
Abstract
Background
Accumulating evidence links the echinoderm microtubule-associated protein-like 4 (EML4)-anaplastic lymphoma kinase (ALK) rearrangement to venous thromboembolism (VTE) in non-small cell lung cancer (NSCLC) patients. However, the corresponding mechanisms remain unclear.
Method
High-throughput sequencing analysis of H3122 human ALK-positive NSCLC cells treated with ALK inhibitor/ dimethyl sulfoxide (DMSO) was performed to identify coagulation-associated differential genes between EML4-ALK fusion protein inhibited cells and control cells. Sequentially, we confirmed its expression in NSCLC patients’ tissues and in the plasma of a subcutaneous xenograft mouse model. An inferior vena cava (IVC) ligation model was used to assess clot formation potential. Additionally, pathways involved in tissue factor (TF) regulation were explored in ALK-positive cell lines H3122 and H2228. Statistical significance was determined by Student t-test and one-way ANOVA using SPSS.
Results
Sequencing analysis identified a significant downregulation of TF after inhibiting EML4-ALK fusion protein activity in H3122 cells. In clinical NSCLC cases, TF expression was increased especially in ALK-positive NSCLC tissues. Meanwhile, H3122 and H2228 with high TF expression exhibited shorter plasma clotting time and higher TF activity versus ALK-negative H1299 and A549 in cell culture supernatant. Mice bearing H2228 tumor showed a higher concentration of tumor-derived TF and TF activity in plasma and the highest adjusted IVC clot weights. Limiting EML4-ALK protein phosphorylation downregulated extracellular regulated protein kinases 1/2 (ERK1/2)-activating the protein-1(AP-1) signaling pathway and thus attenuated TF expression.
Conclusion
EML4-ALK fusion protein may enhance venous thrombogenicity by regulating coagulation factor TF expression. There was potential involvement of the pERK1/2-AP-1 pathway in this process.
Similar content being viewed by others


Introduction
In recent years, accumulating studies have indicated a close link between genetic alterations and venous thromboembolism (VTE) occurrence in patients with malignant tumors [1,2,3]. Our team also explored the relationship between driver oncogene alterations and VTE event occurrence in non-small cell lung cancer (NSCLC) patients through a prospective cohort study. The results showed that the anaplastic lymphoma kinase (ALK) gene rearrangement in NSCLC conferred a significant increase in VTE risk [4, 5], which was consistent with previous retrospective studies. Sequentially, it was also verified again by professor Hanny et al. in a cohort study [6]. However, little is known about the specific mechanism of ALK rearrangement in NSCLC cells regulating VTE occurrence.
ALK rearrangement is the first somatic oncogene translocation discovered in lung cancer. In 2007, a Japanese team identified a fusion gene caused by inversion within chromosome 2p, comprising portions of the echinoderm microtubule-associated protein-like 4 (EML4) gene located in p21 and the ALK gene located in p23 in NSCLC cells [7, 8]. Basically, the full-length EML4 protein consists of the N-terminal coiled-coil trimerization domain and the C-terminal of the ALK protein, which contains the kinase domain. These domains are able to form a dimer without ligand binding, leading to activation of the ALK protein [9, 10]. And EML4-ALK fusion seems to be unique to NSCLC [11, 12]. Sequentially, a transgenic mouse model specifically expressing EML4-ALK in lung alveolar epithelial cells has been established to confirm its potent oncogenic activity [13]. Upon transcription of EML4-ALK, Mitogen-Activated Protein Kinase (MAPK), Janus Kinase with Signal Transducer And Activator Of Transcription (JAK-STAT) and Phosphoinositide-3-Kinase with VAkt Murine Thymoma Viral Oncogene Homolog (PI3K-AKT) are constitutively activated. There is evidence that these signaling pathways enhance proliferation, survival and angiogenesis in cancer cells [9, 14].
Besides, the corresponding targeted small molecule tyrosine kinase inhibitors (TKIs) have also led to unprecedented survival benefits in NSCLC patients with ALK rearrangement [15, 16]. In 2011, the first-generation ALK-TKI, crizotinib was approved for treatment of advanced ALK-positive NSCLC, which is a small molecule ATP-competitive ALK inhibitor [17]. In order to overcome crizotinib resistance, the second-generation ALK-TKIs, including ceritinib, alectinib and brigatinib were developed [18]. ALK TKIs have the potential to inhibit ALK phosphorylation and downstream signalling, leading to cell cycle arrest in the G1-S phase and apoptosis of cancer cells [19].
Typically, VTE occurs as a result of blood stasis, endothelial or vessel wall injury, and hypercoagulability. While cancer patients’ blood stasis and endothelial injury could be shared with non-cancer patients, the hypercoagulability driven by malignancy-specific pathways is probably unique to cancer. Previous studies have reported various mechanisms of hypercoagulation in malignancy, including indirect regulatory mechanisms such as expressing proteins by tumor cells that could alter circulating cells [20,21,22,23], and direct regulatory mechanisms such as expressing procoagulant proteins (TF, podoplanin (PDPN)) which directly activate the coagulation cascade/platelets [24,25,26,27].
Whether EML4-ALK fusion protein in NSCLC cells also enhances venous thrombogenicity through the mechanisms mentioned above is still unclear. We performed the following experiments to address this issue further and explore possible new targets for anticoagulation therapy in EML4-ALK-rearranged NSCLC patients.
Materials and methods
Cells and reagents
The ALK-positive human NSCLC cell lines H3122 and H2228 (purchased from Jiangsu KeyGEN BioTECH Co., Ltd, Jiangsu, China) and ALK-negative human NSCLC cell lines H1299 and A549 (purchased from FuHeng Biology, Shanghai, China) were maintained under a humidified atmosphere of 5% CO2 at 37 °C in RPMI medium 1640 supplemented with 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin (0.1 mg/ml). Alectinib (catalog #S2762, Selleckchem, Houston, USA), SCH772984 (catalog #S7101, Selleckchem, Houston, USA), and T-5224 (catalog #GC16165, GlpBio, Montclair, USA) were each dissolved in dimethyl sulfoxide (DMSO). All reagents were stored at -20 °C or -80 °C.
Western blot analysis
H3122 and H2228 cells were immediately lysed before use in RIPA Lysis Buffer (Solarbio, Beijing, China) supplied with protease inhibitor cocktails (KeyGEN BioTECH, Jiangsu, China) and phosphatase inhibitor cocktail (KeyGEN BioTECH, Jiangsu, China). Quantification of protein was determined by a Bicinchoninic Acid Assay (Thermo Fisher Scientific, Waltham, USA). Equal amounts of protein were subjected to 10% SDS PAGE and then transferred onto PVDF membranes. After incubated in blocking solution (NCM Biotech, Suzhou, China), specific antibodies for phospho-ALK (catalog #3341S, rabbit, 1:500, Cell Signaling, Beverly, USA), ALK (catalog #3633T, rabbit, 1:1000, Cell Signaling, Beverly, USA), TF (catalog #228,968, rabbit, 1:1000, Abcam, Cambridge, USA), phospho-ERK1/2 (catalog #4370, rabbit, 1:1000, Cell Signaling, Beverly, USA), ERK1/2 (catalog #9102, rabbit, 1:1000, Cell Signaling, Beverly, USA), c-FOS (catalog #2250, rabbit, 1:500, Cell Signaling, Beverly, USA) and GAPDH (catalog #5174S, rabbit, 1:3000, Cell Signaling, Beverly, USA) were used to incubate blots overnight at 4℃, then followed by incubating in HRP-conjugated goat anti-rabbit antibody for 1 h at room temperature and detecting the bands with the ECL system (Millipore Sigma).
