
Abstract
Aromaticity and reactivity are two deeply connected concepts. Most of the thermally allowed cycloadditions take place through aromatic transition states, while transition states of thermally forbidden reactions are usually less aromatic, if at all. In this work, we perform a numerical experiment to discuss the change of aromaticity that occurs along the reaction paths that connect two antiaromatic units of cyclobutadiene to form cubane and two aromatic rings of benzene to yield hexaprismane. It is found that the aromaticity profile along the reaction coordinate of the [4+4] cycloaddition of two antiaromatic cyclobutadiene molecules goes through an aromatic highest energy point and finishes to an antiaromatic cubane species. Up to our knowledge, this represents the first example of a theoretically and thermally forbidden reaction path that goes through an intermediate aromatic region. In contrast, the aromaticity profile in the [6+6] cycloaddition of two aromatic benzene rings show a slow steady decrease of aromaticity from reactants to the highest energy point and from this to the final hexaprismane molecule a plunge of aromaticity is observed. In both systems, the main change of aromaticity occurs abruptly near the highest energy point, when the distance between the centers of the two rings is about 2.2 Å.
Graphical contents entry

Similar content being viewed by others

Introduction
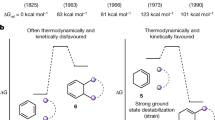
The [n]-prismanes are an attractive class of polycyclic hydrocarbons, constructed of an even number of methine units of general formula (CH)2n and Dnh symmetry [1–3]. These compounds have attracted the interest of chemists for many years because of their compact shape and the expectation of novel structural properties and unusual chemical reactivity. The first [n]-prismane structure was synthesized in 1964 by Eaton and Cole [4], the [4]-prismane or cubane, while [3]-prismane [5] or triprismane and [5]-prismane [6] or pentaprismane were synthesized later in 1973 and 1981, respectively. The next step in the chain, [6]-prismane or hexaprismane has not yet been synthesized [1, 3, 7].
Among [n]-prismanes, the most symmetric is cubane. This molecule consists of eight CH units bound together to form a cube (see Scheme 1), which gives its name. It is exceptionally strained and rehybridization away from the usual sp3 hybridization of tetravalent carbon is required to accommodate the geometry of the system. As it could be synthesized [4], different experimental (i.e., X-ray diffraction and electron-diffraction) [8, 9] and also theoretical techniques (ab initio methods) [10–12] have allowed a complete characterization of this molecule. On the other hand, hexaprismane is composed of twelve identical methine units arranged at the corner of a regular hexagonal prism, the two parallel 6-membered rings (6-MRs) being cojoined by six 4-MRs (see Scheme 2). This system has been studied only by ab initio methods [2] and it is especially relevant because it derives from the benzene dimer, the simplest prototype of π–π stacking interactions [13–17], which are fundamental in diverse areas of science (supramolecular chemistry, drug design, protein folding...).

Schematic representation of reactants and product of the cycloaddition reaction between two cyclobutadiene molecules

Schematic representation of reactants and product of the cycloaddition reaction between two benzene molecules
An interesting property of [n]-prismanes is aromaticity of the cage. In a previous work [18], Schleyer and coworkers analyzed the aromaticity of cubane and hexaprismane among other cage hydrocarbons using the nucleus-independent chemical shift (NICS) [19, 20] indicator of aromaticity. These authors showed that the cubane cage appears to be “super-σ-antiaromatic” with the face centers of the 4-MR being strongly deshielded as correspond to antiaromatic rings. On the other hand, the 4-MRs of hexaprismane are moderately deshielding, while the NICS values in the center of the 6-MRs are close to zero indicating nonaromatic character.
