
Abstract
Millions of lives have been infected since the SARS-CoV-2 outbreak in 2019. The high human-to-human transmission rate has warranted a need for a vaccine to protect people. Although some vaccines are in use, due to the high mutation rate in the SARS-CoV-2 multiple variants, the current vaccines may not be sufficient to immunize people against new variant threats. One of the emerging concern variants is B1.1.529 (Omicron), which carries ~ 30 mutations in the Spike protein (S) of SARS-CoV-2 and is predicted to evade antibody recognition even from vaccinated people. We used a structure-based approach and an epitope prediction server to develop a Multi-Epitope based Subunit Vaccine (MESV) involving SARS-CoV-2 B1.1.529 variant spike glycoprotein. The predicted epitope with better antigenicity and non-toxicity was used for designing and predicting vaccine construct features and structure models. In addition, the MESV construct In silico cloning in the pET28a expression vector predicted the construct to be highly translational. The proposed MESV vaccine construct was also subjected to immune simulation prediction and was found to be highly antigenic and elicit a cell-mediated immune response. Therefore, the proposed MESV in the present study has the potential to be evaluated further for vaccine production against the newly identified B1.1.529 (Omicron) variant of concern.
Similar content being viewed by others

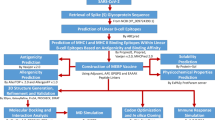
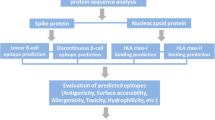
Introduction
The world witnessed a fatal outbreak of a novel coronavirus (COVID-19) infection, also known as severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), at the end of the year 2019 [1, 2]. It was preliminary identified as a causative agent for a series of unusual pneumonia in Wuhan City, China, and later led to the sporadic inter-continental pandemic [1, 3, 4]. On March 2020, as the infection has crossed most international borders, WHO designated the pathogen as SARS-CoV-2 and coronavirus disease 2019 (COVID-2019) (WHO.int). By January 2022, more than > 31.8 million confirmed cases with > 5.5 million death reported in 208 countries (WHO.int). The pandemic outbreak affected millions of lives globally due to mandatory isolations/quarantines and lockdown and travel restrictions resulting in economic mayhem (Socio-economic impact of COVID-19 | United Nations Development Programme 2020) [5]. In humans, COVID-19 infections lead to respiratory tract infections presenting a vast range of pathophysiological symptoms ranging from mild, such as fever, coughing, and shortness of breath, and to severe symptoms, such as pneumonia, multiple organ failure (kidney, liver, heart, and CNS), and severe acute respiratory failure leading to fatality [1, 6].
The coronaviruses (CoVs) are termed due to crown-like spikes on their surface and are classified into four main types α, β, γ, and δ, infecting wide ranges of hosts ranging from mammals to aves [7]. The SARS-CoV-2 is a β‐coronavirus with a nucleocapsid of helical symmetry and belongs to the subfamily Orthocoronavirinae, in the family Coronaviridae, order Nidovirales [8,9,10]. The genome of SARS-CoV-2 is a ( +) sense single-stranded RNA as the genetic component which encodes a set of viral replicase proteins and structural proteins including spike protein (S), membrane, envelope, nucleocapsid, and proteases which cleaves polyproteins and several uncharacterized proteins [10]. The SARS-CoV-2 genomes encode for 14 ORFs, including ORF1a and ORF1ab, which encodes for 16 non-structural proteins (NSP1-16) and four essential structural proteins, including S glycoprotein, envelope (E) protein, membrane (M) protein, and nucleocapsid (N) protein which are vital for viral assembly and invasion of SARS-CoV-2 [11]. The polyproteins 1a and 1ab (pp1a and pp1ab) are auto-proteolytically cleaved by SARS-CoV-2 proteases to release 11 (pp1a) and 5 (pp1ab) functional proteins necessary for viral replication, survival and proliferation [12,13,14]. The homotrimer S protein constitutes the spikes on the viral envelope responsible for attachment to host cells. The three transmembrane domains of M protein determine the virion shape, facilitate membrane curvature, and bind to nucleocapsid. The E protein plays a role in virion assembly, pathogenesis, and release, and the N protein is essential for binding with the viral RNA genome [15].
The mode of entry of SARS-CoV-2 is identical to SARS-CoV, using human angiotensin-converting enzyme 2 (ACE2) and CLEC4M/DC-SIGNR receptor for attachment to the cell membrane [16,17,18]. The ACE2 is also expressed in the mucosa of the epithelial cell and the oral cavity of the tongue, type II alveolar cells (AT2), among others, acting as entry routes for viruses through endosomal pathways [19]. The SARS-CoV-2 S protein is a homotrimer class I type fusion protein that enables viral entry to human cells via ACE2 receptor [16]. The S protein is a 1273 residue glycoprotein with three conformational states: pre-fusion native state, pre-hairpin intermediate state, and post-fusion hairpin state (spike protein) [16]. The S protein comprises two subunits, S1, which is required for receptor recognition and S2, required for membrane fusion. The S1 subunit has C-terminal RBD (receptor-binding domain), primarily interacting with the human ACE2 receptor [3]. The S protein undergoes a conformational change when it fuses with the ACE2 receptor destabilizing the pre-fusion trimer, resulting in the discharge of the S1 subunit, permitting the transition of the S2 subunit to a steady post-fusion state [20]. Then, the host cell-mediated S protein priming by a cellular serine protease TMPRSS2 is responsible for the virus entering the host cell [13, 16]. After the virus has entered the host cell, it follows the typical replication cycle, infection and proliferation of a ( +) sense RNA virus such as MERS-CoV and SARS-CoV [21].
So far, since the pandemic breakout, the S glycoprotein is known to have a pivotal role in viral attachment and infection, in the induction of neutralizing-antibody and T-cell responses, and protective immunity, thus designating it as a top candidate for vaccine production. A vaccine based on the S protein and its epitope could induce antibodies in response to block virus binding and fusion [21, 22]. RNA viruses are known to develop high mutation rates related to the virus’s high proliferation, infection, and evasion of the host immune system. These high mutation rates are due to the low fidelity of the RNA-dependent RNA polymerase (RdRp) [23]. It also has become evident that the high mutation frequency in the S protein is correlated with specific variants of concern that have caused significant mortality in certain geographical regions of the globe [24, 25]. Since the COVID-19 pandemic, viral genome sequencing has been accelerated and shared at an unprecedented rate to identify the lineages and trace the virus evolution (SARS-CoV-2 Variant Classifications and Definitions 2021). More than one million SARS-CoV-2 sequences are available via the Global Initiative on Sharing All Influenza Data (GISAID), enabling real-time surveillance of the pandemic unfolding [26]. Since late 2020, SARS-CoV-2 evolution has witnessed numerous mutations, labeling the new variants as variants of concern, changing the virus characteristics, such as transmissibility and antigenicity [27]. A large number of SARS-CoV-2 variants have been recently revealed through genome sequencing projects, and the characteristics mutations of these variants are, unfortunately, the targets of most of the currently licensed and used COVID-19 vaccines [28]. Recent reports have summarized most of the SARS-CoV-2 variants identified and their mutational landscape [27,28,29,30,31,32].