High-throughput sequencing
High-throughput sequencing was performed to detect different mRNA expressions between H3122 treated with Alectinib (100nmol/L, 6 h) and DMSO. CapitalBio Technology (Beijing, China) used an Illumina NovaSeq 6000 sequencer with a pair end 150-bp reading length (Illumina, San Diego, USA) to sequence mRNA and performed the final data analysis. The screening criteria for differential genes were: |log2FC|>=1 and p-Value < = 0.05. Cluster software was used to depict a heatmap for gene clustering.
RNA isolation, reverse transcription, and quantitative RT-PCR
Total RNA extraction was performed by decomposing cells in TRIzol (Tiangen Biotech, Beijing, China). A reverse Transcription Kit (Takara Bio, Beijing, China) with random primers was used to synthesize cDNA. Quantitative real-time PCR (RT-PCR) analysis was performed by SYBR Green I master (Roche, Basel, Switzerland) and LightCycler480 II (Roche, Basel, Switzerland). The primer sequences were listed as follows: Human F3: forward primer 5’-GGCGCTTCAGGCACTACAAA-3’ and reverse primer 5’- CGTGCCAAGTACGTCTGCTT-3’; Human cfos: forward primer 5’- CACTCCAAGCGGAGACAGAC-3’ and reverse primer 5’- AGGTCATCAGGGATCTTGCAG-3’; Human HPRT-1: forward primer 5’-CCTGGCGTCGTGATTAGTGAT-3’ and reverse primer 5’- AGACGTTCAGTCCTGTCCATAA-3’. The results were calculated using the 2−ΔΔCT method, and HPRT-1 served as a reference gene. Each sample was assayed at least in triplicate.
Immunohistochemical analysis of human specimens
Human NSCLC tissues were obtained from patients at the Beijing Chao-Yang Hospital, Capital Medical University, with the approval of the institutional review board. For all patients involved in this study, amplification refractory mutation system polymerase chain reaction (ARMS-PCR) and Ventana immunohistochemistry were performed to detect ALK rearrangement. And the patients with ALK-rearrangement were confirmed by fluorescence in situ hybridization (FISH). The patients’ demographic and clinical data are presented in Table 1. The cancer tissues were formalin-fixed and paraffin-embedded. Anti-Tissue Factor antibody (catalog #228,968, 1:500; Abcam, Cambridge, USA) was used to detect TF. Images were obtained using a Leica TCS SP5 (Wetzlar, Hesse, Germany), and immunohistochemical staining of TF was determined using Images-Pro Plus version 6.0 software (IPP6) to assess the integrated optical density (IOD) and mean density of the immunohistochemical staining section. Finally, the mean IOD and density of cancer tissue immunohistochemical staining from five randomly selected fields (magnification, ×400) were recorded and analyzed.
Flow cytometry and immunofluorescence
Cells (H3122, H2228, H1299 and A549) were surface-stained with BD Pharmingen™ PE Mouse anti-human CD142 (catalog #550,312, BD Bioscience, Franklin Lakes, USA) for 20 min at 4 °C and washed with PBS. Then cells were resuspended in a cell buffer solution at a concentration of 1 × 106/mL for flow cytometric analysis (FACSCanto II; BD Bioscience) and analyzed using BD FCSDiva Software and FCS Express 5 software (De Novo Software, Los Angeles, USA).
H3122 cells in 6 wells were fixed in 4% paraformaldehyde and incubated with immunofluorescence mAbs against TF (Affinity Biosciences, Cincinnati, USA) overnight at 4 ℃, then followed by incubating in Alexa Fluor® 488-labeled goat anti-rabbit IgG for 1 h at room temperature and DAPI solution for 7 min. Images were obtained using a Leica TCS SP5 (Wetzlar, Hesse, Germany).
Plasma clotting assay
Plasma clotting assay was performed to measure the plasma clotting time induced by lung cancer cells supernatant. Lung cancer cells (1 × 106) were cultured in a complete medium for 24 h. Then, the cell culture supernatant was collected in an Eppendorf and centrifuged (3000 rpm for 10 min at 4℃) to remove cell debris. 200 ul of cell culture supernatant and 200 ul of 25 mM/L CaCl2 were added to 200 ul of citrated human plasma (healthy volunteers) at 37℃ to initiate the plasma clotting process. Samples contained both TF, pro-coagulants-bearing extracellular vesicles and tumor-secreted soluble pro-coagulants. Clotting time was recorded visually by noting when the liquid formed a semisolid gel that did not flow during tube turning over [24].
Mouse model
The animal experiments were approved by the Institutional Animal Care and Use Committee of the Beijing Chaoyang Hospital, the Capital Medical University. Four-week old male athymic nude mice (BALB/c Nude, Vital River Laboratory Animal Technology, Beijing, China) were used to prepare the xenograft model. Human lung cancer cells (H2228, H1299) at a concentration of 1 × 107 cells in 200uL of suspension were injected using 1 ml syringe into the subcutaneous tissue on the backs of mice (n = 6). And the control group was injected with PBS (n = 6). The tumor size was measured weekly until the volume was 200mm3 (about 3–4 weeks). When the tumor volume reached 200mm3, IVC model was developed. The inferior vena cava (IVC) ligation model was performed as described in the previous study [28, 29]. IVC was separated from the aorta after the laparotomy, and was ligated distal to the renal veins by using a 6 − 0 silk suture. To induce thrombus formation within the IVC, the tributaries surrounding the IVC were also ligated to create a total stasis environment. And 48 h later, clots were collected from the IVC and weighed. The clot weight was adjusted by body weight (clot weight/body weight).
Blood collected from the orbital sinus of mice was pipetted into sodium citrate tubes and placed in a centrifuge at 3000 rpm for 10 min at 4℃ to separate the plasma. Blood samples were not performed to remove pro-coagulants-bearing extracellular vesicles and tumor-secreted soluble pro-coagulants.
The experiments above were repeated three times.
TF activity assay
TF procoagulant activity was assessed using a Tissue Factor Chromogenic Activity Kit (catalog #CT1002b, ASSAYPRO, Missouri, USA) as the manufacturer’s protocol.
Statistics
Summary statistics are presented as mean ± SEM (standard error of the mean). A Student t-test and one-way ANOVA followed by LSD or Tamhane test were performed to analyze statistical comparisons between groups. IBM SPSS Statistics 22 software (IBM SPSS Statistics, IBM, Chicago, IL, USA) was used for data analysis and Graph Pad Prism 7 software (San Diego, CA, United States) was used for graphing. Statistical significance was assessed at the P <0.05 level.
Results
Targeting phosphorylated EML4-ALK fusion protein activity inhibits coagulation factor TF expression
The schematic figure of the study is shown in Fig. 1.A. Treating H3122 cells, a validated EML4-ALK-positive NSCLC cell line, with Alectinib (100nmol/L, 6 h) [30]. Alectinib is a highly selective ALK inhibitor that showed strong antitumor activity against cancer cells which could achieve target inhibition of EML4-ALK fusion protein autophosphorylation, and substantially change the mRNA expression profile compared with DMSO in high-throughput mRNA sequencing analysis (Fig. 1.B). Besides, among the genes involved in aforementioned tumor-related coagulation [23, 31, 32], F3 gene was significantly downregulated in Alectinib-treated group (logFC =-1.54, P < 0.001) (Fig. 1.B). Subsequentially, using RT-PCR and immunoblotting, we confirmed that F3 mRNA expression and TF protein were much lower in H3122 cells after being treated with Alectinib (Fig. 1.C and Fig. 1.D).

