
Abstract
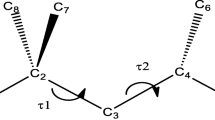
The molecular structure, conformational behaviour, vibrational spectra and electronic (hyper)polarizabilities of tellurophene and 2,2′-bitellurophene rotamers were determined in gas by correlated ab initio and density functional theory calculations. The torsional potential for the rotation around the C2–C2′ inter-ring bond shows two minima corresponding to anti-gauche and syn-gauche structures and three maxima to planar anti and syn forms and to perpendicular conformation. The potential energy curve is rather flat over the entire 0°–180° twisting range and free rotation cannot be excluded. The IR and Raman spectra of the gauche structures are rather similar to each other, vibrational transitions being scarcely helpful for an unambiguous identification of the rotamers. The dipole moment and the first-order hyperpolarizability increase on passing from the anti-gauche to the syn-gauche conformation by a factor of five and four, respectively. The second harmonic generation nonlinear optical process can be useful to identify the 2,2′-bitellurophene rotamers. On the other hand, the electronic polarizabilities of these structures are much more closer to each other, being predicted to be within 2–13 %.






Similar content being viewed by others

References
Zerbi G, Veronelli M, Martina S, Schlüter A-D, Wegner G (1994) π-Electron delocalization in conformationally distorted oligopyrroles and ploypyrrole. Adv Mater 6:385–388
Hernandez V, Castiglioni C, Del Zoppo M, Zerbi G (1994) Confinement potential and π-electron delocalization in polyconjugated organic materials. Phys Rev B 50:9815–9823
Brédas JL, Street GB, Thémans B, André J-M (1985) Organic polymers based on aromatic rings (polyparaphenylene, polypyrrole, polythiophene): evolution of the electronic properties as a function of the torsion angle between adjacent rings. J Chem Phys 83:1323–1329
Brédas JL, Silbey R (eds) (1991) Conjugated polymers, novel science and technology of conducting and nonlinear active materials. Kluver, New York
Skotheim TA (ed) (1986) Handbook of conducting polymers. Marcel Dekker, New York
Ortí E, Viruela PM, Sánchez-Marín J, Tomás F (1995) Ab initio determination of the geometric structure and internal rotation potential of 2,2′-bithiophene. J Phys Chem 99:4955–4963
Millefiori S, Alparone A, Millefiori A (2000) Conformational properties of thiophene oligomers. J Heterocycl Chem 37:847–853
Raos G, Famulari A, Marcon V (2003) Computational reinvestigation of the bithiophene torsion potential. Chem Phys Lett 379:364–372
Karpfen A, Ho Choi C, Kertesz M (1997) Single-bond torsional potentials in conjugated systems: a comparison of ab initio and density functional results. J Phys Chem A 101:7426–7433
Ortí E, Sánchez-Marín J, Merchán M, Tomás F (1987) Theoretical approach to the molecular conformation of nonfused biheterocycles. Bifurans and furylpyrroles. J Phys Chem 91:545–551
Sánchez-Sanz G, Alkorta I, Elguero J (2011) A theoretical study of the conformation of 2,2′-bifuran, 2,2′-bithiophene, 2,2′-bitellurophene and mixed derivatives: Chalcogen–chalcogen interactions or dipole–dipole effects? Comput Theor Chem 974:37–42
Millefiori S, Alparone A (1998) Theoretical investigation of the structure and conformational behaviour of small selenophene oligomers. Synth Met 95:217–224
Roncali J (1992) Conjugated poly(thiophenes): synthesis, functionalization, and applications. Chem Rev 92:711–738
Samuelsen EJ, Mardalen J (1997) Conductive polymers: spectroscopy and physical properties. In: Nolwa HS (ed) Handbook of organic and conductive molecules and polymers, vol 3. Wiley, New York
Glenis S, Benz M, LeGoff E, Schindler JL, Kannewurf CR, Kanatzidis MG (1993) Polyfuran: a new synthetic approach and electronic properties. J Am Chem Soc 115:12519–12525
Lopez Navarrete JT, Hernandez V, Zotti G, Veronelli M, Zerbi G (1994) Infrared spectrum of photoexcited polyfuran. Acta Polym 45:124–126
Brown RD, Croft JG (1973) The microwave spectrum of tellurophene. Chem Phys 1:217–219
Inoue S, Jigami T, Nozoe H, Otsubo T, Ogura F (1994) 2,2′-Bitellurophene and 2,2′:5′,2″-tertellurophene as novel high homologues of tellurophene. Tetrahedron Lett 35:8009–8012
Otsubo T, Inoue, Nozoe H, Jigami T, Ogura F (1995) Electrical properties of polymer composties: conducting polymer-polyacene quinone radical polymer. Synth Met 69:357–358
Distefano G, Pignataro S, Innorta G, Fringuelli F, Marino G, Taticchi A (1973) Ionization energies of selenophen, tellurophen and some of their derivatives. Chem Phys Lett 22:132–136