A recent emerging cluster of cases reported in South Africa identified new variants of SARS-CoV-2 by a team led by Tulio de Oliveira at the University of KwaZulu-Natal. The variant carries many mutations also found in other variants, including Delta, and reports suggest that it is spreading at warp speed across South Africa and anecdotal reports across other countries. Due to the high rate of infectivity, WHO designated the strain as B.1.1.529, a variant of concern naming it Omicron on 26 November 2021. The Omicron joins Delta, Alpha, Beta, and Gamma on the current WHO list of variants of concern (Tracking SARS-CoV-2 variants 2021). The B.1.1.529 (Omicron) variant harbors 30 amino acid substitutions, three small deletions, and one small insertion in the SARS-CoV-2 S protein — the host immune system’s prime target antibodies recognize, potentially dampening their potency. Out of these 30 mutations, some of them were also present in the other strains, such as Delta and Alpha were found to be problematic and linked to high infectivity and enhanced ability to evade antibodies (Science Brief: Omicron (B.1.1.529) Variant | CDC 2021; Tracking SARS-CoV-2 variants, 2021) [30]. The mutational profile of 30 mutations can be found in Table S1. The key amino-acid substitutions in the S protein are A67V, del69-70, T95I, del142-144, Y145D, del211, L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, and L981F (substitutions in RBD region (no. 15 mutations) are highlighted in bold type) (Fig. 1). The RBD domain of the S protein is the prime target of neutralizing antibodies generated following infection by SARS-CoV-2 [33], and the same is the component of currently used mRNA and adenovirus-based vaccines licensed for use and others awaiting regulatory approval [34].

SARS-CoV-2 S glycoprotein (B.1.1.529 Omicron) trimer structure model in different representations: top view (a), surface representation (b), cartoon representation (c). The protein chain is colored in gray, and mutations in the B.1.1.529 Omicron variant are colored in cyan, orange and green, respectively, for three chains in the trimer model. (d) Structural superimposition of RBD domain from S protein of Wuhan reference (gray) and B.1.1.529 Omicron variant (blue). The mutations in B.1.1.529 Omicron S protein are labeled with residue and number in the yellow box
Public vaccination is the best approach and tool for controlling and eliminating the pandemic virus, and the same vaccine development is urgently needed [35]. The diagnosis of COVID-19 is nowadays reliable, efficient, and quick, though public vaccination is required to prevent COVID-19 and its severe repercussions [36]. Since the outbreak of COVID-19, numerous vaccine candidates have been proposed, deployed, or in the development stage. A total of 139 and 194 vaccine candidates are in clinical and pre-clinical development, respectively (WHO.int). Diverse vaccine candidates include but are not limited to live-attenuated or inactivated whole virus, RNA and DNA-based vaccines, subunit vaccines, viral vectors, self-assembling virus-like particle vaccines, and others, with peculiar advantages and disadvantages. Subunit vaccines constructed on S protein have been highly effective for eliciting immunogenicity against the previous coronavirus breaks out, such as SARS and MERS [37, 38]. One promising approach includes designing a subunit vaccine with numerous and diverse antigenic variables which can produce an inclusive spectrum of native viral antigens [42]. A typical vaccine development cycle takes a minimum of ten years from lab research to its approved use after all clinical assessments, making traditional approaches time-consuming and labor intensive [39, 40]. The advent of genome sequencing and high-performance computational technology has made immunoinformatics predictions cost-effective and convenient [41,42,43]. In silico immuno-informatics approaches have reduced the number of experiments needed for vaccine development, and it has been demonstrated to identify protein immunogenic attributes with high efficiency [44,45,46]. Numerous researchers have employed immunoinformatics approaches for the development of novel and potential multi-epitope vaccines against several virulent pathogens including dengue and Ebola virus, Streptococcus parauberis, and SARS-CoV-2 [43,44,45,46,47,48,49,50]. The design of a multi-epitope vaccine candidate targeting different non-structural and structural proteins of SARS-CoV-2 have been recently reported by researchers globally with the potential to be an effective vaccine candidate [51,52,53,54,55,56,57,58]. Recently, researchers have also attempted to identify epitopes against Omicron variants of SARS-CoV-2 and linked them with linkers to finally generate what is known as multi-epitope based peptide vaccine [59,60,61].
In this work, we employed immunoinformatics to predict multiple immunogenic proteins from the SARS-CoV-2 proteome and thereby design a multi-epitope vaccine. In the present study, we used a computational-based immunoinformatics screening approach to identify and construct multi-subunit vaccine candidates. We used SARS-CoV-2 S protein (B.1.1.529 variant) to identify antigenic elements generating B-cell and T-cell immunity. We predicted B-cell and T-cell epitopes and evaluated their further toxicity, allergenicity, and antigenicity properties. We also performed a population coverage analysis and estimated the broad coverage globally. Furthermore, B-cell and T-cell epitopes were linked with appropriate adjuvants to generate a multi-subunit epitope vaccine (MESV). We also performed immune simulations to estimate the possible immune response generated by identified MESV. Finally, to assess the suitability of recombinant MESV production, we also performed in silico cloning.
Methodology
Data retrieval, structural, and physicochemical analysis of SARS-CoV-2 S protein
The protein sequence of S protein (B.1.1.529 variant) was derived from the GISAID (https://www.gisaid.org/) Database (Accession ID EPI_ISL_6980876). For sequence level comparison with wild-type strain, the reference strain (NC 045,512.2) was compared with B.1.1.529 variant S protein for sequence similarity using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) [62]. For tertiary structure prediction of B.1.1.529 S protein, the amino-acid sequence was submitted to the I-TASSER (https://zhanggroup.org/I-TASSER/) [63]. For structure refinement, the GalaxyRefine (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) [75] and ModRefiner (https://zhanggroup.org/ModRefiner/) [64] programs were used and to assess the Ramachandran statistics, the SAVES server (https://saves.mbi.ucla.edu/) was utilized as reported in our earlier studies [65, 66]. The GRAVY (grand average of hydropathicity), half-life, sub-atomic weight, instability index, aliphatic record, and amino acid atomic composition of the protein sequence were computed through ProtParam (http://web.expasy.org/protparam/).
Prediction of B-cell linear and discontinuous epitopes
To identify the linear B-cell epitope areas in the antigen sequence, the ABCpred (http://crdd.osdd.net/raghava/abcpred/) [68] was utilized. ABCpred operates based on artificial neural networks, which are collections of mathematical models that imitate some of the recognized aspects of the biological nervous system and draw on analogies of adaptive biological learning. The putative epitopes are then tested for antigenicity using Vaxijen 2.0 (http://www.ddg-pharmfac.net/vaxijen/) [69]. We also predicted the discontinuous epitopes with much more substantial attributes than the linear epitopes. The identification of discontinuous B-cell epitopes is a significant difficulty in vaccine design. The DiscoTope (https://services.healthtech.dtu.dk/service.php?DiscoTope-2.0) web server [70] was used to predict the surface area accessibility as well as amino acids that create discontinuous B-cell epitopes as identified through X-ray crystallography of antigen/antibody protein structures.