One may consider that the cubane molecule is the result of approaching two cyclobutadiene molecules until four new C–C bonds between the original 4-MRs are formed. In this case, starting from two units of the prototypical antiaromatic species cyclobutadiene (C4H4) one reaches a super-σ-antiaromatic system (C8H8) through a [4+4] cycloaddition reaction (see Scheme 1). Although some thermal [4+4] cycloadditions are known in the literature [21–24], dimerization of cyclobutadiene molecules does not occur through a [4+4] cycloaddition. Rather, experimentally, the dimerization takes place spontaneously even at very low temperature to give the syn dimer in which only two new C–C bonds have been formed in a formally [4+2] cycloaddition reaction [25]. Our main goal, however, is not the study of the reactivity of these species but to discuss how aromaticity changes along a symmetric linear transit that goes from two units of cyclobutadiene to cubane. Similarly, hexaprismane can be considered the result of the [6+6] cycloaddition reaction between two benzene molecules as shown in Scheme 2. In this latter reaction, one moves from the quintessential aromatic species to a non-aromatic molecule. In the present study, we have undertaken a detailed investigation of these two cycloaddition reactions. In both cases, we have investigated the Woodward–Hoffmann (W–H) thermal-symmetry-forbidden concerted supra–supra process [26]. A salient characteristic of these reactions is the correlation between occupied reactant and unoccupied product orbitals and vice-versa. Given this orbital crossing, the use of multiconfigurational correlated methods is mandatory [27, 28]. However, since the size of the systems treated in the present work prevents the use of multiconfigurational correlated levels of theory and also because we are not particularly interested in a quantitative description of the energetics of the two reactions, we have limited our calculations to the correlated second-order Møller-Plesset perturbation theory (MP2) [29] with the frozen core orbitals approximation. It is also worth noting that, for technical reasons, most of the aromaticity descriptors used in this work (see next section) cannot be computed with multiconfigurational methods, yet. Previous studies of cycloaddition reactions using the MP2 method in conjunction with the 6-31+G(d) or similar basis set reported results which must be considered at least of semiquantitative value [30–32].
With the present study, we aim at two objectives. First, our main purpose is to analyze the evolution of the local aromaticity of the cyclobutadiene and benzene rings when cubane and hexaprismane are formed from cyclobutadiene and benzene dimers, respectively. In particular, we discuss whether the loss or gain of aromaticity occurs gradually along the reaction coordinate or abruptly at a certain point of the reaction. With this analysis we intend to get an insight into the relationship between reactivity and aromaticity in pericyclic reactions. In previous works, it has been shown that usually thermally allowed cycloadditions take place through aromatic transition states (TSs) [33], while TSs of thermally forbidden reactions are less aromatic or antiaromatic [34–36]. As recommended by several authors [20, 37–41], in the present work the local aromaticity of the 4- and 6-MRs has been measured using a set of differently based aromaticity criteria, to take into account the multidimensional character of aromaticity [42–45]. As a second goal, we discuss the validity of the different descriptors of local aromaticity that are used to analyze aromaticity changes in these particular processes. It is worth emphasizing that the present study is a numerical experiment on reaction pathways that are not experimental reaction channels. However, their study provides further insight into the relation between two deeply connected concepts: reactivity and aromaticity.
Methodology
Relaxed potential energy surfaces (PESs), as a function of the intermonomer distance for the sandwich configuration of the cyclobutadiene dimer and the benzene dimer were computed using the MP2 [29] method with the 6-31+G(d) basis set [46–48]. In both cases the center-to-center distance (R) was systematically varied from 1.4 to 4.2 Å. D4h and D6h symmetry were preserved along the full reaction paths of the [4+4] and [6+6] cycloadditions, respectively, except for the initial (D2d symmetry) and final (Oh symmetry) steps of the [4+4] cycloaddition.
