lineage sorting and more recent hybridization, as discussed by Twyford and Ennos (2012).
Genetic conservation of the three Japanese Abies species
The three Abies species are not used in forestry, but are important in the temperate forest ecosystem. In this study, we identified important A. firma populations in Chugoku district, which have higher mtDNA diversity and probably represent a refugial population during the ice age. We also detected populations derived from an original ancient hybridization between different Abies species. In A. firma, we can recognize five genetic groups based on the mtDNA haplotypes: two northern populations (A. veitchii mtDNA type), three eastern populations (A. homolepis mtDNA type), populations in Chugoku district (higher mtDNA diversity), the Yakushima Island population, and others. In A. homolepis, there are two genetic groups: populations in central Japan and the Kii Peninsular-Shikoku Island group (A. firma mtDNA type). In A. veitchii, there are two genetic groups: the Shikoku Island population and the others. These genetic groups are recognized as different genetic lineages and thus should be conserved.
Data availability
All genetic data were deposited to the DNA database of DDBJ and Dryad.
References
Abbott R, Albach D, Ansell S, Arntzen JW, Baird SJE, Bierne N, Boughman JW, Brelsford A, Buerkle CA, Buggs R, Butlin RK, Dieckmann U, Eroukhmanoff F, Grill A, Cahan SH, Hermansen JS, Hewitt G, Hudson AG, Jiggins C, Jones J, Keller B, Marczewski T, Mallet J, Martinez-Rodriguez P, Most M, Mullen S, Nichols R, Nolte AW, Parisod C, Pfennig K, Rice AM, Ritchie MG, Seifert B, Smadja CM, Stelkens R, Szymura JM, Vainola R, Wolf JBW, Zinner D (2013) Hybridization and speciation. J Evol Biol 26(2):229–246. https://doi.org/10.1111/j.1420-9101.2012.02599.x
Aizawa M, Iwaizumi MG (2020) Natural hybridization and introgression of Abies firma and Abies homolepis along the altitudinal gradient and genetic insights into the origin of Abies umbellata. Plant Species Biol 35(2):147–157. https://doi.org/10.1111/1442-1984.12269
Aizawa M, Yoshimaru H, Saito H, Katsuki T, Kawahara T, Kitamura K, Shi F, Sabirov R, Kaji M (2009) Range-wide genetic structure in a north-east Asian spruce (Picea jezoensis) determined using nuclear microsatellite markers. J Biogeogr 36(5):996–1007. https://doi.org/10.1111/j.1365-2699.2008.02074.x
Anderson EC, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160(3):1217–1229
Bryant D, Moulton V (2004) Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21(2):255–265. https://doi.org/10.1093/molbev/msh018
Burnham KP, Anderson DR (2002) Model selection and multimodel inference : a practical information-theoretic approach, 2nd edn. Springer, New York
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144(4):2001–2014. https://doi.org/10.1093/genetics/144.4.2001
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
Elith J, Graham CH, Anderson RP, Dudik M, Ferrier S, Guisan A, Hijmans RJ, Huettmann F, Leathwick JR, Lehmann A, Li J, Lohmann LG, Loiselle BA, Manion G, Moritz C, Nakamura M, Nakazawa Y, Overton JM, Peterson AT, Phillips SJ, Richardson K, Scachetti-Pereira R, Schapire RE, Soberon J, Williams S, Wisz MS, Zimmermann NE (2006) Novel methods improve prediction of species’ distributions from occurrence data. Ecography 29(2):129–151. https://doi.org/10.1111/j.2006.0906-7590.04596.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Farjon A, Rushforth K (1988) A classification of Abies miller (Pinaceae). Misc Publ Univ Utrecht Herb 3(1):59–79
Feliner GN (2014) Patterns and processes in plant phylogeography in the Mediterranean Basin. A review. Perspect Plant Ecol 16(5):265–278. https://doi.org/10.1016/j.ppees.2014.07.002
Gent PR, Danabasoglu G, Donner LJ, Holland MM, Hunke EC, Jayne SR, Lawrence DM, Neale RB, Rasch PJ, Vertenstein M, Worley PH, Yang ZL, Zhang MH (2011) The community climate system model version 4. J Clim 24(19):4973–4991. https://doi.org/10.1175/2011jcli4083.1
Goudet J (2001) FSTAT; a program to estimate and test gene diversities and fixation indices. Accessed version 2.9.3, current version 2.9.4 available at https://www2.unil.ch/popgen/softwares/fstat.htm. Accessed 5 Sept 2023
