
Abstract
Organoboron compounds are important building blocks in organic chemistry for a variety of key transformations in the production of compounds in the pharmaceutical and agricultural industries. Direct C–H borylation provides many advantages over more traditional transformation via halide groups that lead to stoichiometric waste. In the direct C(sp2)–H borylation of arenes, Ir-bipyridine systems have shown excellent performance. However, to make the formation of borylated products more benign and greener, transformations catalyzed by heterogeneous catalysts are appealing as they provide easier recovery and reuse of the catalyst. In this study, two different porous organic polymers (POPs) based on polystyrene-bearing bipyridine (bpy) ligands were synthesized. These POPs can, upon metal ligation in situ create an active catalyst that is capable of borylation twice per B2pin2 molecule. Our Ir systems were tested with different arenes, and a preliminary mechanistic investigation was performed. The system was recyclable for up to three consecutive recycles, albeit, the polymer backbone had indications of being borylated during the reaction.
Similar content being viewed by others
1 Introduction
Organoboron compounds have played a key role in the development of organic chemistry and have a central role in the industry [1,2,3,4,5]. However, the formation of the C–B bond has classically been through halides with the potential use of stoichiometric lithium or magnesium, resulting in poor atom efficiency. Thus, the benign production of organoboron compounds directly from C–H bonds is highly attractive. In organic chemistry, the direct, selective functionalization of alkyl, alkenyl, and aryl C–H bonds is demanding. A lot of focus has therefore been on the catalyzed borylation reactions of C–H bonds in alkanes, alkenes, and arenes in high yields and with high selectivity. A challenge has been the functionalization of arenes without the need for directing groups. Functionalization of arenes without directing groups can provide products with complementary regioselectivity [6]. Furthermore, it provides a single-step functionalization of the arenes compared to the more classical multistep synthesis. Additionally, the borylation of arenes eliminates the need for halogenated arene precursors to arylboron compounds. Overall, the borylation generates less waste and reduces the need of prehalogenation in line with the principles of green chemistry [6]. Hartwig et al. demonstrated the first direct C(sp2)–H via a photocatalytic Re complex and later, Miyaura and Hartwig et al. showcased the potential of an Ir/bpy-complex without the need for light [7, 8]. Their work has led to many different derivatives of Ir-bpy catalytic systems over the past two centuries [9,10,11,12,13,14,15,16,17,18,19]. Especially the report from Smith et al. has truly derived many important trends in the Ir-catalyzed borylation of arenes [20].
Heterogeneous catalysts in organic synthesis are highly attractive in the industry due to their easier recovery and reuse [21,22,23,24,25,26,27,28,29]. Heterogeneous molecular catalysts have demonstrated high potential in the direct C(sp2)–H borylation of arenes with examples of silica grafting, metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and porous organic polymers (POPs) [30,31,32,33,34,35,36,37,38,39,40,41,42] Especially, the formation of molecular catalysts using POPs has been an appealing strategy in many transformations, in particular with their ability to mimic homogeneous catalysts while still keeping the property of easy separation known from heterogeneous catalysts [43,44,45,46]. Due to the swelling properties of POP-based catalysts in certain organic solvents, they can provide a quasi-homogeneous catalyst environment while still displaying the benefits of a solid catalyst, such as easy removal by filtration [43,44,45,46]. This is unlike other types of solid catalysts. Sawamura et al. [47] have already demonstrated the potential of swollen POPs in the direct C(sp2)–H functionalization of alkenes. Yet, the reports of direct borylation of arenes are more sparse, and especially polystyrene (PS)-based POPs have been reported to be completely inactive in the transformation [34]. Nevertheless, we would like to report examples of in situ-created molecular Ir-bpy catalysts anchored to polystyrene-based POPs. Two different POP materials were synthesized. One is based on ordinary styrene, and the other is based on the bulkier 4-tert-butyl styrene as a building block. The latter could constrain the bpy moiety, which has been shown to improve the catalytic performance [20]. The systems demonstrate good catalytic performance as they are capable of borylation twice per B2pin2 molecule and borylate different arenes. The reaction pathway was examined by competitive experiments, kinetic isotope effect, change of boron source, and a time study. The system was recyclable up to three times without a significant drop-in activity observed.