Targeting phosphorylated EML4-ALK fusion protein activity inhibits coagulation factor TF expression. (A) Schematic figure of the study. (B) Next-generation mRNA sequencing showed Treatment of ALK rearrangement human lung cancer cell line H3122 with Alectinib (100nmol/L, 6 h) substantially change the mRNA expression profile compared with DMSO-treated (vehicle-treated) H3122 cells; Among the genes involved in tumor-related coagulation, F3 gene was significantly downregulated in Alectinib-treated group (logFC =-1.54, P < 0.001). (C) mRNA expression of F3 gene in H3122 cells treated with Alectinib /DMSO was quantified by quantitative RT-PCR (n = 5). (D) Western blot of TF in H3122 cells treated with Alectinib /DMSO (n = 3). (E) H3122 cells stained with immunofluorescent anti-TF mAb to identify the localization of TF. (F) Flow cytometric analysis of TF expression at H3122 cell surface. (G) The mean fluorescence intensity (MFI) of anti-TF antibody in flow cytometry significantly decreased in Alectinib-treated group (P = 0.038) (n = 3). Data are presented as means ± SEM. *p < 0.05, ***p < 0.001, determined by Student t test
To further investigate the expression and localization of TF in H3122 cells, immunofluorescence and flow cytometry were performed, and the results indicated that TF was mainly presented in the cell membrane (Fig. 1.E), and the mean fluorescence intensity (MFI) of anti-TF antibody in H3122 cells was also significantly decreased in Alectinib-treated group (P = 0.038) (Fig. 1.F and Fig. 1.G).
ALK-positive lung cancer cell with high TF expression enhances clot formation
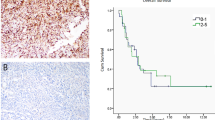
Given the effect of Alectinib on regulating TF expression, we further sought to explore the expression of TF in ALK-positive and ALK-negative NSCLC patients and cell lines. First, we determined the expression of TF in NSCLC patients, and the characteristics of the patients were listed in Table 1. Ten ALK-positive NSCLC patients and 17 ALK-negative NSCLC patients were included, and the immunohistochemical staining results showed that TF was predominantly expressed at the tumor site in ALK-positive NSCLC (Fig. 2.A). Furthermore, quantifying the immunochemical staining results with IPP6, we also observed that ALK-positive NSCLC presented a higher IOD value than ALK-negative NSCLC (Fig. 2.B, P = 0.001), which was proportional to the total amount of expression of TF. However, there was no significant difference in the mean optical density value (Fig. 2.B, P = 0.096), which reflects the intensity of TF protein.

ALK-positive lung cancer cell with high TF expression enhances clot formation. (A) Primary lung cancer tissues obtained from ALK positive and ALK negative lung cancer patients. Tissue sections were stained immunohistochemically with anti-TF mAb. ALK positive NSCLC cancer cells express TF protein mainly on the cell membrane (upper panel); TF protein were negatively expressed in most ALK negative NSCLC cancer cells (lower panel). (B) Quantifying the immunochemical staining results with IPP6. ALK positive NSCLC tissues presented higher IOD value (left panel, P = 0.001). There was no significant difference in mean optical density value (right panel, P = 0.096). (C) The mRNA expression of F3 gene in ALK positive NSCLC lung cancer cell lines (H3122 and H2228) and negative NSCLC lung cancer cell lines (H1299 and A549) was quantified by quantitative RT-PCR (n = 3). (D) The expression of TF protein in ALK positive and negative NSCLC cell lines was quantified by Flow cytometry analysis. (E) TF activity of culture supernatant obtained from cell line cultures. (F) TF activity in plasma of mice bearing NSCLC cell lines and blank controls. (G) Clot weight in mice bearing different NSCLC cells and blank control after adjusted body weight (each n = 3 to 6). Data are presented as means ± SEM. *p < 0.05, **p < 0.01, NS, no significance, determined by Student t test or one-way ANOVA followed by Bonferroni test
Next, TF expression was evaluated in ALK-positive NSCLC lung cancer cell lines (H3122 and H2228) and ALK-negative NSCLC lung cancer cell lines (H1299 and A549). RT-PCR showed that ALK-positive NSCLC cell lines exhibited a higher level of F3 mRNA expression, especially the H2228 cell line, whose expression level was about 625-fold higher than that of the H1299 cell line (Fig. 2.C). Consistent with the RT-PCR results, flow cytometry also confirmed that the H2228 cell line expressed the highest TF protein (Fig. 2.D). Similarly, the TF activity of culture supernatant obtained from H2228 cultures was the highest (79.2 ± 5.0pM), followed by H3122 (22.9 ± 3.5pM), A549 (12.4 ± 1.1pM), and H1299 (10.5 ± 0.4pM) (Fig. 2.E). In addition, we also performed a plasma clotting assay using the culture supernatant of each NSCLC cell line to assess their prothrombotic potent, as TF in the culture supernatant can activate coagulation factors in platelet-poor plasma and finally lead to insoluble fibrin formation. Consistent with the level of TF expression in NSCLC cell lines, the plasma clotting time of culture supernatant obtained from H2228 cultures was the shortest (45.3 ± 1.9s), followed by H3122 (121.7 ± 9.6s), A549 (602 ± 2s) and H1299 (621.7 ± 26.3s) (Supplementary Fig. 1.A).
Next, the expression of tumor-derived TF in circulation was further examined. As soon as the xenograft model tumor volume reached 200 mm3, plasma was collected for ELISA detection of tumor-derived TF concentration. Meanwhile, TF activity (tumor-derived and mice-derived) in the plasma of mice bearing H2228 is highest (H2228 vs. H1299, P = 0.021; H2228 vs. blank control, P = 0.006), followed by H1299 and blank control (H1299 vs. blank control, P < 0.001) (Fig. 2.F). After that, the IVC ligation model was also developed on blank mice and mice bearing H1299 and H2228 tumors. The results indicated that clot weight increased in mice with NSCLC when compared to blank mice after adjusting body weight (clot weight/body weight) (blank control vs. H1299, P = 0.025; blank control vs. H2228, P < 0.001). Also, consistent with the level of TF concentration and activity of NSCLC cell lines, the clot weight in mice bearing H2228 tumors was significantly higher than those bearing H1299 tumors (H2228 vs. H1299, P = 0.005) (Fig. 2G).
EML4-ALK fusion protein regulates TF expression through the pERK1/2-AP-1 pathway in NSCLC cells
The promoter region of the F3 gene contains multiple elements for diverse transcription factors binding [33], and phosphorylated EML4-ALK fusion protein in NSCLC cells could activate multiple downstream pathways [15]. We further analyzed the results of high-throughput mRNA sequencing for transcription factors that may regulate F3 gene expression and KEGG pathways in which EML4-ALK fusion protein is involved. The results implied that cfos mRNA expression, whose protein product was one of the subunits comprising F3 gene transcription factor AP-1 [34,35,36], was also down-regulated along with the F3 gene. Besides, the upper regulatory pathway of AP-1 overlapped with the downstream pathway of EML4-ALK at the node of ERK1/2 [37, 38].
The results verified that cfos gene expression was downregulated with F3 in Alectinib-treated H3122 cells through RT-PCR (Fig. 3.A, left panel). Further, pERK1/2 and cfos protein expression were also decreased in Alectinib-treated H3122 using Western Blot (Fig. 3.A, right panel).