The MP2 level of theory is able to account for most of the dispersion energy in π-stacked species, although it tends to overestimate the binding energy in van der Waals clusters [15, 49–51]. CCSD(T) is required to accurately evaluate dispersion interaction, however, due to its high computational cost, it is often unaffordable. It has been reported that MP2 in conjunction with small basis sets tends to exhibit a fortuitous cancellation of errors [14]: small basis sets underestimate binding, whereas MP2 overestimates it, in general. Polarization and diffuse functions have been recommended to better describe the polarizability of the monomers and the dispersion interaction between them [15, 52, 53]. On the other hand, current density functional theory (DFT) methods do not, in general, properly describe dispersion interactions [54–57]. For instance, the sandwich configuration of benzene is not bound with the PW91, BLYP, and B3LYP exchange-correlation functionals [58]. In this sense, the MP2/6-31+G(d) method represents a good compromise between efficiency and reliability for the kind of systems analyzed.
The analysis of aromaticity has been carried out by means of the magnetic based NICS [19, 20], the electronic based para-delocalization index (PDI) [59] and the fluctuation aromatic index (FLU) [60], and the geometry based harmonic oscillator model of aromaticity (HOMA) [61, 62] measures. NICS is defined as the negative value of the absolute shielding computed at a ring center or at some other interesting point of the system. Rings with large negative NICS values are considered aromatic. The GIAO method [63] has been used to perform calculations of NICS at ring centers (NICS(0)) determined by the non-weighted average of the heavy atoms coordinates. In order to complement the NICS analysis, we have also calculated NICS(1) values, which are the NICS measured at 1 Å above or below the center of the ring taken into analysis [64–66]. PDI is obtained employing the delocalization index (DI) [67] as defined in the framework of the AIM theory of Bader [68]. The PDI is an average of all DI of para-related carbon atoms in a given 6-MR. The DI value between atoms A and B, δ(A,B), is obtained by double integration of the exchange-correlation density ( \( \Gamma _{XC} \left( {\vec{r}}_1 ,{\vec{r}}_2 \right) \)) over the basins of atoms A and B, which are defined from the zero-flux gradient condition applied to the one-electron density, ρ(r):
δ(A,B) provides a quantitative idea of the number of electron pairs delocalized or shared between atoms A and B. Finally, the FLU index is based on the fact that aromaticity is related to the cyclic delocalized circulation of π electrons, and it is constructed by considering the amount of electron sharing between contiguous atoms, which should be substantial in aromatic molecules, and also taking into account the similarity of electron sharing between adjacent atoms. It is defined as:
where the summation runs over all adjacent pairs of atoms around the ring, n is equal to the number of atoms of the ring, V(A) is the global delocalization of atom A, δ(A,B) and δ ref(A,B) are the DI values for the atom pairs A and B and its reference value (δ ref(A,B) = 1.4 e for C–C bonds, which is obtained from benzene at the HF/6-31G(d) level of theory [60]), respectively, and
Consequently, FLU is close to 0 in aromatic species; the higher the FLU values, the lower the aromaticity.
NICS and HOMA have been calculated at the MP2/6-31+G(d) level, whereas for PDI and FLU the HF/6-31+G(d)//MP2/6-31+G(d) method has been used. PDI and FLU have not been calculated at the MP2 level because it is computationally very demanding. Although it has been previously shown that the introduction of Coulomb correlation causes a major localization of the atomic basins, hence lower DIs [67, 69, 70], the DIs obtained from HF calculations yield the same qualitative trends than those derived from correlated wave functions. All calculations have been performed by means of the Gaussian03 [71] and AIMPAC [72] packages of programs.
Results and discussion
The present section is organized as follows: first, we present the results for the cyclobutadiene dimer to cubane reaction and, second, we discuss the benzene dimer to hexaprismane reaction. For each case, the geometries and energies along the PES are reported first, and then the corresponding analysis of aromaticity is presented.