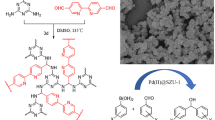
The formation of styrene (bpy-PS-POP) and 4-tert-butyl styrene (bpy-tBuPS-POP) based polymer materials containing bpy was done by radical polymerization. The radical polymerization required the formation of vinylated bpy. Fortunately, the formation of 4,4′-divinyl-2,2′-bipyridine proceeded effortlessly by a Suzuki coupling with potassium trifluorovinylborate and commercially available 4,4′-dibromo-2,2′-bipyridine. An emulsion methodology was applied to successfully create spherical polymer particles using either styrene or 4-tert-butylstyrene as a co-building block showcased with the full synthesis in Scheme 1 and with corresponding images and SEM images in Figure S1 and Figure S4. We have previously shown that similar materials possess excellent swelling properties that can improve the catalytic performance, and these POPs were no exception as they swelled in benzene, toluene, xylene, THF, and dioxane [48, 49]. These swelling properties made them promising candidates for direct arene borylation with the formation of a quasi-homogeneous environment in the swollen POP. A reference material was made as well without the addition of bpy to the polymer backbone (abbreviated PS-POP).

Synthetic pathway for the formation of bpy-POPs
2 Experimental
2.1 Materials
All chemicals were reagent grade and used as received without further purification. NaCl and benzene were purchased from TCI. B2pin2 and 4,4′-dibromo-2,2′-bipyridine were obtained from Fluorochem. Toluene was obtained from a solvent purification system (Puresolv MD-7). The remaining chemicals were obtained from Sigma Aldrich; [Ir(cod)Cl]2, [Ir(cod)OMe]2, HBpin, anisole, trifluorotoluene, fluorobenzene, chlorobenzene, iodobenzene, 1-methylpyrrole, benzofuran, d6-benzene, potassium vinyltrifluoroborate, PPh3, Pd(OAc)2, Cs2CO3, acacia gum, styrene, divinylbenzene, 4-tert-butylstyrene, and 0.2 M AIBN in toluene.
Scanning Electron Microscopy (SEM) images were taken on a Quanta 200 ESEM FEG microscope operating at 20 kV. Samples were placed on carbon tape and coated with gold for 60 s at 20 nA current in an argon atmosphere. X-ray photoelectron spectroscopy (XPS) analysis was done with a spot size of 400 µm with an Al alpha X-ray source with 10 scans per element. X-ray Fluorescence (XRF) analysis was conducted on a PANalytical Epsilon3 system. N2-physisorption was conducted on a Micrometrics 3Flex instrument at 77 K. Samples for N2-physisorption were degassed overnight before the analysis in a Micrometrics VacPrep 061 Sample Degas System at room temperature. Thermogravimetric Analyzer (TGA) was performed on a Mettler Toledo TGA/DSC 1 STARe System under a synthetic gas atmosphere. Liquid 1H NMR was measured on a Bruker Ascend 400 (400 Hz). 11B MAS NMR experiments were conducted using a Bruker Avance III spectrometer (B0 = 11.75 T, νL(11B) = 192.546 MHz) equipped with a 4 mm CP/MAS Probe. The spectra were acquired with the Hahn echo pulse sequence (π/2–τ–π–τ–ACQ) using a spinning frequency of 8000 Hz, a “solid π/2” pulse of 3 us (π/2(NaBH4) = 6 μs), and an interscan delay of 30 s. High-power SPINAL64 1H decoupling was applied during the acquisition. Chemical shifts are reported relative to F3B·O(C2H5) (δiso = 0 ppm) using neat NaBH4 as a secondary reference (δiso = − 42.06 ppm). An 11B spectrum of an empty rotor has been subtracted for all presented spectra to eliminate any background signals from the probe. The Topspin module SOLA was used for simulating the quadrupolar line shapes.