Schaefer W, Schweig A, Gronowitz S, Taticchi A, Fringuelli F (1973) Reversal in the sequence of two highest occupied molecular orbitals in the series thiophen, selenophen, and tellurophen. J Chem Soc Chem Commun 541–542. http://pubs.rsc.org/en/content/articlelanding/1973/c3/c39730000541#!divAbstract
Fringuelli F, Marino G, Taticchi A, Colonna FP, Distefano G, Pignataro S (1976) Photoelectron spectra of the -substituted derivatives of furan, thiophen, selenophen, and tellurophen. A comparative study of the molecular orbital energies. J Chem Soc Perkin Trans 2:276–279
Modelli A, Jones D, Distefano G, Irgolich KJ, French K, Pappalardo G (1984) Electron transmission spectra of selenophene and tellurophene and Xα computations of electron affinities for chalcophenes. Chem Phys 88:455–461
Varsányi G, Nyulászi L, Veszpremi T (1982) Vibronic analysis and symmetry of the lowest energy ultraviolet transition of thiophen. J Chem Soc Perkin Trans 2:761–765
Katsumi Y, Yasutaka K, Takelora S, Keichi K (1985) Electrical and optical properties of electrochemically prepared polyselenophene film. Synth Met 10:319–326
Bryce M (1991) Recent progress on conducting organic charge-transfer salts. Chem Soc Rev 20:355–390
Cowan DO, Mays M, Lee M, McCullough R, Bailey A, Lerstrup K, Wiygul F, Kistenmacher T, Poehler T, Chiang LJ (1985) Tellurium containing organic metals. Mol Cryst Liq Cryst 125:191–204
Cowan DO, McCullough R, Bailey A, Lerstrup K, Talham D, Herr D, Mays M (1992) Organic metals containing tellurium. Phosphorus Sulphur Silicon Relat Elem 67:277–294
Tamura R, Nagata Y, Shimizu H, Matsumoto A, Ono N, Kamimura A, Ori K (1993) Novel tellurium-containing p-terphenoquinone analogues: preparation and unique redox properties of paramagnetic tellurium-centered radical cation complexes. Adv Mater 5:719–721
Møkved EH, Faccin G, Manfrotto D, Kjøsen H, Becker JY, Shapiro L, Ellern A, Berstein J, Khodorkovsky V (1997) Synthesis and properties of novel components for organic metals: dihydrotellurophene derivatives. J Mater Chem 7:1697–1700
Robinson DW, Abdel-Halim H, Inoue S, Kimura M, Cowan DO (1989) Hyperpolarizabilities of 4-amino-4′-nitrodiphenyl sulfide and all of its chalcogen analogues. J Chem Phys 90:3427–3429
Blenkle M, Boldt P, Brauchle C, Grahn W, Ledoux I, Nerenz H, Stadler S, Wichern J, Zyss J (1996) Chalcogens as electron donors for selected nonlinear optic phores. J Chem Soc Perkin Trans 2:1377–1384
Li DQ, Ratner MA, Marks TJ (1988) Molecular and macromolecular nonlinear optical materials. Probing architecture/electronic structure/frequency doubling relationships via an SCF-LCAO MECI.pi. electron formalism. J Am Chem Soc 110:1707–1715
Irgolic KJ (1990) In: Klamann D (ed) Organotellurium compounds. Georg Thieme, Stuttgart
Fringuelli F, Marino G, Taticchi A (1977) Tellurophene and related compounds. Adv Heterocycl Chem 21:119–173
Millefiori S, Alparone A (1998) (Hyper)polarizability of chalcogenophenes C4H4X (X = O, S, Se, Te). Ab initio and density functional theory study. J Mol Struct (Theochem) 431:59–78
Millefiori S, Alparone A (2000) Theoretical determination of the vibrational and electronic (hyper)polarizabilities of C4H4X (X = O, S, Se, Te) heterocycles. Phys Chem Chem Phys 2:2495–2501
Kamada K, Ueda, Nagao H, Tawa K, Sugino T, Shmizu Y, Ohta K (2000) Molecular design for organic nonlinear optics: polarizability and hyperpolarizabilities of furan homologues investigated by ab initio molecular orbital method. J Phys Chem A 104:4723–4734
Jansik B, Schimmelpfennig B, Norman P, Macak P, Ågren H, Ohta K (2003) Relativistic effects on linear and non-linear polarizabilities of the furan homologues. J Mol Struct 633:237–246
Becke AD (1993) A new mixing of Hartree-Fock and local density-functional theories. J Chem Phys 98:1372–1377
Lee C, Yang AD, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Wilson PJ, Bradley TJ, Tozer DJ (2001) Hybrid exchange-correlation functional determined from thermochemical data and ab initio potentials. J Chem Phys 115:9233–9242
Dunning TH Jr, Hay PJ (1977) In: Schaefer HF III (ed) Methods of electronic structure theory, vol 2. Plenum Press, New York
Hay PJ, Wadt WR (1985) Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J Chem Phys 82:284–298
Check CE, Faust TO, Bailey JM, Wright BJ, Gilbert TM, Sunderlin LS (2001) Addition of polarization and diffuse functions to the LANL2DZ basis set for p-block elements. J Phys Chem A 105:8111–8116