Hansen OK, Vendramin G, Sebastiani F, Edwards K (2005) Development of microsatellite markers in Abies nordmanniana (Stev.) Spach and cross-species amplification in the Abies genus. Mol Ecol Notes 5(4):784–787. https://doi.org/10.1111/j.1471-8286.2005.01062.x
Hayashi Y (1960) Taxonomical and phytogeographical study of Japanese conifers. Norin-Shuppan, Tokyo, 246 pp
Hiraoka K, Tomaru N (2009) Genetic divergence in nuclear genomes between populations of Fagus crenata along the Japan Sea and Pacific sides of Japan. J Plant Res 122(3):269–282. https://doi.org/10.1007/s10265-009-0217-9
Hofreiter M, Stewart J (2009) Ecological change, range fluctuations and population dynamics during the Pleistocene. Curr Biol 19(14):R584–R594. https://doi.org/10.1016/j.cub.2009.06.030
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23(2):254–267. https://doi.org/10.1093/molbev/msj030
Isoda K, Shiraishi S, Watanabe S, Kitamura K (2000) Molecular evidence of natural hybridization between Abies veitchii and A. homolepis (Pinaceae) revealed by chloroplast, mitochondrial and nuclear DNA markers. Mol Ecol 9(12):1965–1974
Iwaizumi MG, Tsuda Y, Ohtani M, Tsumura Y, Takahashi M (2013) Recent distribution changes affect geographic clines in genetic diversity and structure of Pinus densiflora natural populations in Japan. For Ecol Manage 304:407–416. https://doi.org/10.1016/j.foreco.2013.05.026
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5(1):187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kimura MK, Uchiyama K, Nakao K, Moriguchi Y, San Jose-Maldia L, Tsumura Y (2014) Evidence for cryptic northern refugia in the last glacial period in Cryptomeria japonica. Ann Bot 114:1687–1700
King RA, Ferris C (2000) Chloroplast DNA and nuclear DNA variation in the sympatric alder species, Alnus cordata (Lois.) Duby and A. glutinosa (L.) Gaertn. Biol J Linn Soc 70(1):147–160. https://doi.org/10.1111/j.1095-8312.2000.tb00205.x
Kitamura K, Uchiyama K, Ueno S, Ishizuka W, Tsuyama I, Goto S (2020) Geographical gradients of genetic diversity and differentiation among the southernmost marginal populations of Abies sachalinensis revealed by EST-SSR polymorphism. Forests 11(2):233
Knowles LL (2001) Did the Pleistocene glaciations promote divergence? Tests of explicit refugial models in montane grasshopprers. Mol Ecol 10(3):691–701. https://doi.org/10.1046/j.1365-294x.2001.01206.x
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15(5):1179–1191. https://doi.org/10.1111/1755-0998.12387
Lian C, Goto S, Hogetsu T (2007) Microsatellite markers for Sachalin fir (Abies sachalinensis Masters). Mol Ecol Notes 7(5):896–898
Liu T-S (1971) A monograph of the genus Abies. National Taiwan University, Taipei
Matsumoto A, Kawahara T, Kanazashi A, Yoshimaru H, Takahashi M, Tsumura Y (2009) Differentiation of three closely related Japanese oak species and detection of interspecific hybrids using AFLP markers. Botany 87(2):145–153. https://doi.org/10.1139/B08-121
Mayr H (1890) Monographie der Abirtineen des japanischen Reiches (Tannen, Fichten, Tsugen, Larchen und Kiefern). Gustav Himmer, Munich, Germany
McKinnon GE, Jordan GJ, Vaillancourt RE, Steane DA, Potts BM (2004a) Glacial refugia and reticulate evolution: the case of the Tasmanian eucalypts. Philos T R Soc B 359(1442):275–284. https://doi.org/10.1098/rstb.2003.1391
McKinnon GE, Vaillancourt RE, Steane DA, Potts BM (2004b) The rare silver gum, Eucalyptus cordata, is leaving its trace in the organellar gene pool of Eucalyptus globulus. Mol Ecol 13(12):3751–3762. https://doi.org/10.1111/j.1365-294X.2004.02364.x
Mogensen HL (1996) The hows and whys of cytoplasmic inheritance in seed plants. Am J Bot 83(3):383–404. https://doi.org/10.2307/2446172
Momohara A, Yoshida A, Kudo Y, Nishiuchi R, Okitsu S (2016) Paleovegetation and climatic conditions in a refugium of temperate plants in central Japan in the Last Glacial Maximum. Quatern Int 425:38–48. https://doi.org/10.1016/j.quaint.2016.07.001
Nagamitsu T, Uchiyama K, Izuno A, Shimizu H, Nakanishi A (2020) Environment-dependent introgression from Quercus dentata to a coastal ecotype of Quercus mongolica var. crispula in northern Japan. New Phytol 226(4):1018–1028. https://doi.org/10.1111/nph.16131