2.2 Vinylation of 4,4′-dibromo-2,2′-bipyridine
Synthesis adapted from Nie et al. [50], 4,4′-dibromo-2,2′-bipyridine (0.32 mmol, 100 mg), potassium vinyltrifluoroborate (1.27 mmol, 170 mg), Pd(OAc)2 (0.0064 mmol, 1.4 mg), PPh3 (0.020 mmol, 5.0 mg), and Cs2CO3 (0.96 mmol, 311 mg) were weighed into a Schlenk tube where a magnetic stir bar was added. The mixture was made inert by 3 vacuum/N2 cycles. Next, degassed THF (4.8 mL) and degassed H2O (0.2 mL) were added to the solids, and inside a N2-filled plastic bag, the Schlenk tube was sealed. The reaction mixture was heated to 85 °C for 48 h behind a blast shield. Next, the mixture was cooled down to rt., and H2O (15 mL) was added before the organic phase was extracted with dichloromethane (20 mL) four times. The organic phase was washed with brine (50 mL) and dried over MgSO4. The solvent was removed, and the product was purified over silica gel chromatography in a mixture of hexane, EtOAc, and Et3N (90:10:2), providing a white powder (48 mg, 70% yield).
2.3 Polymerization procedure
Polymerization methodology adapted from Iwai et al. [51], H2O (60 mL), NaCl (26 mmol, 1.5 g), and acacia gum (2.4 g) were mixed into a 250 mL round-bottom flask with a magnetic stir bar. The mixture was stirred to create a clear solution and before being bubbled with N2. In a 10 mL round-bottom flask, 4,4′-divinyl-2,2′-bipyridine (0.31 mmol, 65 mg), styrene (15 mmol, 1.71 mL, 1.55 g), or 4-tert-butylstyrene (15 mmol, 2.75 mL, 2.4 g), divinylbenzene (0.31 mmol, 43.5 µL, 39.7 mg), and chlorobenzene (3 ml) were mixed, and a magnetic stir bar was added. The mixture was sealed with a rubber septum and bubbled with N2. After being bubbled, 0.2 M AIBN in toluene (0.3 mmol, 1.5 mL) was added, and the whole content was mixed with the aqueous mixture in the 250 mL round-bottom flask by a syringe. The combined mixture was emulsified by stirring the mixture at 600 rpm for 1 h before being heated to 80 °C in an oil bath to start the polymerization. The mixture was left to stir overnight at 80 °C. The next day, the POP was collected on a glass funnel and washed 3 × times with H2O (50 mL), methanol (50 mL), THF (50 mL), and toluene (50 mL) before being dried in a vacuum oven. The procedure yielded white/off-white small beads, see figure S1.
In the polymerization of PS-POP, 4,4′-divinyl-2,2′-bipyridine is replaced with divinylbenzene (to a total of 0.62 mmol, 87 µL, 79.4 mg) to ensure the same cross-linking effect.
3 Result and Discussion
3.1 Characterization
TGA analysis was performed to evaluate the thermal stability of the materials. The POPs showed high thermal stability with no mass loss up to 300 °C (Figure S2). N2-physisorption analysis isotherms revealed no inherent porosity demonstrating the requirement of swelling to make the bpy-sites accessible (Figure S3) [48, 49]. Table S1 provides a summary of textural properties for the POPs calculated from the physisorption, confirming very low surface areas and total pore volumes of < 5 m2/g and < 0.1 cm3/g, respectively, for all synthesized materials.
3.2 Catalytic Activity
Under optimized reaction conditions, the active catalysts were formed by in-situ ligation of [Ir(cod)Cl]2 using 5 mol% bpy with B2pin2 (0.1 mmol) and applying benzene as solvent (0.1 M) at 60 °C for 6 h (see Table S2 for optimization). The combination of [Ir(cod)Cl]2 and a bpy-based ligand has shown to be very robust towards moisture, ligand loading, and highly active with B2pin2 as the boron source is ideal for further investigation [9,10,11,12,13,14,15,16,17,18,19]. Under these conditions, the reaction proceeds smoothly, providing quantitative yields with two borylated products per B2pin2. Albeit, substituted arenes required a reaction time of 24 h to reach adequate yields, as seen in Table 1. Toluene and anisole proceeded smoothly using both POPs favoring borylation in the meta-position (Table 1, entries 1–4). Fluorobenzene provided excellent yield for bpy-PS-POP and more sluggish for bpy-tBuPS-POP where the borylation occurred both in ortho, meta, and para position (entries 5–6). Chlorobenzene gave high yields with a product distribution favoring borylation meta to the chloro group (entries 7–8). A similar trend was seen for iodobenzene, albeit with lower yields (entries 9–10). Applying m-xylene as a substrate required a temperature increase to 100 °C to proceed with a decent yield for bpy-PS-POP. These results showcase that the bulkier bpy-tBuPS-POP is less active than bpy-PS-POP, and the introduction of more bulkiness at the bpy site does not give any regioselectivity as the product distribution follows the substrate rather than the POP.