(A) [4+4] Cycloaddition reaction to yield cubane from cyclobutadiene sandwich
The optimized structural parameters of the rectangular singlet ground state (X1Ag) of cyclobutadiene with D2h symmetry and of the first triplet excited state (13A2g) with square geometry of D4h symmetry are given in Table 1. Although the experimental structure of cyclobutadiene is not available, our results are close to those obtained at the CCSD(T)/cc-pVTZ level of theory [73] (X1Ag: C–C = 1.566 Å, C=C = 1.343 Å, C–H = 1.074 Å; 13A2g: C–C = 1.439 Å, C–H = 1.073 Å). The square structure of minimum energy of cyclobutadiene, with D4h symmetry, corresponds to a triplet state which is 9.2 kcal mol−1 (9.9 kcal mol−1 if one uses Gibbs energies at 298.15 K) higher in energy than the D2h one. This adiabatic energy difference between the D2h singlet and the D4h triplet states obtained at the MP2/6-31+G(d) level differs only by 0.2 kcal mol−1 from a previously calculated CCSD(T) value [74] and it is not far from the 13.2 kcal mol−1 obtained using the MR-AQCC/SS-CASSCF/cc-pVDZ method [75]. Thus, the MP2/6-31+G* level reproduces qualitatively well both geometries and the energy difference between the D2h singlet and the D4h triplet states of cyclobutadiene.
Cubane presents Oh symmetry (see Table 1) with the hydrogen atoms getting out of the plane as compared to the planar monomers. The optimized MP2 C–C and C–H bond lengths found for cubane are very close to experimental data obtained by electron diffraction (C–C = 1.571(2), C–H = 1.098(6) Å) [9, 76] and microwave spectroscopy (C–C = 1.5708, C–H = 1.097 Å) [77], as well as previous theoretical data [10, 11]. The [4+4] concerted thermal cycloaddition is exothermic by 70.8 kcal mol−1 (ΔG o = −48.5 kcal mol−1), which may be attributed in part to the instability of the antiaromatic cyclobutadiene ring according to the [4n+2] Hückel’s rule [78–81].
The PES of the [4+4] cycloaddition reaction obtained by approaching the two cyclobutadiene molecules is depicted in Fig. 1. This reaction path was followed by a linear transit procedure in which the distance between the center of mass of the two cyclobutadiene units was chosen as the reaction coordinate. The cyclobutadiene sandwich dimer is located at 4.097 Å, and is 1.3 kcal mol−1 (0.2 kcal mol−1 with BSSE correction) more stable than the two separated monomers, although it does not correspond to a minimum with respect to horizontal displacement and in plane rotation of one of the cyclobutadiene units. The lowest energy point (LEP) found in the linear transit at 2.3 Å presents five imaginary frequencies associated with the breaking of the D4h symmetry. The barrier height for the formation of cubane is 5.8 kcal mol−1, calculated as the difference between the reactants at R = 4.2 Å and the highest energy point (HEP) in the linear transit at 1.9 Å. We must emphasize that this point does not correspond to a real TS as it has more than one imaginary frequency. The energy barrier, calculated as the difference between the HEP in the linear transit and the above LEP at 2.3 Å, is 51.4 kcal mol−1. On the other hand, Fig. 2 shows the evolution of the most important geometrical parameters of this reaction along the linear transit. For R shorter than 3.4 Å, the system acquires D4h symmetry with C–C bond lengths in the initial cyclobutadiene species equal and intermediate between those of typical single and double bonds. For R shorter than 1.9 Å, the C–C bond lengths in the initial cyclobutadiene units correspond to single C–C bonds. It is also noticeable the lost of planarity of the C4H4 units from reactants to cubane by observing the dihedral angle between a hydrogen atom and the corresponding cyclobutadiene ring [25]. On the basis of geometrical parameters, the higher the degree of bond alternation and the deviation from planarity, the lower the aromaticity is. This point will be analyzed further below. Finally, Fig. 