Prediction of CTL and HTL epitopes
The IEDB MHC I binding prediction methods (http://tools.iedb.org/mhci) were used to predict S glycoprotein CD8 + T-cell epitopes from the sequence. This method unifies epitope prediction restricted to many MHC class I alleles and proteasomal C-terminal cleavage using complicated artificial neural network applications. For predicting the CD4 + T-cell epitopes (peptides), the MHC II (http://tools.iedb.org/mhcii/) was used. Briefly, the epitopes showing the highest binding diversity with the various HLA serotypes were chosen. Furthermore, these epitopes were submitted to Vaxijen 2.0 server for assessing their antigenicity at a 0.7 value threshold. All the top-scoring epitopes identified from each tool mentioned above were then submitted to the IEDB T-cell class I Immunogenicity predictor (http://tools.iedb.org/immunogenicity/). After the determination of the HLA-confined CD8 + and CD4 + T-cell epitopes, the epitope toxicity and allergenicity were predicted using the ToxinPred server (http://crdd.osdd.net/raghava/toxinpred/) [71] and AlgPred (http://crdd.osdd.net/raghava/algpred/) [67] server respectively. Finally, the non-allergenic epitopes predicted from the above servers were chosen as T-cell epitopes.
Population coverage analysis
The IEDB Population Coverage tool (http://tools.iedb.org/population/) was used to test chosen epitopes from the HLA class I and class II families and their corresponding binding leukocytes antigens. With a known HLA background, this tool computed the distribution or fraction of persons projected to respond to the selected epitopes. Likewise, the IEDB device processes the usual number of epitope hits/HLA allele blends perceived by the all-out populace, just as the most extreme and least number of epitope hits are perceived by 90% of the chosen populace. The HLA genotypic frequencies are estimated, and T-cell epitopes are searched by region, ethnicity, and country.
Design, structural modeling, and validation of multi-epitope vaccine construct
The most efficient epitopes likely to generate subunit immunization should have the qualities such as exceptionally antigenic, immunogenic, non-allergenic, and non-toxic. Those epitopes with the following qualities were chosen further to construct multi-epitope subunit vaccine (MESV). An adjuvant was also appended with the EAAAK linker to the primary cytotoxic T lymphocytes (CTL) epitope. The different epitopes were connected with AAY, GPGPG, and KK linkers. β-defensin has been used as an adjuvant since it is a 45 amino acids long peptide that is an immunomodulator and an antimicrobial agent. The 6X His-tag was inserted into the vaccine constructs at the C-terminal end for easy purification using affinity chromatography. For checking the sequence homology of the MESV constructs with the human proteome, the MESV sequence was submitted for the BLAST sequence homology tool. Physicochemical properties such as half-life, theoretical isoelectric point (pI), hydropathy, and aliphatic index of the MESV construct were assessed using the Protparam (https://web.expasy.org/protparam/) tool [72]. The MESV construct’s allergenicity was assessed using the AllerTOP v.2.0 (https://www.ddg-pharmfac.net/AllerTOP/) [73, 74]. For evaluation of the secondary structure component of the MESV construct, PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) [76] was used for the identification of secondary structural elements such as alpha-helices, extended chains, the number of beta turns, and random coils.
The homology model of MESV was constructed using I-TASSER (https://zhanggroup.org/I-TASSER/) webserver. The GalaxyRefine [63] and ModRefiner [75] servers were used to energy minimize the predicted MESV 3D structure. The Ramachandran plot statistical evaluation was performed using the SAVES web server, followed by structural validation analysis utilizing the PROSA (https://prosa.services.came.sbg.ac.at/prosa.php) webserver. For estimating the conformational B-cell epitopes of the proposed MESV, the ElliPro tool (http://tools.iedb.org/ellipro/) was used [77]. It estimates the residual protrusion index (PI), protein structure, and neighbour residue clustering to predict epitopes.
Molecular docking of the MESV and human immune receptors
Studying the binding affinity of the epitopes, which characterizes their molecular interaction with the human HLA class I and II molecules, is one of the finest approaches to gaining insight into their immune response. The CASTp server (http://sts.bioe.uic.edu/castp/) [78] was used to estimate the binding pockets on HLA-class I and II molecules as well as the human immunological receptor (TRL3, PDB: 3ULV, and TLR5, PDB: 3J0A). The CASTp server provides an extensive and detailed quantitative assessment of a protein’s topographic characteristics. The molecular docking of MESV against TLR3 structure and TLR5 structure (PDB: 3J0A) was carried out using GalaxyPepDock (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=PEPDOCK) [79]. The exact output was re-submitted to ClusPro (https://cluspro.bu.edu/) for further refinement [80]. The entire docking process has been previously validated and followed as mentioned in our earlier studies on different targets and using different approaches [80,81,82,83,84,85,86].
MM/GBSA-based evaluation of docked pose
The best scoring dock complex was retrieved from the above docking process, and the top pose was assessed through molecular mechanics/generalized born surface area (MM/GBSA) calculations computed by the HawkDock Server (http://cadd.zju.edu.cn/hawkdock/) [87]. HawkDock investigates the docked pose on a per residue basis across electrostatic potentials, Van der Waal potentials, polar solvation free energy, and solvation free energy using empirical models. The default parameters were used to minimize the docked pose complex for 5000 steps, including steepest descents (2000 cycles) and conjugate gradient minimizations (3000 cycles) through an implicit solvent model, the ff02 force field.
MD simulation and immune simulation of the multi-epitope vaccine
The WAXSIS web server (http://waxsis.uni-goettingen.de/) was utilized for the small-and wide-angle X-ray scattering (SWAXS) to study the properties of the peptide vaccine construct [88]. An accurate prediction of the generated curves was required. The predictions are complicated due to the scattering contribution from the hydration layer and the impact of temperature fluctuations. The MD simulation provides a complete model of the hydration layer and solvent. The Guinier analysis evaluated the protein compactness and the radius of gyration. The Vaccine-TLR5 complex was simulated and investigated; the deformability, B factor, eigenvalues, and elastic network of the docked complex were also analyzed.
To evaluate the immunogenicity and immune response characteristics, the MESV was subjected to immune simulation using a C-ImmSim online server (http://150.146.2.1/C-IMMSIM/index.php) [89]. C-ImmSim predicts related immunological reactions using a machine learning approach for three anatomical compartments: (i) bone, where hematopoietic stem cells are activated, and myeloid cells are created, (ii) the lymphatic organ, and (iii) the thymus, where naive T lymphocytes are selected to avoid autoimmune conditions. The simulations were carried out to administer two-dose injections of MESV at six-week intervals on days 0 and 42. Each time step was positioned at 100 based on the default values, indicating that each time step is 8 h long and time step 1 is the injection provided at time zero. As a result, two injections were given at 6-week intervals. The T-cell memory will be evaluated constantly in this situation. The plot analysis generated a graphical interpretation of the Simpson index.
In silico cloning and codon optimization
For maximizing vaccine expression in the host E. coli K12 system, codon optimization was carried out using the Java Codon Adaptation Tool (JCat) (http://www.jcat.de/) [90]. The restriction enzymes, prokaryotic ribosome binding site, and rho-independent transcription termination were kept in the default state. The acquired codon adaptation index (CAI) value and GC content of the adapted sequence against a certain threshold were assessed. The improved nucleotide was then cloned into the pET28a ( +) vector prototype using the SnapGene 4.2 tool.