Neale DB, Sederoff RR (1988) Inheritance and evolution of conifer organelle genomes. In: Hanover J, Kethley D (eds) Genetic manipulation of woody plants. Springer, pp 251–264
Otto-Bliesner BL, Marsha SJ, Overpeck JT, Miller GH, Hu AX, Mem CLIP (2006) Simulating arctic climate warmth and icefield retreat in the last interglaciation. Science 311(5768):1751–1753. https://doi.org/10.1126/science.1120808
Palmé AE, Su Q, Palsson S, Lascoux M (2004) Extensive sharing of chloroplast haplotypes among European birches indicates hybridization among Betula pendula, B. pubescens and B. nana. Mol Ecol 13(1):167–178. https://doi.org/10.1046/j.1365-294X.2003.02034.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research - an update. Bioinformatics 28(19):2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Petit RJ, Pineau E, Demesure B, Bacilieri R, Ducousso A, Kremer A (1997) Chloroplast DNA footprints of postglacial recolonization by oaks. Proc Natl Acad Sci U S A 94(18):9996–10001. https://doi.org/10.1073/pnas.94.18.9996
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90(4):502–503. https://doi.org/10.1093/jhered/90.4.502
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. https://doi.org/10.1093/genetics/155.2.945
R Development Core Team (2012) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rieseberg LH, Soltis D (1991) Phylogenetic consequences of cytoplasmic gene flow in plants. Evolutionary Trends in Plants 5(1):5–84
Rozas J, S nchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19(18):2496–2497. https://doi.org/10.1093/bioinformatics/btg359
Saito Y, Lian CL, Hogetsu T, Ide Y (2005) Development and characterization of microsatellite markers in Abies firma and interspecific amplification in other Japanese Abies species. Mol Ecol Notes 5(2):234–235. https://doi.org/10.1111/j.1471-8286.2005.00887.x
San Jose-Maldia L (2010) Evaluation of genetic diversity in natural populations of Japanese larch for its conservation and breeding. Ph. D. Dissertation, Nagoya University
Semerikova S, Semerikov V (2014) Mitochondrial DNA variation and reticulate evolution of the genus Abies. Russ J Genet 50:366–377
Semerikova SA, Semerikov VL, Lascoux M (2011) Post-glacial history and introgression in Abies (Pinaceae) species of the Russian Far East inferred from both nuclear and cytoplasmic markers. J Biogeogr 38(2):326–340
Semerikova SA, Khrunyk YY, Lascoux M, Semerikov VL (2018) From America to Eurasia: a multigenomes history of the genus Abies. Mol Phylogenet Evol 125:14–28
Senjo M, Kimura K, Watano Y, Ueda K, Shimizu T (1999) Extensive mitochondrial introgression from Pinus pumila to P. parviflora var. Pentaphylla (Pinaceae). J Plant Res 112(1105):97–105. https://doi.org/10.1007/Pl00013867
Shao Y-Z, Xiang Q-P (2015) Species delimitation and phylogeography of the Abies chensiensis complex inferred from morphological and molecular data. Bot J Linn Soc 177(2):175–188
Suyama Y, Tsumura Y, Ohba K (1997) A cline of allozyme variation in Abies mariesii. J Plant Res 110(1098):219–226. https://doi.org/10.1007/Bf02509310
Suyama Y, Yoshimaru H, Tsumura Y (2000) Molecular phylogenetic position of Japanese Abies (Pinaceae) based on chloroplast DNA sequences. Mol Phylogenet Evol 16(2):271–277. https://doi.org/10.1006/mpev.2000.0795
Swets JA (1988) Measuring the accuracy of diagnostic systems. Science 240(4857):1285–1293. https://doi.org/10.1126/science.3287615
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595. https://doi.org/10.1093/genetics/123.3.585
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. https://doi.org/10.1093/molbev/msr121
Tanaka N, Nakao K, Tsuyama I, Higa M, Nakazono E, Matsui T (2012) Predicting the impact of climate change on potential habitats of fir (Abies) species in Japan and on the East Asian continent. Procedia Environ Sci 13:455–466
Tanaka N (2012) Phytosociological Relevé Database of Japan (PRDB). In: Dengler J, Oldeland J, Jansen F et al. (eds) Vegetation databases for the 21st century, Biodivers Ecol 4:308–308. https://doi.org/10.7809/b-e.00104
Tani N, Tomaru N, Araki M, Ohba K (1996) Genetic diversity and differentiation in populations of Japanese stone pine (Pinus pumila) in Japan. Can J for Res 26(8):1454–1462. https://doi.org/10.1139/x26-162