Hehre WJ, Random L, Schleyer PvR, Pople JA (1986) Ab initio molecular orbital theory. Wiley, New York
Pulay P, Fogarasi G, Pongor G, Boggs JE, Vargha A (1983) Combination of theoretical ab initio and experimental information to obtain reliable harmonic force constants. Scaled quantum mechanical (QM) force fields for glyoxal, acrolein, butadiene, formaldehyde, and ethylene. J Am Chem Soc 105:7037–7047
Rauhut G, Pulay P (1995) Transferable scaling factors for density functional derived vibrational force fields. J Phys Chem 99:3093–3100
Johnson RD III (2013) NIST computational chemistry comparison and benchmark database, NIST standard reference database number 101, release 16a. http://cccbdb.nist.gov/. Accessed Aug 2013
Paliani G, Cataliotti R, Poletti A, Fringuelli F, Taticchi A, Giorgini MG (1976) Vibrational analysis of tellurophene and its deuterated derivatives. Spectrochim Acta A 32:1089–1104
Yanai T, Tew D, Handy NC (2004) A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393:51–57
Sadlej AJ (1988) Medium-size polarized basis sets for high-level correlated calculations of molecular electric properties. Collect Czechoslov Chem Comm 53:1995–2016
Sadlej AJ (1992) Medium-size polarized basis sets for high-level-correlated calculations of molecular electric properties. Theor Chim Acta 81:339–354
Eckart U, Fülscher MP, Serrano-Andrés L, Sadlej AJ (2000) Static electric properties of conjugated cyclic ketones and thioketones. J Chem Phys 113:6235–6244
Eckart U, Ingamells VE, Papadopoulos MG, Sadlej AJ (2001) Vibrational effects on electric properties of cyclopropenone and cyclopropenethione. J Chem Phys 114:735–745
Alparone A, Millefiori S (2005) Gas and solution phase electronic and vibrational (hyper)polarizabilities in the series formaldehyde, formamide and urea: CCSD(T) and DFT theoretical study. Chem Phys Lett 416:282–288
Alparone A (2011) Comparative study of CCSD(T) and DFT methods: electronic (hyper)polarizabilities of glycine. Chem Phys Lett 514:21–25
Alparone A (2013) Electron correlation effects and density analysis of the first-order hyperpolarizability of neutral guanine tautomers. J Mol Model 19:3095–3102
Alparone A (2013) The effect of secondary structures on the NLO properties of single chain oligopeptides: a comparison between β-strand and α-helix polyglycines. Phys Chem Chem Phys 15:12958–12962
Jacquemin D, Perpète EA, Medved’ M, Scalmani G, Frisch MJ, Kobayashi R, Adamo C (2007) First hyperpolarizability of polymethineimine with long-range corrected functional. J Chem Phys 126:191108
Karna SP, Dupuis M (1991) Frequency dependent nonlinear optical properties of molecules: formulation and implementation in the HONDO program. J Comput Chem 12:487–504
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) GAUSSIAN 09 revision A.02. Gaussian Inc., Wallingford
Radom L, Pople JA (1970) Molecular orbital theory of the electronic structure of organic compounds. IV. Internal rotation in hydrocarbons using a minimal Slater-type basis. J Am Chem Soc 92:4786–4795
Salafsky JS (2003) Second-harmonic generation as a probe of conformational change in molecules. Chem Phys Lett 381:705–709
Xu H-L, Li Z-R, Su Z-M, Muhammad S, Gu F-L, Harigaya K (2009) Knot-isomers of Mobius cyclacene: How does the number of knots influence the structure and first hyperpolarizability? J Phys Chem C 113:15380–15383
Lipiński J, Bartkowiak W (1999) Conformation and solvent dependence of the first and second molecular hyperpolarizabilities of charge-transfer chromophores. Quantum-chemical calculations. Chem Phys 245:263–276
Bartkowiak W, Lipiński J (1998) Conformation and solvent dependence of the first molecular hyperpolarizability of pyridinium-N-phenoxide betaine dyes. Quantum chemical calculations. J Phys Chem A 102:5236–5240
Kang H, Facchetti A, Jiang H, Cariati E, Righetto S, Ugo R, Zuccaccia C, Macchioni A, Stern CL, Liu Z, Ho S-T, Brown EC, Ratner MA, Marks TJ (2007) Ultralarge hyperpolarizability twisted π-electron system electro-optic chromophores: synthesis, solid-state and solution-phase structural characteristics, electronic structures, linear and nonlinear optical properties, and computational studies. J Am Chem Soc 129:3267–3286
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Alparone, A. Structural, torsional, vibrational and response electric properties of 2,2′-bitellurophene rotamers. An ab initio and density functional theory investigation. Struct Chem 25, 959–968 (2014). https://doi.org/10.1007/s11224-013-0370-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-013-0370-6