Results
Data retrieval, structural, and physicochemical analysis of SARS-CoV-2 S protein
The protein-coding sequence of S protein (B.1.1.529 variant) was retrieved from the GISAID database in FASTA format. The sequence homology of S protein B.1.1.529 variant with reference strain (NC 045,512.2) was 97% (Supplementary Fig. S1). The tertiary structure of full-length S protein (B.1.1.529 variant) was generated with I-TASSER with a C-score of − 1.60 (− 5 to 2, the higher the better) and an estimated TM-score of 0.52 ± 0.15. Ideally the homology model prediction having C-score of > − 1.50 and a TM-score > 0.5 is considered as a model of correct global topology [62, 63]. The Ramachandran statistics of the 3D model were 73.4% in the core, 20.4% in allowed, 3.5% in generously allowed, and 2.7% in the disallowed region (Fig. 2). After rounds of energy refinement, the Ramachandran statistics were improved to 86.7% in the core, 9.7% in allowed, 1.6% in generously allowed, and 1.9% in the disallowed region. The physiochemical attributes of the S protein (B.1.1.529 variant) are as follows: no. of amino acids (1270), Mol. Wt. 14,1300 Da, pI 7.14, estimated half-life > 10 h in prokaryotes and 30 h in eukaryotes, instability index 34.12 classifying the protein as stable. The estimated aliphatic index 84.95 indicating higher thermostability of the protein [90]. The negative GRAVY score of − 0.080 indicates the protein is hydrophilic and will interact with the water molecules [91].

SARS-CoV-2 S glycoprotein (B.1.1.529 Omicron) trimer structure model with each color representing three different chains
Linear and discontinuous B-cell epitopes
B-cell epitopes play a significant role in viral infection resistance. The amino acid screening methods analyzed possible B-cell epitopes from the S protein (B.1.1.529 variant) sequence. For predicting possible B-cell epitopes, a consensus-based technique was utilized. Three potential linear B-cell epitopes with non-allergenic, non-toxicity, and antigenic properties were identified after stringent criteria selection (Table 1). The peptide “HVTYVPAQEKNFTTAP” exhibited the most significant antigenic index compared to the other predicted B-cell epitope candidates due to its higher antigenicity score of 0.8375 which is above the virus threshold of 0.4 [68]. Our results with B-cell epitope prediction had higher antigenicity score compared to another recent report of Omicron S protein epitope prediction [60]. The linear B-cell epitopes were mapped on the trimeric S glycoprotein model, as shown in Fig. 3. The discontinuous B-cell epitopes were predicted by Discotope 2.0 using the trimeric S protein (B.1.1.529) model. The positions of discontinuous epitopes were mapped on the surface of the model structure of S protein (Fig. 4). All the discontinuous B-cell epitopes were mapped on the fully-exposed “spike head” region (Fig. 4), and no discontinuous B-cell epitopes were found on the less-exposed “spike stem” and the “spike root” region of the S glycoprotein (Fig. 4). Predicted discontinuous epitopes were selected from Omicron’s S glycoprotein’s complete protein chain component and graded according to their propensity score (Table 2).

SARS-CoV-2 S glycoprotein (B.1.1.529 Omicron) trimer structure model with mapped B-cell (yellow) and T-cell (green) predicted epitopes. The novel mutations of the B.1.1.529 Omicron variant in the predicted epitopes are red

SARS-CoV-2 S glycoprotein (B.1.1.529 Omicron) trimer structure model with mapped discontinuous B-cell (green) epitopes
T-cell (THTL and TCTL) epitope prediction
The S protein (B.1.1.529) sequence was tested against several HLA class 1 alleles. The peptides were chosen depending on their rate rankings and the number of alleles they possibly bind. Also, the peptides were exposed to antigenicity tests utilizing Vaxijen 2.0. Nine epitopes were chosen for the subsequent analysis regarding the antigenicity scores. The main peptides have high restricting energy to HLA class I atoms and show a non-allergenic property. Before considering vaccine design, allergenicity prediction is critical, as vaccination candidates may induce a type II hypersensitivity reaction. Allergen 1.0 and ToxinPred assessed the epitope’s non-allergenicity and non-toxicity properties. The S protein (B.1.1.529) sequence was also searched through many MHC-II alleles for the HLA class II T-cell epitopes. Three epitopes were chosen for their antigenic characteristics. These non-allergenic epitopes can elicit an immunological response by activating either or all of the IFN-γ and IL-4 cytokines. Therefore, an ideal peptide anticipated as an epitope should have high antigenicity and capacity to bind with many alleles, thus having a high potential to initiate a strong defense response upon immunization. A total of 9 MHC class-I allele binding peptides and 3 MHC class-II allele binding peptides were obtained using the above stringent criteria virus threshold of 0.4 [68] (Tables S2A and S2B). The T-cell epitopes (MHC class I and II) were mapped on the trimeric S glycoprotein model, as shown in Fig. 3.
The non-allergenic peptides were as follows: “IPFAMQMAY” restricted to HLA-B*35:01,HLA-B*53:01, HLA-B*51:01 HLA allele; “LPFFSNVTW” restricted to HLA-B*35:01, HLA-B*51:01, HLA-B*57: 01, HLA-B*07:02, HLA-B*58:01 with an antigenicity score of 1.0808; “SVYAWNRKR” attaches to 7 alleles HLA-A*31:01, HLA-A*33:01, HLA-A*68:01, HLA-A*03:01, HLA-A*68:01, HLA-A*11:01, HLA-A*30:01; “EILPVSMTK” restricted to HLA-A*68:01, HLA-A*11:01, HL-A*11:01, HLA-A*03:01,HLA-A*33:01, HLA-A*33:01; the peptide “TEILPVSMTK” would be able to bind with 3 alleles HLA-A*11:01, HLA-A*03:01, HLA-A*68:01; “PFAMQMAYR” restricted to HLA-A*33:01; “IPFAMQMAYR” restricted to HLA-A*33:01, HLA-A*35:01; “KFGAISSVL” restricted to HLA-A*24:02, HLA-A*23:01, “PFAMQMAYRF” restricted to HLA-A*23:01, HLA-A*24:02 (Table S2A).
The peptide “PYRVVVLSFELLHAP,” with an antigenicity score of 0.7072, attaches to 8 HLA alleles: HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DRB1*08:02, HLA-DRB4*01:01, HLA-DRB5*01:01. The epitope “RVVVLSFELLHAPAT,” with an antigenicity score of 0.7485, is also restricted to 8 HLA alleles: HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DRB1*08:02, HLA-DQA1*03:01/DQB1*03:02, HLA-DQA1*04:01/DQB1*04:02, HLA-DQA1*05:01/DQB1*02:01. The peptide “YRVVVLSFELLHAPA” binds to allele HLA-DPA1*03:01/DPB1*04:02, HLA-DPA1*02:01/DPB1*01:01, and HLA-DPA1*01:03/DPB1*02:01 with the antigenicity score of 0.7072. The HLA-DRB1 is the most common and versatile MHC-II molecule.
Population coverage of CTL and HTL epitopes
The distribution of HLA alleles varies among heterogeneous ethnic groups and geographical regions. Therefore, while constructing a viable epitope-based vaccine, it is necessary to consider population coverage. The selected MHC-I class epitopes showed higher individual percentage cover when examined with the entire global population. A total of 76.38% of the world’s individuals are capable of responding to the median of one MHC-I epitope (Table 3).