3 shows the corresponding orbital correlation diagram, which is typical of a W–H thermally forbidden reaction. The HOMO and HOMO-2 π-orbitals of the cyclobutadiene dimer correlate with unoccupied MOs of cubane, while the LUMO and LUMO+2 of the cyclobutadiene dimer are connected with occupied MOs of cubane. The relatively low barrier height obtained with the MP2/6-31+G(d) method (5.8 kcal mol−1) for this cycloaddition is somewhat unexpected for such a hypothetically W–H thermally forbidden reaction. Additional B3LYP/6-31+G(d) and CASSCF(8,8)/6-31G(d) calculations for the same attack provide the same qualitative shape of the PES of the [4+4] cycloaddition reaction. These results support the validity of the PES obtained at the MP2/6-31G+(d) level. Our interpretation of the low barrier height obtained is that this particular [4+4] cycloaddition is less forbidden (more affordable) than expected due to, first, the antiaromatic character of the two reactants and, second, the fact that the two initial reactants have alternate single and double bonds. In this case, the approach of the two units with one rectangular cyclobutadiene molecule rotated 90° with respect to the other so that C–C single bonds in one monomer face the C=C double bonds of the other unit and vice versa partially breaks the symmetry of the system and makes the barrier height lower than expected on the basis of the W–H rules. This kind of symmetry breaking is not possible in the [2+2] cycloaddition between two ethene molecules or in the [6+6] cycloaddition of two benzene molecules, both reactions showing high energy barriers (vide infra) as expected in W–H thermally forbidden reactions. Shaik et al. [82], on the other hand, have attributed the high reactivity of cyclobutadiene to the presence of a very low-lying triplet state.

Potential energy curve for the cycloaddition reaction of two cyclobutadiene molecules as a function of the ring-to-ring separation (R). Energy differences with respect to two cyclobutadiene molecules separated by 4.2 Å are depicted

Evolution of the C–C and C=C bond lengths, and the HCCC dihedral angle along the cycloaddition reaction between two cyclobutadiene molecules

Orbital correlation diagram for the [4+4] cycloaddition reaction between two cyclobutadiene molecules
With respect to the aromaticity analysis, Fig. 4 shows the evolution of the structural HOMA and the electronic FLU (PDI cannot be applied to 4-MRs) [59] along the reaction path. As can be seen both criteria give the same tendency. Departing from the dimer, aromaticity increases sharply at 3.4 Å, after it remains constant, and it decreases drastically at 1.9 Å. This trend given by both measures is expected on chemical grounds, as we start from antiaromatic 4-MRs with localized single and double bonds, at 3.4 Å it turns into aromatic rings with delocalized double bonds, and finally at 1.9 Å we end with antiaromatic 4-MRs. Up to our knowledge, this represents the first example of a thermally forbidden pathway that goes through an intermediate aromatic region. A previous example of a transition structure with a low activation energy and antiaromatic character has been reported [83]. The above situation can be corroborated with Fig. 5, which shows the evolution of the delocalization index (DI) for the most important C–C bonds along the path. In the dimer δ(C–C) is close to 1 e, corresponding to the electron pair of a single C–C bond, whereas δ(C=C) is close to 2 e, corresponding to a double C=C bond. When the square cyclobutadiene dimer is formed (3.4 Å), equivalent DIs for C–C and C=C of around 1.2 e are obtained. After, at 1.9 Å these DIs decrease to around 1 e because of the single C–C bond in cubane. Finally, the intermolecular DI, δ(C···C), also shows the formation of the single new C–C bond in cubane, with a sharp increase at 1.9 Å, thus being the main electron-pair reorganization next to the HEP.