Tani N, Tomaru N, Tsumura Y, Araki M, Ohba K (1998) Genetic structure within a Japanese stone pine (Pinus pumila Regel) population on Mt. Aino-Dake in Central Honshu, Japan. J Plant Res 111(1101):7–15. https://doi.org/10.1007/Bf02507145
Tani N, Maruyama K, Tomaru N, Uchida K, Araki M, Tsumura Y, Yoshimaru H, Ohba K (2003) Genetic diversity of nuclear and mitochondrial genomes in Pinus parviflora Sieb. & Zucc.(Pinaceae) populations. Heredity 91(5):510–518
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680. https://doi.org/10.1093/nar/22.22.4673
Thuiller W, Araujo MB, Lavorel S (2003) Generalized models vs. classification tree analysis: predicting spatial distributions of plant species at different scales. J Veg Sci 14(5):669–680. https://doi.org/10.1111/j.1654-1103.2003.tb02199.x
Tsumura Y, Suyama Y (1998) Differentiation of mitochondrial DNA polymophisms in populations of five Japanese Abies species. Evolution 52(4):1031–1042. https://doi.org/10.1111/j.1558-5646.1998.tb01831.x
Tsumura Y, Yoshimura K, Tomaru N, Ohba K (1995) Molecular phylogeny of conifers using RFLP analysis of PCR-amplified specific chloroplast genes. Theor Appl Genet 91(8):1222–1236. https://doi.org/10.1007/BF00220933
Tsuyama I, Nakao K, Higa M, Matsui T, Shichi K, Tanaka N (2014) What controls the distribution of the Japanese endemic hemlock, Tsuga diversifolia? Footprint of climate in the glacial period on current habitat occupancy. J For Res 19(1):154–165. https://doi.org/10.1007/s10310-013-0399-9
Tsuyama I, Higa M, Nakao K, Matsui T, Horikawa M, Tanaka N (2015) How will subalpine conifer distributions be affected by climate change? Impact assessment for spatial conservation planning. Reg Environ Change 15(2):393–404
Twyford AD, Ennos RA (2012) Next-generation hybridization and introgression. Heredity 108(3):179–189. https://doi.org/10.1038/hdy.2011.68
Uchiyama K, Fujii S, Ishizuka W, Susumu G, Tsumura Y (2013) Development of 32 EST-SSR markers for Abies firma (Pinaceae) and their transferability to related species. Appl Plant Sci 1(2):1200464. https://doi.org/10.3732/ajb.1100299
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Watano Y, Kanai I, Tani N (2004) Genetic structure of hybrid zones between Pinus pumila and P. parviflora var. Pentaphylla (Pinaceae) revealed by molecular hybrid index analysis. Am J Bot 91(1):65–72. https://doi.org/10.3732/ajb.91.1.65
Whittemore AT, Schaal BA (1991) Interspecific gene flow in sympatric Oaks. Proc Natl Acad Sci U S A 88(6):2540–2544. https://doi.org/10.1073/pnas.88.6.2540
Xiang Q-P, Wei R, Shao Y-Z, Yang Z-Y, Wang X-Q, Zhang X-C (2015) Phylogenetic relationships, possible ancient hybridization, and biogeographic history of Abies (Pinaceae) based on data from nuclear, plastid, and mitochondrial genomes. Mol Phylogenet Evol 82:1–14. https://doi.org/10.1016/j.ympev.2014.10.008
Acknowledgements
We thank Y. Taguchi and M. Koshiba for technical assistance with the DNA analysis and S. Masuda and Y. Moriguchi for sample collection.
Funding
This study was partly supported by the JSPS KAKENHI program (grant nos. 21H02240, 15K07489, 23H00337).
Author information
Authors and Affiliations
Contributions
YT planned and designed the research. SF, KU, SS, YS, and YT collected the samples. SF, SS, MK, and KU performed the experiments and analysis. IT and NT performed the SDM analysis. YS is responsible for the assistance with data analysis and manuscript revision. KU, IT, and YT wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Data archiving statement
All sequence and genotype data used in this study have been deposited in the DDBJ and Dryad database, and the accession numbers are LC747190-LC749391 and doi:https://doi.org/10.5061/dryad.v15dv420g.
Additional information
Communicated by J. Beaulieu.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Uchiyama, K., Fujii, S., Tsuyama, I. et al. Range shift and introgression of three Japanese Abies species: insights from population genetic data and species distribution modeling. Tree Genetics & Genomes 19, 41 (2023). https://doi.org/10.1007/s11295-023-01617-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-023-01617-7