EML4-ALK fusion protein regulates TF expression through pERK1/2/AP-1 pathway in H3122 cells. (A1) The mRNA expression of cfos gene in H3122 cells treated with Alectinib and DMSO was quantified by quantitative RT-PCR (n = 6). (A2) The expression of pERK1/2 and cfos protein in H3122 cells treated with Alectinib and DMSO was quantified by Western Blot (n = 3). (B1-3) Pretreating H3122 cells with ERK1/2 inhibitor (SCH772984) downregulated ERK1/2 phosphorylation, cfos and TF protein expression compared with DMSO control which identified by Western Blot and flow cytometry (n = 3). (C1-3) Pretreating H3122 cells with AP-1 inhibitor (T-5224) also attenuated TF expression compared with DMSO control which identified by Western Blot and flow cytometry (n = 3). Data are presented as means ± SEM. **p < 0.01, ***p < 0.001, determined by Student t test
Next, we determined TF and cfos expression in H3122 cells after SCH772984 treatment. SCH772984 is a selective inhibitor of ERK1/2, which adopts a unique kinase binding mode in ERK1/2 [39]. Pretreated H3122 cells with SCH772984 (2 μm/L, 24 h) to downregulate ERK1/2 phosphorylation, and compared cfos and TF protein expression with DMSO control. The results showed that cfos and TF proteins were downregulated when ERK1/2 phosphorylation was inhibited (Fig. 3B). Also, in line with the ERK1/2 inhibitor, pretreated H3122 cells with T-5224 (400nmol/L, 24 h), a small molecule selective inhibitor that selectively inhibits c-Fos/AP-1 binding to DNA [40], TF expression was downregulated compared to DMSO control (Fig. 3C).
After evaluating the mechanism of EML4-ALK regulating TF through the ERK1/2 /AP-1 axis in H3122 cells, we further verified these observations in H2228 cells. When H2228 was pretreated with the same concentration of Alectinib, flow cytometry analysis showed that TF expression was significantly down-regulated at 18 h and even more evident at 24 h (Fig. 4.A, left two panels). Western Blot results indicated that pERK1/2 and cFOS proteins were decreased with TF downregulation (Fig. 4A, middle panel). Also, mRNA expression of cFOS and F3 genes was both reduced after Alectinib treatment (Fig. 4A, right panel). In addition, ERK1/2 and AP-1 inhibitors also reduced the expression of TF mRNA and protein in H2228 cells (Fig. 4B and C).

EML4-ALK fusion protein regulates TF expression through pERK1/2/AP-1 pathway in H2228 cells. (A1-2) Pretreating H2228 with the same concentration of Alectinib in H3122. Flow cytometry analysis showed that TF expression was significantly down regulated at 18 h and even more obvious at 24 h (n = 3). (A3) Western Blot was observed decreased pERK1/2 and cFOS along with TF down regulation (n = 3). (A4) F3 gene and cFOS mRNA expression were similarly reduced compared with DMSO control which quantified by quantitative RT-PCR (n = 4). (B1-4) Pretreating H2228 cells with ERK1/2 inhibitor (SCH772984, 2 μm/L)) downregulated ERK1/2 phosphorylation, cfos and TF protein expression compared with DMSO control both at RNA (n = 6) and protein level (n = 3). (C1-4) Pretreating H2228 cells with AP-1 inhibitor (T-5224, 400 μm/L) also attenuated TF expression compared with DMSO control both at RNA (n = 6) and protein level (n = 3). Data are presented as means ± SEM. **p < 0.01, ***p < 0.001, determined by Student t test
Discussion
In recent decades, an increasing number of studies have found that ALK-rearranged NSCLC patients have a higher risk of VTE occurrence [41, 42]. Besides, a large proportion of these VTE events are developed in newly diagnosed NSCLC patients, supporting the underlying cancer-specific biology as a causal factor. According to previous clinical studies, the association between EML4-ALK rearrangement and VTE occurrence is verified. Thus, in this study, we firstly demonstrated that TF might be the key regulator of thrombus formation in EML4-ALK rearranged NSCLC in both in vitro and vivo experiments. Meanwhile, we also observed in vitro that EML4-ALK fusion protein in NSCLC cells enhanced venous thrombogenicity via pERK1/2 mediating AP-1-TF signaling. Consequentially, tumor-derived TF in the circulation triggered a coagulation cascade (Fig. 5).

Summary scheme graphic showing an association between EML4-ALK fusion protein mediated cellular pathway and thrombosis. Phosphorylated EML4-ALK fusion protein activated downstream the ERK1/2 pathway, inducing cFOS expression which was one of subunit of transcription factor AP-1. Thus, in turn result in upregulation of TF expression. Consequentially, tumor-derived TF in circulation triggered coagulation cascade
This research indicated that the EML4-ALK-pERK1/2-AP-1-TF axis might be a potential mechanism of VTE in ALK-rearranged NSCLC patients. The nodes of this axis also play essential roles in cancer initiation and progression [43,44,45], and both of them are therapeutically targetable proteins, which implies an important translational potential for the treatment of cancer-associated VTE.
Cancer-specific mechanisms of VTE in EML4-ALK fusion NSCLC cells
VTE occurrence in malignant patients could be due to multiple factors, including patient characteristics (like age, sex, and co-morbidities), cancer treatment (like chemotherapy, radiotherapy, and anti-vascular therapy), and cancer itself. Recent studies have discovered various cancer-specific mechanisms of VTE in brain, pancreatic, and colon cancer. These mainly included cancer cells expressing proteins that could alter the number of platelets and leukocytes in circulation (such as IL6, MUC1, MUC16, and MUC5AC) [46] and the expression of procoagulant proteins such as TF and PDPN, which could directly activate the coagulation cascade and upregulation of antifibrinolytic/anticoagulation proteins (PAI1 and HPSE) [47,48,49,50].
TF protein is a 47 kDa transmembrane protein that is highly expressed in many human cancers, including glioma, pancreatic, head, neck, lung, cervical, and prostate cancers, as well as leukemia [51]. Besides, an alternative spliced (as) form of TF that lacks the transmembrane domain can be released from cells. But studies about procoagulant activity of asTF are inconsistent [52,53,54]. So, the researches mostly focus on the full-length TF (TF). Studies have shown that tumor growth and angiogenesis are mediated by TF expression, and that TF expression correlates directly with oncogenic status, while circulating TF-positive extracellular vesicle level correlates with oncogenic status as well [55]. And tumor TF expression level is proven to influence cancer prognosis [55]. TF initiates the extrinsic coagulation cascade and can be released into circulation through cell-derived extracellular vesicles [56]. When TF-positive extracellular vesicles shed from cancer cells are associated with coagulation factor VII (FVII), this can trigger the blood coagulation cascade, leading to cancer-associated VTE [55]. There is no evidence that cancer cells-derived TF is activated in the circulation.
In the current research, as cohort studies indicated a heightened risk of VTE occurrence in patients with ALK-rearranged NSCLC [4, 6], we targeted inhibition of EML4-ALK fusion protein in NSCLC cells and observed that TF expression was significantly decreased. And this finding is similar to previous studies that increased cell surface expression of TF was associated with higher procoagulant activity in malignancies and higher circulating levels in vivo [57, 58], which was also shown in breast and ovarian cancer patients [59, 60]. Thus, inhibiting EML4-ALK fusion protein in cancer cells might decrease coagulability. Previous studies detected tumor-derived TF-positive extracellular vesicles (micropaticles or microvesicles) in the plasma of human pancreatic/colorectal tumor cells xenografted mice and examined venous thrombogenicity in a mouse model [61,62,63,64]. In addition, we demonstrated the function of TF by testing the activity of TF in human ALK-positive/ALK-negative NSCLC cell lines and also in mice bearing tumors derived from the corresponding cell lines. Furthermore, EML4-ALK fusion NSCLC cell lines with higher TF expression showed shorter clotting time in their culture supernatant involving plasma clotting assay. As a result, the plasma clotting assay might be influenced by samples containing TF-positive extracellular vesicles and tumor-secreted soluble procoagulants. And further experiments should be performed to verify this result.