These conditions demonstrate good robustness as the reaction could be scaled up to 2.0 mmol B2pin2 reaching up to 83% of isolated yield using toluene with a statistic distribution of meta- and para- borylated products (2:1) (see Figures S5 and S6). A more significant drop in yield was seen for bpy-tBuPS-POP with 47% isolated yield.
Competition experiments between benzene and different mono-substituted arenes demonstrate that electron-deficient arenes react faster than benzene, whereas electron rich have a slower reaction rate than benzene (Table 2). These observations are similar to other reported examples of Ir-bpy-based systems [8, 38, 39].
As a consequence of B2pin2 being capable of reacting twice per molecule, the release of hydrogen should occur. Thus, the headspace volume of a reaction was analyzed by a GC-FID to verify the presence of molecular hydrogen (Figs. 1a and S7). To confirm that the released hydrogen does not react with the POP and thereby drives the reaction to completion, XPS analysis of the C1s of fresh and recovered POP was carried out. The XPS spectra did not showcase any significant change between the fresh and used POP (Figure S8). Similarly, the reaction proceeded nicely with the use of homogeneous 2,2′-bipyridine, demonstrating that the POP is not a requirement for the reaction to occur under these conditions (Fig. 1b).

a Experiment confirming the presence of hydrogen released from the reaction. b The reaction proceeds when bpy-POP is replaced with homogeneous bpy
Replacing B2pin2 either partially or completely with HBpin proceeded smoothly for bpy-PS-POP whereas a small loss in yield was observed for bpy-tBuPS-POP (Figure S9). However, it has been demonstrated previously, that catalytic systems with [Ir(cod)Cl]2 and bpy perform better with B2pin2 than HBpin [20].
A simple time study using bpy-PS-POP revealed an induction period of approximately 1.3 h where no catalytic activity was observed matching other reports (Figure S10) [8, 20]. The reaction profile is sigmoidal, indicating that the catalytic active species is formed in situ. For deuterated benzene, a primary kinetic isotope effect of 3.7 was observed, which is identical to that previously reported [8]. These observations combined with the detection of H2, the success of using B2pin2 and HBpin as reagents, and the competition studies suggest that the reaction proceeds through an IrIII/IrV catalytic cycle where the IrI-precatalyst ([Ir(cod)Cl]2] is activated initially. A similar pathway has been reported for other Ir/bpy-based catalytic systems [2, 3, 20, 33].
The Ir-bpy-PS-POP system was recovered and recycled inside an argon-filled glovebox, where it was possible to successfully reuse the catalyst three times with quantitative yields (Table 3). However, upon further usage, the catalytic activity dropped, and no activity was observed at the fifth consecutive reaction. The deactivation could be due to exposure to atmospheric air or moisture which is known to influence some iridium catalysts for the borylation reaction [20]. The potential deactivation by metal leaching was analyzed by XRF, which revealed an iridium concentration of 0.09 mM and could potentially explain the diminishing activity upon multiple reactions, which corresponds to around 9% of the original iridium concentration. To demonstrate that the activity does not originate from the leached iridium, a filtration experiment was done where a reaction mixture was filtered after 1 h and 40 min and stirred at 60 °C for an additional 1 h and 20 min. Fortunately, the activity stopped completely with the removal of the POP from the reaction.