It was inferred that North America would possibly show a significant response to the selected HLA-I class-restricted epitopes out of the selected continents and subcontinents. East Asia, Oceania, West Indies, West Africa, Northeast Asia, and Southeast Asia had the highest population coverage of 83.65%, 82.01%, 81.85%, 79.54%, 77.90%, and 77.45%, respectively, while the Central America population had the lowest population coverage at 6.44%. The MHC-I class epitope’s population coverage has been considerably higher than the MHC-II class epitopes (Table 4) (Figs. 5 and 6).

Population coverage of MHC class I and II epitopes in selected geographical regions

a 3D structure model of MESV shown in cartoon representation, and b secondary structure prediction of the MESV
Structural model and physicochemical properties of multi-epitope subunit vaccine (MESV)
The constructed multi-epitope subunit vaccine (MESV) comprises 280 amino acids from 15 antigenic B-cell and T-cell epitopes linked with an immunoadjuvant. The tertiary structure of the multiple epitope vaccine was constructed using the I-TASSER server with a C-score of − 4.11 (C-score range: − 5 to 2, the higher the better) and an estimated TM-score of 0.20 ± 0.05 and was further structurally validated using Ramachandran statistics and the ProSA-web server. Ideally, the homology model prediction having C-score of > − 1.50 indicates a model of correct global topology [62]. The Ramachandran statistics of the 3D model were 66.5% in the core, 26.4% in allowed, 5.0% in generously allowed, and 2.1% in the disallowed region. After rounds of energy refinement, the Ramachandran statistics were improved to 86.8% in the core, 10.7% in allowed, 0.4% in generously allowed, and 2.1% in the disallowed region (Fig. S4). In addition, the MESV structural model had a Z-score of − 2.32, indicating that it was a relatively acceptable model. The physiochemical attributes of MESV are as follow: no. of amino acids (280), Mol. Wt. 31,277.77 Da, pI 10.19, estimated half-life 30 h in eukaryotes, instability index 36.51 classifying the protein as stable. The estimated aliphatic index 71.61 indicating higher thermostability of the protein [90]. The negative GRAVY score of − 0.139 indicates the protein is hydrophilic and will interact with the water molecules [91].
Molecular docking between the vaccine and the toll-like receptor (TLR5)
TLR5 was chosen for its immunomodulatory potential to induce IFN-gamma. It was demonstrated in the study that our chosen CD4 + epitopes induced both Th1 and Th2 cytokines. In addition, the vaccine’s molecular interaction with TLR5 (PDB: 3J0A) was studied, considering their refined binding energies and numerous interacting residues. The possible orientation and interactions between the MESV and TLR5 are shown in Fig. 7a and b, respectively. The initial pose of MESV and TLR5 docked complex was first generated using GalaxyPepDock and then, the pose with the highest docking score (lowest free energy) was subjected to ClusPro server. The predicted binding free energy of the complex in the cluster center was − 48.61 (kcal/mol) as predicted by HawkDock server. The docking score of MESV-TLR5 complex was also compared to a recent report on MESV prediction using immuninformatics in Omicron spike protein [60]. The docking score for MESV-TLR5 was reported to be − 48.61 (kcal/mol), which is better than the reported in similar study with a score of − 35.16 kcal/mol.

a Docked complex of MESV (red) and TLR5 receptor (blue); b, c docked complex of MESV (blue) and TLR5 receptor (red) in surface representation at different angles
MD simulation and immune simulation of the MESV
The radius of gyration shows the rigidity of the vaccine, and the Rg value of the vaccine-TLR5 construct was 36.275. The eigenvalue of the vaccine-TLR5 complex was 1.697699e-06; the value is low, which shows the easier deformation of the complex, which means that the vaccine-TLR5 construct will activate the immune cascade. At some points, the B factor value of the complex is high, which directly corresponds to the higher deformability. The MD simulation results are shown in Fig. 8. The immune simulation of the vaccine was also carried out to simulate the immune response post-vaccination. At administering every dose of the MESV peptide vaccine, there is a sharp increase in the antibody response and a co-currently decrease in the antigen level. The antibody response was significantly higher, i.e., IgM > IgG humoral response highlighting the seroconversion (Fig. 9a). Also, following the second dose, IFN-γ response was higher (accompanying both CD8 + T-cell and CD4 + Th 1 response), and IL-10 and TGF-b cytokines response related to T-reg phenotype (Fig. 9b). Overall, the immune simulation results showed that each dose simultaneously increased the immune response. B-cell population per cell analysis revealed that the overall B-cell population and B-cell memory responses were higher and stable, showing minimal decay for over 300 days (Fig. 9c). After the second dose, there was a simultaneous rise in Th effector cell phenotype and a lower Th-memory readout (Fig. 9d). In addition, there was a concomitant increase in natural killer cell activities (Fig. 9e) throughout the simulation. To summarize the results, the points mentioned above are a good performance indicator of the vaccine construct, showing its capability to stimulate the correct immunological compartment for an effective response.

The MD simulation results of the MESV vaccine. a SAWXS net intensity of MSEV-TLR5 complex; and molecular dynamics simulation of the MESV-TLR5 complex, showing b deformability; c eigenvalue; d variance; e covariance matrix indicates coupling between pairs of residues (red), uncorrelated (white), or anti-correlated (blue) motions; f elastic network analysis which defines which pairs of atoms are connected by springs; and g radius of gyration plot showing the calculated values vs Guinier fit

Results of MESV immune simulations: a the plot of antigen level along with immunoglobin profile; b induced array of cytokines; c B-cell population over the time; d Th cell population over the time; e Tc cell population over the time; f natural killer cells (total count)
In silico codon optimization and cloning
The optimal vaccine codon sequence was 840 nucleotides long. The GC content of the cDNA sequence and adaptive codon index was calculated to be 52.14%, which is still within the recommended range of 30–70% for effective translational efficiency. The computed codon adaptability index was 1, within the range of 0.8–1.0, indicating that the vaccine designs were effectively expressed in Escherichia coli. The optimized nucleotide sequence was cloned into the pET28a ( +) vector with NdeI and EagI restriction enzymes (Supplementary Fig. S2).
Discussion
With the emergence of SARS-CoV-2 and, as a result, the World Health Organization (WHO) declaration that COVID-19 was a pandemic, researchers began working to develop vaccines to prevent the disease [92]. Despite the high mutational rate in SARS-CoV-2, new variants are emerging. Some variants may evade the current vaccines. This study was carried out because of the rapid rise of the omicron variant and its fear of eluding the vaccine. Traditional vaccine development has a high success rate, but the immunoinformatics approach is instrumental because it saves time and eliminates various biosafety concerns because of the immunogenic reaction. Compared to traditional vaccines, multieptiope vaccines have an advantage. The traditional method of vaccine development which involves the entire organisms or proteins can lead to unavoidable and unnecessary antigenic load along with allergenic responses. The said issue can be circumvent by peptide based vaccines comprising of multiple short immunogenic peptide with the capability to elicit targeted immune responses [22, 53, 54]. Designing a multi-epitope vaccine entails screening the viral genome and simulating a targeted immune response that accurately identifies immunogenic epitopes. Several vaccines against SARS-CoV-2 have been developed and reported [93,94,95]. Due to the vast information available for SARS-CoV-2 proteomics and genomics, the in silico approach for vaccine development is helpful for SARS-CoV-2. Few of the corporation involved with vaccine development have also demonstrated the use of immunoinformatics techniques to identify the most antigenic epitopes from plethora of candidates to final vaccine candidate development [43, 96].