Also, several studies within different cancer types indicated that increased tumor-derived TF-positive microvesicles upregulated the VTE incidence and venous clot weight in IVC stenosis or ligation mouse models [28, 29, 63]. In addition, Zwicker et al. proved that tumor-derived TF-positive microvesicles are associated with VTE in cancer patients [65]. Our findings are consistent with these results. Additionally, the present study has intensively considered oncogenic mutation and demonstrated that oncogenic mutation is a significant factor influencing cancer hypercoagulation.
We further identified TF expression in ALK-positive and ALK-negative NSCLC tissues and observed that ALK-positive NSCLC tissues exhibited higher TF expression. This finding was also indicated in a previous retrospective study, and the ALK-positive NSCLC patients in this cohort with higher TF expression showed a greater possibility of developing VTE compared to patients with EGFR mutation and both negative [66]. Collectively, the findings mentioned above suggest TF as the specific mechanism of EML4-ALK fusion in NSCLC-associated VTE.
EML4-ALK-pERK1/2-AP-1-TF axis in EML4-ALK fusion NSCLC cells
The EML4-ALK fusion protein is the product of the EML4-ALK fusion gene caused by chromosome translocation. The EML4 locus located in the short arm of chromosome 2 is broken, inversed, and fused with the ALK locus located on the same chromosome in somatic cells [7]. Thus, the fusion protein is composed of the amino-terminal half of EML4 protein ligating to the intracellular region of the receptor-type protein tyrosine kinase ALK. This action leads to dimerization and autophosphorylation of the ALK kinase domain and thus abnormally activates downstream signaling pathways, such as PI3K/AKT, JAK/STAT3, and RAS/ERK [67, 68], finally acquiring tumor-formation activity [13].
The regulatory mechanism of TF expression has been reported in many cancer models containing brain, breast, and colorectal cancer [2, 24, 69,70,71,72]. These studies revealed that multiple signaling pathways, transcription factors, and microRNAs, such as the Raf-MEK-ERK signaling pathway, transcription factor AP-1, and nuclear factor κB (NF-κB) could regulate TF expression in cancer cells [70]. And the mammalian target of rapamycin (mTOR) kinase pathway was identified in human pancreatic neuroendocrine tumor cell lines [73].
In the current study, targeted inhibiting EML4-ALK fusion protein in NSCLC cells showed downstream suppression of pERK1/2 and reduction of cFOS, a subunit of AP-1, accompanied by downregulation of TF. It is similar to a previous study within breast cancer cells, in which ERK1/2 kinase activity was measured in nuclear extracts and shown to be upregulated in MDA-MB-231 breast cancer cells with higher expression of TF mRNA [70]. Also, both AP-1 and NF-κB are important transcription factors for TF expression. However, compared to NF-κb, MDA-MB-231 nuclear extracts contain a molar excess of AP-1 [70]. And in our study, targeting ERK1/2, we also observed the downregulation of the subunit of AP-1 and TF. Finally, after pretreating with an AP-1 inhibitor, there is a significant reduction of TF expression both in H3122 and H2228 cell lines. Nevertheless, the former study only investigated the regulation of TF expression in non-specific breast cancer, and in this study, we linked EML4-ALK oncogenic alteration, the activation of downstream signaling pathways, and the coagulation cascade. Overall, the current study implies that EML4-ALK fusion protein in NSCLC cells may regulate the expression of TF through the pERK1/2-AP-1 axis.
The translational potential of the EML4-ALK-pERK1/2-AP-1-TF axis in the treatment of cancer-associated VTE
Thromboprophylaxis and anticoagulation therapy for cancer patients need to consider the risk of bleeding, the impact of the anti-tumor treatment process, and the additional sense of futility and burden caused by such treatment because it seems this action does not extend survival. Despite anticoagulation therapy, the incidence of recurrent pulmonary embolism (PE) remains relatively high [32, 74]. Hence, identifying patients at high risk of VTE is a current research focus. Notably, many studies suggested that high levels of TF expression were observed in different types of cancer, and the level of TF expression was associated with tumor progression and hypercoagulability [55]. Thus, TF expression might be a marker of cancer prognosis. However, little research on individualized anticoagulation therapy based on specific cancer types. This study aims to identify a non-anticoagulant target for VTE treatment in NSCLC with a specific oncogenic mutation.
This regulatory axis may be an appealing target for cancer-associated VTE in EML4-ALK fusion NSCLC patients. The nodes of this axis have essential roles in the occurrence and development of malignancy, and they all have corresponding specific targeted inhibitors. For example, small molecular ATP-competitive ALK inhibitors, which can effectively inhibit the autophosphorylation of ALK protein and suppress downstream signal activation [75, 76], have led to unprecedented survival benefits in EML4-ALK fusion NSCLC patients [77,78,79]. Although targeted ERK1/2 inhibitors are still in preclinical/clinical research [80, 81], inhibitors targeting upstream signaling protein MEK (RAS-RAF-MEK-ERK1/2 pathway) have been approved by FDA in the USA for the treatment of various solid tumors and have achieved excellent results in clinical [82,83,84]. Hrustanovic, Gorjan, and Tanizaki, J et al. [85, 86] found that EML4-ALK fusion NSCLC cells specifically depend on the MAPK pathway, and their sensitivity to MEK inhibitors is similar to that of KRAS or BRAF-positive lung adenocarcinoma cells. However, ALK inhibitors are not entirely effective, and single-drug treatment for long-term use can induce the reactivation of the downstream MAPK pathway and lead to drug resistance. Thus, a combined treatment strategy of initial ALK and MEK inhibitors is recommended to improve the survival rate of patients [87].
Now that the current data support the role of the axis in the pathogenesis of cancer-associated VTE in EML4-ALK fusion NSCLC cells, the node inhibitors may fulfill a dual purpose in patients with EML4-ALK fusion NSCLC. And the results suggest that clinicians should give careful consideration to providing thromboprophylaxis to NSCLC patients harboring EML4-ALK rearrangement. A non-anticoagulant target for VTE treatment might offer a new reference for thromboprophylaxis and anticoagulation therapy for cancer patients, and it might also provide more theoretical support for a combination treatment strategy of ALK and MEK inhibitors.
Limitations
This study has the following limitations. First, the mouse model used in this study is a subcutaneous ectopic lung cancer model, which cannot fully represent the real pulmonary environment with rich blood circulation. This limitation can be addressed by building an orthotropic model in the future. However, due to differences in the immune response and tumour microenvironment between mice and humans, animal models may not fully recapitulate human disease physiology. Additionally, the complex heterogeneity and microenvironmental factors present in clinical settings may not be fully reflected in the results obtained from the limited cell line experiments. Thus, further in vivo experiments and clinical studies should be performed to confirm these results. And these results should be verified in more types of cell lines. Also, there is a lack of evaluation of TF concentration in peripheral blood of lung cancer patients in the current study, limited by the fact that the number of enrolled ALK-rearranged NSCLC patients is too small. A comprehensive analysis will be carried out after further expanding the sample size in the future.
Conclusion
In summary, we have uncovered that EML4-ALK fusion protein in lung cancer cells enhances venous thrombogenicity through the pERK1/2-AP-1-tissue factor axis. This finding provides more theoretical support for a combination treatment strategy of ALK and MEK inhibitors and offers a new reference for thromboprophylaxis and anticoagulation therapy for cancer patients.