A potential drawback in the usage of PS-based POPs is the possibility of the borylation occurring on the polymer backbone instead of the substrate. Thus, recovered POP was analyzed by 11B MAS NMR (see Figure S12 for the spectra). The analyses revealed boron species that did not originate from B2pin2 and indicate undesired borylation of the backbone. However, the intensity was higher for bpy-PS-POP than bpy-tBuPS-POP showcasing that the bulkier POP is less exposed.
4 Conclusion
Here, we have reported the first example of a polystyrene-based POP active in the direct C(sp2)–H borylation of arenes. The in situ-made system was active using different arenes, and competition studies showed that electron-deficient arenes react faster than electron rich. A possible IrIII/IrV catalytic cycle was believed to occur for the system where the IrI-precatalyst ([Ir(cod)Cl]2] had to be activated, explaining the observed induction period. The system was recyclable and provided quantitative yields over the first three consecutive reactions, albeit the activity was lost upon further usage, believed to originate from metal leaching.
References
Hall DG (2005) Boronic Acids: Preparation Applications in Or-ganic Synthesis and Medicine. Wiley-VCH Verlag GmbH & Co. KGaA, pp 1–99
Hartwig JF (2011) Chem Soc Rev 40:1992–2002
Xu L, Wang G, Zhang S, Wang H, Wang L, Liu L, Jiao J, Li P (2017) Tetrahedron 73:7123–7157
Wang M, Shi Z (2020) Chem Rev 120:7348–7398
Wang F, Mielby J, Richter FH, Wang G, Prieto G, Kasama T, Weidenthaler C, Bongard H-J, Kegnæs S, Fürstner A, Schüth F (2014) Angew Chem Int Ed 53:8645–8648
Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF (2010) Chem Rev 110:890–931
Chen H, Hartwig JF (1999) Angew Chem Int Ed 38:3391–3393
Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF (2002) J Am Chem Soc 124:390–391
Tran LD, Daugulis O (2012) Angew Chem Int Ed 51:5188–5191
Sun WW, Cao P, Mei RQ, Li Y, Ma YL, Wu B (2014) Org Lett 16:480–483
Lee LC, He J, Yu JQ, Jones CW (2016) ACS Catal 6:5245–5250
He G, Wang B, Nack WA, Chen G (2016) Acc Chem Res 49:635–645
Bisht R, Haldar C, Hassan M-M, Hoque M-E, Chaturvedi J, Chattopadhyay B (2022) Chem Soc Rev 51:5042–5100
Hassan M-M, Mondal B, Singh S, Haldar C, Chaturvedi J, Bisht R, Sunoj R-B, Chattopadhyay B (2022) J Org Chem 87:4360–4375
Haldar C, Hoque M-E, Chaturvedi J, Hassan M-M, Chattopadhyay B (2021) Chem Commun 57:13059–13074
Chaturvedi J, Haldar C, Bisht R, Pandey G, Chattopadhyay B (2021) J Am Chem Soc 143:7604–7611
Hoque M-M, Hassan MM-M, Chattopadhyay B (2021) J Am Chem Soc 143:5022–5037
Hoque M-E, Bisht R, Haldar C, Chattopadhyay B (2017) J Am Chem Soc 139:7745–7748
Bisht R, Chattopadhyay B (2016) J Am Chem Soc 138:84–87
Preshlock SM, Ghaffari B, Maligres PE, Krska SW, Ma-leczka RE, Smith MR (2013) J Am Chem Soc 135:7572–75821
Copéret C, Chabanas M, Saint-Arroman R, Basset J-M (2003) Angew Chem Int Ed 42:157–181
Thomas J, Raja R, Lewis D (2005) Angew Chem Int Ed 44:6465–6482
Astruc D, Lu F, Aranzaes J (2005) Angew Chem Int Ed 44:7852–7872
Kramer S, Hejjo F, Rasmussen KH, Kegnæs S (2018) ACS Catal 8:754–759