In our study, the development of the multi-epitope vaccine elicits a humoral and cell-mediated immune response. MESV contains the epitopes that bind with CTL, HTL, and B-cells and beta-defensin-3 was used as an adjuvant. The S protein of the omicron variant was used to create vaccine epitopes. As an adjuvant, beta-defensin-3 was combined with GPGPG and KK linkers as per the previous report [97]. Antigenicity and allergenicity of predicted CTL and HTL epitopes were assessed. The population analysis of CTL and HTL epitopes in 16 different regions of the world showed that epitopes selected from S protein usually bind to HLA molecules. Immune cells like monocytes, macrophages, and immature dendritic cells express TLR receptors in the plasma membrane, which identifies the S protein of SARS-CoV-2 as a structural component. The binding of COVID19 antigens to TLRs activates NFκB, protein kinase, and cytokine secretion. Molecular modeling can provide detailed information to determine the exact position of the drug/ligand [98].
Here, we used protein–protein molecular docking to study the interaction of vaccine constructs with TLR5. The MESV interacted with TLR5 via two salt bridges between K255-E432 and E229-R29. Three H-bonds were also seen between the complex (H278-S617, R248-D390, and K238-E123). Optimization of the vaccine nucleotide sequence codon was performed to ensure efficient expression in the E. coli host, and its elevation of GC was acceptable for E. coli. Although in silico vaccine studies required entering the clinical phase for confirmation, prediction from immunoinformatics tools confirmed the viability of the vaccine and its role in inducing humoral and cellular immune responses. To eliminate the SARS-CoV-2 infection, there should be a successful interaction with MESV and TLR5, which activates the messenger cascade and triggers the antiviral response. Since in silico analysis has limitations, the in vivo/ in vitro experiments with the clinical trials are required to reinforce the result.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Chan JFW, Yuan S, Kok KH et al (2020) A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395:514–523. https://doi.org/10.1016/S0140-6736(20)30154-9
Wang C, Horby PW, Hayden FG, Gao GF (2020) A novel coronavirus outbreak of global health concern. Lancet 395:470–473
Yuan M, Wu NC, Zhu X et al (2020) A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science (80- )368:630–633. https://doi.org/10.1126/science.abb7269
Zhou P, Yang X-L, Wang X-G et al (2020) Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. bioRxiv 2020.01.22.914952. https://doi.org/10.1101/2020.01.22.914952
Nicola M, Alsafi Z, Sohrabi C et al (2020) The socio-economic implications of the coronavirus pandemic (COVID-19): A review. Int J Surg 78:185–193. https://doi.org/10.1016/j.ijsu.2020.04.018
Hui DS, Azhar EI, Madani TA et al (2020) The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health — The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis 91:264–266
Cui J, Li F, Shi ZL (2019) Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17:181–192. https://doi.org/10.1038/s41579-018-0118-9
Defay T, Cohen FE (1995) Evaluation of current techniques for Ab initio protein structure prediction. Proteins Struct Funct Genet 23:431–445. https://doi.org/10.1002/prot.340230317
Zhu N, Zhang D, Wang W et al (2020) A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 382:727–733. https://doi.org/10.1056/nejmoa2001017
Rota PA, Oberste MS, Monroe SS et al (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science (80- ) 300:1394–1399. https://doi.org/10.1126/science.1085952
Lu R, Zhao X, Li J et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. https://doi.org/10.1016/S0140-6736(20)30251-8
Báez-Santos YM, St. John SE, Mesecar AD (2015) The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res 115:21–38
Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181. https://doi.org/10.1016/j.cell.2020.02.052
Ziebuhr J, Snijder EJ, Gorbalenya AE (2000) Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol 81:853–879
Naqvi AAT, Fatima K, Mohammad T et al (2020) Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim Biophys Acta - Mol Basis Dis 1866:165878
Huang Y, Yang C, Xu X feng et al (2020) Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin 419;41:1141–1149. https://doi.org/10.1038/s41401-020-0485-4
Walls AC, Park YJ, Tortorici MA et al (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281-292.e6. https://doi.org/10.1016/j.cell.2020.02.058
Wan Y, Shang J, Graham R et al (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 94. https://doi.org/10.1128/jvi.00127-20
Xu H, Zhong L, Deng J et al (2020) High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci 12:1–5. https://doi.org/10.1038/s41368-020-0074-x
de Wilde AH, Snijder EJ, Kikkert M, van Hemert MJ (2018) Host factors in coronavirus replication. Curr Top Microbiol Immunol 419:1–42. https://doi.org/10.1007/82_2017_25
Saha RP, Sharma AR, Singh MK et al (2020) Repurposing drugs, ongoing vaccine, and new therapeutic development initiatives against COVID-19. Front Pharmacol 11:1258. https://doi.org/10.3389/fphar.2020.01258
Behmard E, Soleymani B, Najafi A, Barzegari E (2020) Immunoinformatic design of a COVID-19 subunit vaccine using entire structural immunogenic epitopes of SARS-CoV-2. https://doi.org/10.1038/s41598-020-77547-4
Duffy S (2018) Why are RNA virus mutation rates so damn high? PLoS Biol 16:e3000003. https://doi.org/10.1371/journal.pbio.3000003
Rees-Spear C, Muir L, Griffith SA et al (2021) The effect of spike mutations on SARS-CoV-2 neutralization. Cell Rep 34. https://doi.org/10.1016/j.celrep.2021.108890
Toyoshima Y, Nemoto K, Matsumoto S et al (2020) SARSCoV-2 genomic variations associated with mortality rate of COVID-19. J Hum Genet 65:1075–1082. https://doi.org/10.1038/s10038-020-0808-9
Meredith LW, Hamilton WL, Warne B et al (2020) Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: a prospective genomic surveillance study. Lancet Infect Dis 20:1263–1272. https://doi.org/10.1016/S1473-3099(20)30562-4
Harvey WT, Carabelli AM, Jackson B et al (2021) SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 19:409–424. https://doi.org/10.1038/s41579-021-00573-0
Jia Z, Gong W (2021) Will mutations in the spike protein of SARS-CoV-2 lead to the failure of COVID-19 vaccines? J Korean Med Sci 26:1–11. https://doi.org/10.3346/JKMS.2021.36.E124
Lauring AS, Hodcroft EB (2021) Genetic Variants of SARS-CoV-2 - What Do They Mean? JAMA - J Am Med Assoc 325:529–531. https://doi.org/10.1001/jama.2020.27124
Tracking SARS-CoV-2 variants (2021) Science Brief: Omicron (B.1.1.529) Variant | CDC, 2021. https://www.cdc.gov/coronavirus/2019-ncov/variants/omicron-variant.html. Updated July 29, 2022