References
Ades S, Kumar S, Alam M, Goodwin A, Weckstein D, Dugan M, Ashikaga T, Evans M, Verschraegen C, Holmes CE (2015) Tumor oncogene (KRAS) status and risk of venous Thrombosis in patients with metastatic Colorectal cancer. J Thromb Haemost 13(6):998–1003
Unruh D, Schwarze SR, Khoury L, Thomas C, Wu MJ, Chen L, Chen R, Liu YX, Schwartz MA, Amidei C, Kumthekar P, Benjamin CG, Song K, Dawson C, Rispoli JM, Fatterpekar G, Golfinos JG, Kondziolka D, Karajannis M, Pacione D, Zagzag D, McIntyre T, Snuderl M (2016) Horbinski C.: Mutant IDH1 and thrombosis in gliomas. Acta Neuropathologica 132(6):917–930
Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martinez-Trillos A, Casetti I, Colomer D, Pieri L, Pratcorona M, Rotunno G, Sant’Antonio E, Bellini M, Cavalloni C, Mannarelli C, Milanesi C, Boveri E, Ferretti V, Astori C, Rosti V, Cervantes F, Barosi G, Vannucchi AM, Cazzola M (2014) Grp Assoc Italiana Ric Canc: clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 124(7):1062–1069
Dou F, Zhang Y, Yi J, Zhu M, Zhang S, Zhang D, Zhang Y (2020) Association of ALK rearrangement and risk of venous thromboembolism in patients with non-small cell Lung cancer: a prospective cohort study. Thromb Res 186:36–41
Su YP, Huo MR, Hua L, Zhang Y, Yi JW, Zhang S, Li J, Zhang YH (2021) Association of Venous Thromboembolism and early mortality in patients with newly diagnosed metastatic non-small cell Lung Cancer. Cancer Manage Res 13:4031–4040
Al-Samkari H, Leiva O, Dagogo-Jack I, Shaw A, Lennerz J, Iafrate AJ, Bendapudi PK, Connors JM (2020) Impact of ALK rearrangement on venous and arterial thrombotic risk in NSCLC. J Thorac Oncol 15(9):1497–1506
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara SI, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell Lung cancer. Nature 448(7153):561–U563
Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Hamada T, Haruta H, Watanabe H, Kurashina K, Hatanaka H, Ueno T, Takada S, Yamashita Y, Sugiyama Y, Ishikawa Y, Mano H (2008) Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell Lung cancer. Cancer Res 68(13):4971–4976
Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G (2008) The anaplastic Lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 8:11–23. https://doi.org/10.1038/nrc2291
Zhang SS, Nagasaka M, Zhu VW, Ou SI (2021) Going beneath the tip of the iceberg. Identifying and understanding EML4-ALK variants and TP53 mutations to optimize treatment of ALK fusion positive (ALK+) NSCLC. Lung Cancer 158:126–136. https://doi.org/10.1016/j.lungcan.2021.06.012
Atherly AJ, Camidge DR (2012) The cost-effectiveness of screening Lung cancer patients for targeted drug sensitivity markers. Br J Cancer 106:1100–1106. https://doi.org/10.1038/bjc.2012.60
Shinmura K, Kageyama S, Tao H, Bunai T, Suzuki M, Kamo T, Takamochi K, Suzuki K, Tanahashi M, Niwa H, Ogawa H, Sugimura H (2008) EML4-ALK fusion transcripts, but no NPM-, TPM3-, CLTC-, ATIC-, or TFG-ALK fusion transcripts, in non-small cell lung carcinomas. Lung Cancer 61:163–169. https://doi.org/10.1016/j.lungcan.2007.12.013
Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, Haruta H, Hamada T, Yamashita Y, Ishikawa Y, Sugiyama Y, Mano H (2008) A mouse model for EML4-ALK-positive Lung cancer. Proc Natl Acad Sci USA 105(50):19893–19897
Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D, Thomas R, Lee J, Richards WG, Sugarbaker DJ, Ducko C, Lindeman N, Marcoux JP, Engelman JA, Gray NS, Lee C, Meyerson M, Jänne PA (2008) EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in Lung cancer. Clin Cancer Res 14:4275–4283. https://doi.org/10.1158/1078-0432.CCR-08-0168
Hallberg B, Palmer RH (2013) Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology (vol 13, pg 685, 2012). Nat Rev Cancer 13(11):821–821
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Mino-Kenudson GL, Wei M, Shreeve GC, Ratain SM, Settleman MJ, Christensen J, Haber JG, Wilner DA, Salgia K, Shapiro R, Clark GI (2010) J. W., Iafrate A. J.: Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363(18):1693–1703
Le T, Gerber DE (2017) ALK alterations and inhibition in Lung cancer. Semin Cancer Biol 42:81–88. https://doi.org/10.1016/j.semcancer.2016.08.007
Golding B, Luu A, Jones R, Viloria-Petit AM (2018) The function and therapeutic targeting of anaplastic Lymphoma kinase (ALK) in non-small cell Lung cancer (NSCLC). Mol Cancer 17:52. https://doi.org/10.1186/s12943-018-0810-4
Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, Yamazaki S, Alton GR, Mroczkowski B, Los G (2007) Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic Lymphoma kinase and c-Met, in experimental models of anaplastic large-cell Lymphoma. Mol Cancer Ther 6:3314–3322. https://doi.org/10.1158/1535-7163.MCT-07-0365
Bevilacqua MP, Pober JS, Majeau GR, Fiers W, Cotran RS, Gimbrone MA Jr. (1986) Recombinant Tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: characterization and comparison with the actions of interleukin 1. Proc Natl Acad Sci U S A 83(12):4533–4537
Clauss M, Gerlach M, Gerlach H, Brett J, Wang F, Familletti PC, Pan YC, Olander JV, Connolly DT, Stern D (1990) Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J Exp Med 172(6):1535–1545
Abdol Razak N, Elaskalani O, Metharom P (2017) : Pancreatic Cancer-Induced Neutrophil Extracellular traps: a potential contributor to Cancer-Associated Thrombosis. Int J Mol Sci 18(3)
Razak NBA, Jones G, Bhandari M, Berndt MC, Metharom P (2018) : Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers 10(10)
Rong Y, Post DE, Pieper RO, Durden DL, Van Meir EG, Brat DJ (2005) PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res 65(4):1406–1413
Khorana AA, Ahrendt SA, Ryan CK, Francis CW, Hruban RH, Hu YC, Hostetter G, Harvey J, Taubman MB (2007) Tissue factor expression, angiogenesis, and Thrombosis in Pancreatic cancer. Clin Cancer Res 13(10):2870–2875
Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C (2009) Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med 206(9):1913–1927
Riedl J, Preusser M, Nazari PMS, Posch F, Panzer S, Marosi C, Birner P, Thaler J, Brostjan C, Lotsch D, Berger W, Hainfellner JA, Pabinger I, Ay C (2017) Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood 129(13):1831–1839
Belghasem M, Roth D, Richards S, Napolene MA, Walker J, Yin WQ, Arinze N, Lyle C, Spencer C, Francis JM, Thompson C, Andry C, Whelan SA, Lee N, Ravid K (2019) Metabolites in a mouse cancer model enhance venous thrombogenicity through the aryl hydrocarbon receptor-tissue factor axis. Blood 134(26):2399–2413
Thomas GM, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C, Wagner DD (2015) Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein Thrombosis in mice. J Thromb Haemost 13:1310–1319. https://doi.org/10.1111/jth.13002
Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, Iafrate AJ, Takeuchi K, Taiji M, Okuno Y, Fujita N, Engelman JA, Shaw AT (2014) Two novel ALK mutations mediate Acquired Resistance to the next-generation ALK inhibitor Alectinib. Clin Cancer Res 20(22):5686–5696
Campello E, Ilich A, Simioni P, Key NS (2019) The relationship between Pancreatic cancer and hypercoagulability: a comprehensive review on epidemiological and biological issues. Br J Cancer 121(5):359–371
Khorana AA, Mackman N, Falanga A, Pabinger I, Noble S, Ageno W, Moik F, Lee AYY (2022) : Cancer-associated venous thromboembolism. Nat Reviews Disease Primers 8(1)
Hisada Y, Mackman N (2019) Tissue factor and Cancer: regulation, Tumor Growth, and Metastasis. Semin Thromb Hemost 45(4):385–395
Felts SJ, Stoflet ES, Eggers CT, Getz MJ (1995) Tissue factor gene transcription in serum-stimulated fibroblasts is mediated by recruitment of c-Fos into specific AP-1 DNA-binding complexes. Biochemistry 34(38):12355–12362
Syrovets T, Jendrach M, Rohwedder A, Schüle A, Simmet T (2001) Plasmin-induced expression of cytokines and tissue factor in human monocytes involves AP-1 and IKKbeta-mediated NF-kappaB activation. Blood 97(12):3941–3950
Liu Y, Pelekanakis K, Woolkalis MJ (2004) Thrombin and Tumor necrosis factor alpha synergistically stimulate tissue factor expression in human endothelial cells: regulation through c-Fos and c-Jun. J Biol Chem 279(34):36142–36147
AGE-RAGE signaling pathway in diabetic complications - Homo sapiens (human) [https://www.genome.jp/kegg-bin/show_pathway?157724228798458/hsa04933.args]
Non-small cell lung cancer - Reference pathway [https://www.genome.jp/pathway/map05223]
Chaikuad A, Tacconi EM, Zimmer J, Liang Y, Gray NS, Tarsounas M, Knapp S (2014) A unique inhibitor binding site in ERK1/2 is associated with slow binding kinetics. Nat Chem Biol 10:853–860. https://doi.org/10.1038/nchembio.1629
Ishida M, Ueki M, Morishita J, Ueno M, Shiozawa S, Maekawa N (2015) T-5224, a selective inhibitor of c-Fos/activator protein-1, improves survival by inhibiting serum high mobility group box-1 in lethal lipopolysaccharide-induced acute kidney injury model. J Intensive Care 3:49. https://doi.org/10.1186/s40560-015-0115-2
Leiva O, Connors JM, Al-Samkari H (2020) : Impact of Tumor genomic mutations on thrombotic risk in Cancer patients. Cancers (Basel) 12(7)
Roopkumar J, Poudel SK, Gervaso L, Reddy CA, Velcheti V, Pennell NA, McCrae KR, Khorana AA (2021) Risk of thromboembolism in patients with ALK- and EGFR-mutant Lung cancer: a cohort study. J Thromb Haemost 19(3):822–829
Samatar AA, Poulikakos PI (2014) Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discovery 13(12):928–
Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, Piccolo S (2015) Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17(9):1218–
Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Hu Z, Barney KA, Degen JL (2007) Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood 110(1):133–141
Suzuki-Inoue K (2019) Platelets and cancer-associated Thrombosis: focusing on the platelet activation receptor CLEC-2 and podoplanin. Blood 134(22):1912–1918
Hermano E, Meirovitz A, Meir K, Nussbaum G, Appelbaum L, Peretz T, Elkin M (2014) : Macrophage polarization in pancreatic carcinoma: role of heparanase enzyme. J Natl Cancer Inst 106(12)
Geddings JE, Mackman N (2013) Tumor-derived tissue factor-positive microparticles and venous Thrombosis in cancer patients. Blood 122(11):1873–1880
Hisada Y, Garratt KB, Maqsood A, Grover SP, Kawano T, Cooley BC, Erlich J, Moik F, Flick MJ, Pabinger I, Mackman N, Ay C (2021) Plasminogen activator inhibitor 1 and venous Thrombosis in Pancreatic cancer. Blood Adv 5(2):487–495
Mir Seyed Nazari P, Riedl J, Pabinger I, Ay C (2018) The role of podoplanin in cancer-associated Thrombosis. Thromb Res 164(Suppl 1):S34–S39
Saidak Z, Soudet S, Lottin M, Salle V, Sevestre MA, Clatot F, Galmiche A (2021) A pan-cancer analysis of the human Tumor coagulome and its link to the Tumor immune microenvironment. Cancer Immunol Immunother 70(4):923–933
Censarek P, Bobbe A, Grandoch M, Schror K, Weber AA (2007) Alternatively spliced human tissue factor (asHTF) is not pro-coagulant. Thromb Haemost 97(1):11–14
Bogdanov VY, Versteeg HH (2015) Soluble Tissue Factor in the 21st Century: definitions, Biochemistry, and pathophysiological role in Thrombus formation. Semin Thromb Hemost 41(7):700–707
Goldin-Lang P, Tran QV, Fichtner I, Eisenreich A, Antoniak S, Schulze K, Coupland SE, Poller W, Schultheiss HP, Rauch U (2008) Tissue factor expression pattern in human non-small cell Lung cancer tissues indicate increased blood thrombogenicity and Tumor Metastasis. Oncol Rep 20(1):123–128
Hisada Y, Mackman N (2021) Tissue factor and extracellular vesicles: activation of Coagulation and Impact on Survival in Cancer. Cancers (Basel) 13. https://doi.org/10.3390/cancers13153839
Gardiner C, Harrison P, Belting M, Boing A, Campello E, Carter BS, Collier ME, Coumans F, Ettelaie C, van Es N, Hochberg FH, Mackman N, Rennert RC, Thaler J, Rak J, Nieuwland R (2015) Extracellular vesicles, tissue factor, cancer and Thrombosis - discussion themes of the ISEV 2014 Educational Day. J Extracell Vesicles 4:26901
Welsh J, Smith JD, Yates KR, Greenman J, Maraveyas A, Madden LA (2012) Tissue factor expression determines tumour cell coagulation kinetics. Int J Lab Hematol 34:396–402. https://doi.org/10.1111/j.1751-553X.2012.01409.x
Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW (2005) Oncogenic events regulate tissue factor expression in Colorectal cancer cells: implications for Tumor progression and angiogenesis. Blood 105:1734–1741. https://doi.org/10.1182/blood-2004-05-2042
Ueno T, Toi M, Koike M, Nakamura S, Tominaga T (2000) Tissue factor expression in Breast cancer tissues: its correlation with prognosis and plasma concentration. Br J Cancer 83:164–170. https://doi.org/10.1054/bjoc.2000.1272
Han LY, Landen CN Jr, Kamat AA, Lopez A, Bender DP, Mueller P, Schmandt R, Gershenson DM, Sood AK (2006) Preoperative serum tissue factor levels are an Independent prognostic factor in patients with ovarian carcinoma. J Clin Oncol 24:755–761. https://doi.org/10.1200/JCO.2005.02.9181
Yu JL, Rak JW (2004) Shedding of tissue factor (TF)-containing microparticles rather than alternatively spliced TF is the main source of TF activity released from human cancer cells. J Thromb Haemost 2(11):2065–2067
Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW (2005) Oncogenic events regulate tissue factor expression in Colorectal cancer cells: implications for Tumor progression and angiogenesis. Blood 105(4):1734–1741
Hisada Y, Ay C, Auriemma AC, Cooley BC, Mackman N (2017) Human pancreatic tumors grown in mice release tissue factor-positive microvesicles that increase venous clot size. J Thromb Haemost 15(11):2208–2217