Witham C, Huang W, Tsung C-K, Somorjai G, Toste F (2010) Nat Chem 2:36–41
Magano J, Dunetz J (2011) Chem Rev 111:2177–2255
Rupp-rechter G (2017) Nat Chem 9:833–834
Buendia MB, Kegnæs S, Kramer S (2020) Adv Synth Catal 362:5506–5512
Poreddy R, García-Suárez EJ, Riisager A, Kegnæs S (2014) Dalton Trans 43:4255–4259
Kawamorita S, Ohmiya H, Hara K, Fukuoka A, Sawamu-ra M (2009) J Am Chem Soc 131:5058–5059
Yamazaki K, Kawamorita S, Ohmiya H, Sawamura M (2010) Org Lett 12:3978–3981
Grüning WR, Siddiqi G, Safonova OV, Copéret C (2014) Adv Synth Catal 356:673–679
Wu F, Feng Y, Jones CW (2014) ACS Catal 4:1365–1375
Waki M, Maegawa Y, Hara K, Goto Y, Shirai S, Yamada Y, Mi-zoshita N, Tani T, Chun WJ, Muratsugu S, Tada M, Fu-kuoka A, Inagaki S (2014) J Am Chem Soc 136:4003–4011
Maegawa Y, Inagaki S (2015) Dalt Trans 44:13007–13016
Li Y, Kanbur U, Cui J, Wang G, Kobayashi T, Sadow A-D, Qi L (2022) Angew Chem 134:e202117394
Mamlouk H, Suriboot J, Manyam P-K, AlYazidi A, Bergbreiter D, Madrahimov S-T (2018) Catal Sci Technol 8:124–127
Manna K, Zhang T, Lin W (2014) J Am Chem Soc 136:6566–6569
Syed ZH, Chen Z, Idrees KB, Goetjen TA, Wegener EC, Zhang X, Chapman KW, Kaphan DM, Delferro M, Farha OK (2020) Organometallics 39:1123–1133
Tahir N, Muniz-Miranda F, Everaert J, Tack P, Heugebaert T, Leus K, Vincze L, Stevens CV, Van Speybroeck V, Van Der Voort P (2019) J Catal 371:135–143
Vardhan H, Pan Y, Yang Z, Verma G, Nafady A, Al-Enizi AM, Alotaibi TM, Almaghrabi OA, Ma S (2019) APL Mater 7:101111
Kimura A, Hayama H, Hasegawa JY, Nageh H, Wang Y, Naga N, Nishida M, Nakano T (2017) Polym Chem 8:7406–7415
Kaur P, Hupp J, Nguyen S (2011) ACS Catal 1:819–835
Zhang Y, Riduan N (2012) Chem Soc Rev 41:2083–2094
Sun Q, Dai Z, Meng X, Xiao F-S (2015) Chem Soc Rev 44:6018–6034
Kramer S, Bennedsen N, Kegnæs S (2018) ACS Catal 8:6961–6982
Iwai T, Harada T, Hara K, Sawamura M (2013) Angew Chem Int Ed 52:12322–12326
Bennedsen NR, Kramer S, Kegnæs S (2020) Catal Sci Technol 10:7697–7705
Bennedsen NR, Christensen DB, Mortensen RL, Wang R, Kramer S, Kegnæs S (2021) ChemCatChem 13:1781–1786
Nie HJ, Yao J, Zhong YW (2011) J Org Chem 76:4771–4775
Iwai T, Harada T, Shimada H, Asano K, Sawamura M (2017) ACS Catal 7:1681–1692
Acknowledgements
The authors are grateful for funding from the Independent Research Fund Denmark (Grant Nos. 6111-00237 and 0217-00146B), from Villum fonden (Grant No. 13158), and from Haldor Topsøe A/S. S. Kramer is deeply appreciative of generous financial support from the Lundbeck Foundation (Grant No. R250-2017-1292) and the Technical University of Denmark. The authors thank Dr. Kasper Enemark-Rasmussen at the DTU NMR center supported by the Villum foundation for aid with solid-state NMR.
Funding
Open access funding provided by Royal Danish Library.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bennedsen, N.R., Yang, F., Goodarzi, F. et al. Direct C(sp2)–H Borylation of Arenes Using Ir-bpy Porous Organic Polymers. Top Catal 66, 1451–1456 (2023). https://doi.org/10.1007/s11244-023-01820-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-023-01820-9