Wahid M, Jawed A, Mandal RK et al (2021) Variants of SARS-CoV-2, their effects on infection, transmission and neutralization by vaccine induced antibodies. Eur Rev Med Pharmacol Sci 25:5857–5864. https://doi.org/10.26355/eurrev_202109_26805
Akkız H (2022) The biological functions and clinical significance of SARS-CoV-2 variants of concern. Front Med 9:849217. https://doi.org/10.3389/fmed.2022.849217
Piccoli L, Park YJ, Tortorici MA et al (2020) Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell 183:1024-1042.e21. https://doi.org/10.1016/j.cell.2020.09.037
Dai L, Gao GF (2021) Viral targets for vaccines against COVID-19. Nat Rev Immunol 21:73–82. https://doi.org/10.1038/s41577-020-00480-0
Ishack S, Lipner SR (2021) Bioinformatics and immunoinformatics to support COVID-19 vaccine development. J Med Virol 93:5209–5211. https://doi.org/10.1002/JMV.27017/FORMAT/PDF
Lu S (2020) Timely development of vaccines against SARS-CoV-2. https://doi.org/10.1080/22221751.2020.1737580
Verma N, Patel D, Pandya A (2020) Emerging diagnostic tools for detection of COVID-19 and perspective. Biomed Microdevices 22:1–18
Wang J, Tricoche N, Du L et al (2012) The adjuvanticity of an O. volvulus-derived rOv-ASP-1 protein in mice using sequential vaccinations and in non-human primates. PLoS One 7:e37019. https://doi.org/10.1371/JOURNAL.PONE.0037019
Zhang N, Channappanavar R, Ma C et al (2016) (2015) Identification of an ideal adjuvant for receptor-binding domain-based subunit vaccines against Middle East respiratory syndrome coronavirus. Cell Mol Immunol 132(13):180–190. https://doi.org/10.1038/cmi.2015.03
Heaton PM (2020) Challenges of developing novel vaccines with particular global health importance. Front Immunol 11:2456. https://doi.org/10.3389/FIMMU.2020.517290/BIBTEX
Papaneri AB, Johnson RF, Wada J et al (2015) Middle East respiratory syndrome: obstacles and prospects for vaccine development. 14:949–962. https://doi.org/10.1586/14760584.2015.1036033
De Groot AS, Moise L, Terry F et al (2020) Better epitope discovery, precision immune engineering, and accelerated vaccine design using Immunoinformatics tools. Front Immunol 11:442. https://doi.org/10.3389/FIMMU.2020.00442/BIBTEX
Bahrami AA, Payandeh Z, Khalili S et al (2019) Immunoinformatics: in silico approaches and computational design of a multi-epitope, immunogenic protein. 38:307–322. https://doi.org/10.1080/08830185.2019.1657426
Oli AN, Obialor WO, Ifeanyichukwu MO et al (2020) <p>Immunoinformatics and vaccine development: an overview</p>. ImmunoTargets Ther 9:13–30. https://doi.org/10.2147/ITT.S241064
Khan MK, Zaman S, Chakraborty S et al (2014) In silico predicted mycobacterial epitope elicits in vitro T-cell responses. Mol Immunol 61:16–22. https://doi.org/10.1016/J.MOLIMM.2014.04.009
Patronov A, Doytchinova I (2012) T-cell epitope vaccine design by immunoinformatics. Open Biol 3. https://doi.org/10.1098/RSOB.120139
Naz RK, Dabir P (2007) Peptide vaccines against cancer, infectious diseases, and conception. Front Biosci 12:1833–1844. https://doi.org/10.2741/2191
Ahmad B, Ashfaq UA, Rahman M, ur, et al (2019) Conserved B and T cell epitopes prediction of ebola virus glycoprotein for vaccine development: an immuno-informatics approach. Microb Pathog 132:243–253. https://doi.org/10.1016/J.MICPATH.2019.05.010
Sabetian S, Nezafat N, Dorosti H et al (2018) Exploring dengue proteome to design an effective epitope-based vaccine against dengue virus 37:2546–2563. https://doi.org/10.1080/07391102.2018.1491890
Bhatnager R, Bhasin M, Arora J, Dang AS (2020) Epitope based peptide vaccine against SARS-COV2: an immune-informatics approach. 39:5690–5705. https://doi.org/10.1080/07391102.2020.1787227
Kim YS, Yoon NK, Karisa N et al (2019) Identification of novel immunogenic proteins against Streptococcus parauberis in a zebrafish model by reverse vaccinology. Microb Pathog 127:56–59. https://doi.org/10.1016/J.MICPATH.2018.11.053
Bhattacharya M, Sharma AR, Patra P et al (2020) A SARS-CoV-2 vaccine candidate: in-silico cloning and validation. Informatics Med Unlocked 20:100394. https://doi.org/10.1016/J.IMU.2020.100394
Devi A, Chaitanya NSN (2021) In silico designing of multi-epitope vaccine construct against human coronavirus infections. J Biomol Struct Dyn 39:6903–6917. https://doi.org/10.1080/07391102.2020.1804460
Dong R, Chu Z, Yu F, Zha Y (2020) Contriving multi-epitope subunit of vaccine for COVID-19: immunoinformatics approaches. Front Immunol 11:1784. https://doi.org/10.3389/FIMMU.2020.01784/BIBTEX
Enayatkhani M, Hasaniazad M, Faezi S et al (2020) Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: an in silico study 39:2857–2872. https://doi.org/10.1080/07391102.2020.1756411
Kar T, Narsaria U, Basak S et al (2020) A candidate multi-epitope vaccine against SARS-CoV-2. Sci Reports 101(10):1–24. https://doi.org/10.1038/s41598-020-67749-1
Lizbeth RSG, Jazmín GM, José CB, Marlet MA (2021) Immunoinformatics study to search epitopes of spike glycoprotein from SARS-CoV-2 as potential vaccine. J Biomol Struct Dyn 39:4878–4892. https://doi.org/10.1080/07391102.2020.1780944
Abraham Peele K, Srihansa T, Krupanidhi S et al (2020) Design of multi-epitope vaccine candidate against SARS-CoV-2: a in-silico study. J Biomol Struct Dyn 40:1–9. https://doi.org/10.1080/07391102.2020.1770127/FORMAT/EPUB
Samad A, Ahammad F, Nain Z et al (2020) Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J Biomol Struct Dyn 1–17. https://doi.org/10.1080/07391102.2020.1792347
Aasim, Sharma R, Patil CR et al (2022) Identification of vaccine candidate against Omicron variant of SARS-CoV-2 using immunoinformatic approaches. Silico Pharmacol 10:12. https://doi.org/10.1007/S40203-022-00128-Y
Khan K, Khan SA, Jalal K et al (2022) Immunoinformatic approach for the construction of multi-epitopes vaccine against omicron COVID-19 variant. Virology 572:28. https://doi.org/10.1016/J.VIROL.2022.05.001
Altschul SF, Gish W, Miller W et al (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Yang J, Yan R, Roy A et al (2014) The I-TASSER suite: protein structure and function prediction. Nat Methods 12:7–8
Xu D, Zhang Y (2011) Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization 101:2525–2534. https://doi.org/10.1016/j.bpj.2011.10.024
Rajkishan T, Rachana A, Shruti S et al (2021) Computer-aided drug designing. Adv Bioinformatics 151–182. https://doi.org/10.1007/978-981-33-6191-1_9
Patel B, Singh V, Patel D (2019) Structural bioinformatics. Essentials of Bioinformatics, vol I. Springer International Publishing, Cham, pp 169–199
Saha S, Raghava GPS (2006) AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 34:W202–W209. https://doi.org/10.1093/NAR/GKL343
Saha S, Raghava GPS (2006) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65:40–48. https://doi.org/10.1002/PROT.21078
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 8:1–7. https://doi.org/10.1186/1471-2105-8-4/TABLES/2