Thomas GM, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C, Wagner DD (2015) Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein Thrombosis in mice. J Thromb Haemost 13(7):1310–1319
Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, Furie B (2009) Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res 15(22):6830–6840
Yang S, Yang L, Wu Y, Zhang C, Wang S, Ma N, Wang L, Wang Q (2020) Anaplastic Lymphoma kinase rearrangement may increase the incidence of venous thromboembolism by increasing tissue factor expression in advanced lung adenocarcinoma. Ann Transl Med 8:1307. https://doi.org/10.21037/atm-20-6619
Bayliss R, Choi J, Fennell DA, Fry AM, Richards MW (2016) Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted Drugs. Cell Mol Life Sci 73(6):1209–1224
Mano H (2008) Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in Lung cancer. Cancer Sci 99(12):2349–2355
Rong Y, Belozerov VE, Tucker-Burden C, Chen G, Durden DL, Olson JJ, Van Meir EG, Mackman N, Brat DJ (2009) Epidermal Growth Factor Receptor and PTEN Modulate Tissue Factor Expression in Glioblastoma through JunD/Activator Protein-1 transcriptional activity. Cancer Res 69(6):2540–2549
Zhou JN, Ljungdahl S, Shoshan MC, Swedenborg J, Linder S (1998) Activation of tissue-factor gene expression in breast carcinoma cells by stimulation of the RAF-ERK signaling pathway. Mol Carcinog 21(4):234–243
Yu JL, Xing R, Milsom C, Rak J (2010) Modulation of the oncogene-dependent tissue factor expression by kinase suppressor of ras 1. Thromb Res 126(1):E6–E10
Bourcy M, Suarez-Carmona M, Lambert J, Francart ME, Schroeder H, Delierneux C, Skrypek N, Thompson EW, Jerusalem G, Berx G, Thiry M, Blacher S, Hollier BG, Noel A, Oury CE, Polette M, Gilles C (2016) Tissue factor Induced by Epithelial-Mesenchymal Transition Triggers a Procoagulant State that drives Metastasis of circulating Tumor cells. Cancer Res 76(14):4270–4282
Lewis CS, Thomas HE, Orr-Asman MA, Green LC, Boody RE, Matiash K, Karve A, Hisada YM, Davis HW, Qi X, Mercer CA, Lucas FV, Aronow BJ, Mackman N, Versteeg HH (2019) mTOR kinase inhibition reduces tissue factor expression and growth of pancreatic neuroendocrine tumors. J Thromb Haemost 17(1):169–182
Kraaijpoel N, Bleker SM, Meyer G, Mahé I, Muñoz A, Bertoletti L, Bartels-Rutten A, Beyer-Westendorf J, Porreca E, Boulon C, van Es N, Iosub DI, Couturaud F, Biosca M, Lerede T, Lacroix P, Maraveyas A, Aggarwal A, Girard P, Büller HR, Di Nisio M (2019) UPE investigators: treatment and long-term clinical outcomes of incidental Pulmonary Embolism in patients with Cancer: an international prospective cohort study. J Clin Oncol 37:1713–1720. https://doi.org/10.1200/JCO.18.01977
Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y (2011) CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 19(5):679–690
Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, McTigue M, Deng YL, Liu W, Brooun A, Timofeevski S, McDonnell SR, Jiang P, Falk MD, Lappin PB, Affolter T, Nichols T, Hu W, Lam J, Johnson TW, Smeal T, Charest A, Fantin VR (2015) PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A 112(11):3493–3498
Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, Ou SHI, Perol M, Dziadziuszko R, Rosell R, Zeaiter A, Mitry E, Golding S, Balas B, Noe J, Morcos PN, Mok T, Investigators ALEX (2017) Trial: Alectinib versus Crizotinib in untreated ALK-Positive non-small-cell Lung Cancer. N Engl J Med 377(9):829–838
Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, Iyer S, Reisman A, Tursi WKD, Blackhall J (2014) Investigators Profile: first-line crizotinib versus chemotherapy in ALK-positive Lung cancer. N Engl J Med 371(23):2167–2177
Huang L, Jiang S, Shi Y (2020) Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol 13(1):143
Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V, Patel MR, Shapiro GI, Mier JW, Tolcher AW, Wang-Gillam A, Sznol M, Flaherty K, Buchbinder E, Carvajal RD, Varghese AM, Lacouture ME, Ribas A, Patel SP, DeCrescenzo GA, Emery CM, Groover AL, Saha S, Varterasian M, Welsch DJ, Hyman DM, Li BT (2018) First-in-Class ERK1/2 inhibitor Ulixertinib (BVD-523) in patients with MAPK mutant Advanced Solid tumors: results of a phase I dose-escalation and expansion study. Cancer Discov 8(2):184–195
Kohler J, Zhao YT, Li JQ, Gokhale PC, Tiv HL, Knott AR, Wilkens MK, Soroko KM, Lin MK, Ambrogio C, Musteanu M, Ogino A, Choi JY, Bahcall M, Bertram AA, Chambers ES, Paweletz CP, Bhagwat SV, Manro JR, Tiu RV, Janne PA (2021) ERK inhibitor LY3214996-Based treatment strategies for RAS-Driven Lung Cancer. Mol Cancer Ther 20(4):641–654
Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T (2011) Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on Colorectal cancer cell lines in vitro and in vivo. Int J Oncol 39(1):23–31
Long GV, Hauschild A, Santinami M, Atkinson V, Mandala M, Chiarion-Sileni V, Larkin J, Nyakas M, Dutriaux C, Haydon A, Robert C, Mortier L, Schachter J, Schadendorf D, Lesimple T, Plummer R, Ji R, Zhang P, Mookerjee B, Legos J, Kefford R, Dummer R, Kirkwood JM (2017) Adjuvant dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N Engl J Med 377(19):1813–1823
Zhao YJ, Adjei AA (2014) The clinical development of MEK inhibitors. Nat Reviews Clin Oncol 11(7):385–400
Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin LP, Neel DS, Sabnis A, Flanagan J, Chan E, Varella-Garcia M, Aisner DL, Vaishnavi A, Ou SHI, Collisson EA, Ichihara E, Mack PC, Lovly CM, Karachaliou N, Rosell R, Riess JW, Doebele RC, Bivona TG (2015) RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive Lung cancer. Nat Med 21(9):1038–
Tanizaki J, Okamoto I, Takezawa K, Sakai K, Azuma K, Kuwata K, Yamaguchi H, Hatashita E, Nishio K, Janne PA, Nakagawa K (2012) Combined effect of ALK and MEK inhibitors in EML4-ALK-positive non-small-cell Lung cancer cells. Br J Cancer 106(4):763–767
Zhou BY, Cox AD (2015) Up-front polytherapy for ALK-positive Lung cancer. Nat Med 21(9):974–975
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 82370058, 31570890 and 82000098), the Reform and Development Program of Beijing Institute of Respiratory Medicine (Grant no. Ggyfz202322), and Clinical Research Incubation Project, Beijing Chao-Yang Hospital, Capital Medical University (Grant no. CYFH202212).
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to conception and design; analysis and interpretation of data, critical writing or revising of the intellectual content, and final approval of the version to be published. All agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, Y., Yi, J., Zhang, Y. et al. EML4-ALK fusion protein in Lung cancer cells enhances venous thrombogenicity through the pERK1/2-AP-1-tissue factor axis. J Thromb Thrombolysis 57, 67–81 (2024). https://doi.org/10.1007/s11239-023-02916-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-023-02916-5