Kringelum JV, Lundegaard C, Lund O, Nielsen M (2012) Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLOS Comput Biol 8:e1002829. https://doi.org/10.1371/JOURNAL.PCBI.1002829
Gupta S, Kapoor P, Chaudhary K et al (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 8:e73957. https://doi.org/10.1371/JOURNAL.PONE.0073957
Gasteiger E, Hoogland C, Gattiker A et al (2005) Protein identification and analysis tools on the ExPASy server. Proteomics Protoc Handb 571–607. https://doi.org/10.1385/1-59259-890-0:571
Dimitrov I, Naneva L, Doytchinova I, Bangov I (2014) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30:846–851. https://doi.org/10.1093/BIOINFORMATICS/BTT619
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014) AllerTOP v.2--a server for in silico prediction of allergens. J Mol Model 20.https://doi.org/10.1007/S00894-014-2278-5
Heo L, Park H, Seok C (2013) GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res 41. https://doi.org/10.1093/NAR/GKT458
Moffat L, Jones DT (2021) Increasing the accuracy of single sequence prediction methods using a deep semi-supervised learning framework. Bioinformatics 37:3744–3751. https://doi.org/10.1093/BIOINFORMATICS/BTAB491
Ponomarenko J, Bui HH, Li W et al (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics 9:514. https://doi.org/10.1186/1471-2105-9-514
Tian W, Chen C, Lei X et al (2018) CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res 46:W363–W367. https://doi.org/10.1093/NAR/GKY473
Lee H, Heo L, Lee MS, Seok C (2015) GalaxyPepDock: a protein–peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res 43:W431–W435. https://doi.org/10.1093/NAR/GKV495
Desta IT, Porter KA, Xia B et al (2020) Performance and its limits in rigid body protein-protein docking. Structure 28:1071-1081.e3. https://doi.org/10.1016/J.STR.2020.06.006
Sharma A, Vora J, Patel D et al (2020) Identification of natural inhibitors against prime targets of SARS-CoV-2 using molecular docking, molecular dynamics simulation and MM-PBSA approaches. J Biomol Struct Dyn 40:1–16. https://doi.org/10.1080/07391102.2020.1846624
Chaudhari A, Chaudhari M, Mahera S et al (2020) In-silico analysis reveals lower transcription efficiency of C241T variant of SARS-CoV-2 with host replication factors MADP1 and HNRNP-1. bioRxiv
Patel D, Athar M, Jha PCC (2022) Computational investigation of binding of chloroquinone and hydroxychloroquinone against PLPro of SARS-CoV-2. J Biomol Struct Dyn 40:1–11
Patel B, Patel D, Parmar K et al (2018) L. donovani XPRT: molecular characterization and evaluation of inhibitors. Biochim Biophys Acta - Proteins Proteomics 1866:426–441. https://doi.org/10.1016/j.bbapap.2017.12.002
Manhas A, Patel D, Lone MY, Jha PC (2019) Identification of natural compound inhibitors against Pf DXR: a hybrid structure-based molecular modeling approach and molecular dynamics simulation studies. J Cell Biochem 120:14531–14543. https://doi.org/10.1002/jcb.28714
Patel D, Athar M, Jha PC (2021) Exploring ruthenium-based organometallic inhibitors against plasmodium falciparum calcium dependent kinase 2 (PfCDPK2): a combined ensemble docking, QM/MM and molecular dynamics study. ChemistrySelect 6:8189–8199. https://doi.org/10.1002/SLCT.202101801
Chen F, Liu H, Sun H et al (2016) Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys Chem Chem Phys 18:22129–22139. https://doi.org/10.1039/C6CP03670H
Knight CJ, Hub JS (2015) WAXSiS: a web server for the calculation of SAXS/WAXS curves based on explicit-solvent molecular dynamics. Nucleic Acids Res 43:W225–W230. https://doi.org/10.1093/NAR/GKV309
Stolfi P, Castiglione F (2021) Emulating complex simulations by machine learning methods. BMC Bioinformatics 22:1–14. https://doi.org/10.1186/S12859-021-04354-7/METRICS
Grote A, Hiller K, Scheer M et al (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33https://doi.org/10.1093/NAR/GKI376
Doytchinova IA, Flower DR (2007) Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine 25:856–866. https://doi.org/10.1016/J.VACCINE.2006.09.032
Cucinotta D, Vanelli M (2020) WHO declares COVID-19 a pandemic. Acta Biomed 91:157–160. https://doi.org/10.23750/ABM.V91I1.9397
Ahmad S, Navid A, Farid R et al (2020) Design of a novel multi epitope-based vaccine for pandemic coronavirus disease (COVID-19) by vaccinomics and probable prevention strategy against avenging zoonotics. Eur J Pharm Sci 151. https://doi.org/10.1016/J.EJPS.2020.105387
Khairkhah N, Aghasadeghi MR, Namvar A, Bolhassani A (2020) Design of novel multiepitope constructs-based peptide vaccine against the structural S, N and M proteins of human COVID-19 using immunoinformatics analysis. PLoS ONE 15:e0240577. https://doi.org/10.1371/JOURNAL.PONE.0240577
Mukherjee S, Tworowski D, Detroja R et al (2020) Immunoinformatics and structural analysis for identification of immunodominant epitopes in SARS-CoV-2 as potential vaccine targets. Vaccines 8:1–17. https://doi.org/10.3390/VACCINES8020290
Chakraborty C, Sharma AR, Bhattacharya M, Lee SS (2021) Lessons learned from cutting-edge immunoinformatics on next-generation COVID-19 vaccine research. Int J Pept Res Ther 27:2303–2311. https://doi.org/10.1007/S10989-021-10254-4/FIGURES/3
Kavoosi M, Creagh AL, Kilburn DG, Haynes CA (2007) Strategy for selecting and characterizing linker peptides for CBM9-tagged fusion proteins expressed in Escherichia coli. Biotechnol Bioeng 98:599–610. https://doi.org/10.1002/BIT.21396
Khanmohammadi S, Rezaei N (2021) Role of Toll-like receptors in the pathogenesis of COVID-19. J Med Virol 93:2735–2739. https://doi.org/10.1002/JMV.26826
Funding
The authors acknowledge the Gujarat State Biotechnology Mission (GSBTM), Department of Science and Technology (DST), Gujarat, for financial support and resources for project grant GSBTM/JD(R&D)/610/20–21/351. The authors also acknowledge the Institute of Advanced Research, Gandhinagar, for the resources and infrastructure facilities. The authors also thank the Gujarat Council on Science and Technology (GUJCOST), Gujarat, for the PARAM SHAVAK supercomputing facility and the Science and Engineering Research Board (SERB), Department of Science and Technology (DST) for a project grant, EMR/2016/003025.
Author information
Authors and Affiliations
Contributions
DP: conceptualization, methodology, investigation, analysis, writing — original draft preparation. MP: writing — original draft preparation, methodology, analysis. RT: methodology, analysis. JS: methodology, analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Parmar, M., Thumar, R., Sheth, J. et al. Designing multi-epitope based peptide vaccine targeting spike protein SARS-CoV-2 B1.1.529 (Omicron) variant using computational approaches. Struct Chem 33, 2243–2260 (2022). https://doi.org/10.1007/s11224-022-02027-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-022-02027-6