Evolution of HOMA and FLU aromaticity criteria along the cycloaddition reaction of two cyclobutadiene molecules

Evolution of relevant delocalization indices (in electrons) along the cycloaddition reaction of two cyclobutadiene molecules
On the contrary, the magnetic NICS(0), calculated at the center of the cyclobutadiene ring plane, presents the increase of aromaticity at 3.4 Å (see Fig. 6), but also shows an increase between 3.4 Å and 1.9 Å, and a sudden decrease at 1.9 Å, although it increases again at the product. This strange behavior of NICS(0) is not unexpected, and has been previously attributed to magnetic couplings between superimposed rings [17, 84], the cyclobutadiene rings in this case. In order to analyze further this particular behavior of NICS, we have also calculated NICS(1) values, which are calculated at 1 Å above the center of the ring (NICS(1)out, outside the sandwich) and below (NICS(1)in, inside the sandwich). It is found that NICS(1)in follows the same tendency as NICS(0), both showing a large increase of aromaticity from 3.4 Å to 1.9 Å. The increase of local aromaticity in superimposed aromatic rings indicated by NICS(0) and NICS(1)in is not real but the result of the coupling between the magnetic fields generated by the two superimposed rings. On the contrary, NICS(1)out values give a trend closer to those afforded by HOMA and FLU, except for the last small increase of aromaticity next to the product. Finally, Fig. 6 also shows the out-of-plane component of the NICS tensor, NICSzz, which is considered to be a better magnetic descriptor of aromaticity than NICS itself [64, 85–87]. The different NICSzz values perfectly reproduce the trend obtained from HOMA and FLU. Finally, it may be pointed out that the present aromaticity analysis could be positively complemented in the future by the dissection of the NICS in σ and π contributions [88, 89].

Evolution of different measures of NICS (in ppm) along the cycloaddition reaction between two cyclobutadiene molecules
(B) [6+6] Cycloaddition reaction to yield hexaprismane from benzene sandwich
The geometries of benzene and hexaprismane were also optimized and characterized at the MP2 level, constraining the D6h symmetry (see Table 2 for details). The MP2/6-31+G(d) optimization of benzene leads to a C–C bond length of 1.399 Å to be compared with the experimental [90] and CCSD/TZ2P [91] results of 1.390 and 1.392 Å, respectively. At variance with the [4+4] dimerization of cyclobutadiene, the thermal cycloaddition of two benzene molecules to give hexaprismane is highly endothermic (117.7 kcal mol−1) because of the high stability of benzene. This value agrees with previous ab initio calculations by Dailey that predicted that the inverse reaction was exothermic by about 115 kcal mol−1 [2]. The sandwich structure of benzene dimer corresponds to an intermediate of the [6+6] cycloaddition, with an equilibrium intermonomer distance (3.85 Å) very close to that obtained at the CCSD(T) level (3.9 Å).[15] In contrast, the binding energy at the MP2 level (−3.9 kcal mol−1 (or −1.06 kcal mol−1 with BSSE correction)) differs somewhat from the more accurate CCSD(T) values of about −2.0 kcal mol−1 [13, 15, 52] for the benzene sandwich (or face-to-face) dimer. So, in agreement with previous studies, equilibrium geometries can be accurately predicted using small basis sets at the MP2 level, although binding energies in van der Waals complexes without including BSSE complexes are clearly overestimated. It is worth noticing that the benzene sandwich dimer presents four imaginary frequencies [92], one of them corresponding to the breaking of the D6h symmetry to give the parallel-displaced configuration [15, 52, 93]. In fact, this last structure is a true minimum (R = 3.81 Å) with a MP2 binding energy of 5.9 kcal mol−1 (1.7 kcal mol−1 after BSSE correction). This parallel-displaced benzene dimer and the T-shaped conformer are nearly isoenergetic [50, 52, 53, 94].
Figure 7 depicts the linear transit of this [6+6] cycloaddition. The energy changes in the linear transit are not completely smooth because it is constructed keeping the D6h constraint. The HEP of the linear transit analyzed is located at 2.2 Å and it is not a real transition state. It shows an energy barrier of 203.9 kcal mol−1 indicating that the concerted process to yield hexaprismane is highly unfavorable, as expected from a W–H thermally forbidden reaction. In fact, although many diverse synthetic strategies have been developed and significant progress toward the synthesis of hexaprismane has been made, it has not been synthesized so far [1, 3, 7]. On the other hand, the evolution of the C–C bond distance along the reaction coordinate depicted in Fig. 8 starts with the aromatic bond length of benzene (∼1.4 Å) that is kept constant until R = 2.2 Å. After that, it increases until the new single C–C bonds in hexaprismane are formed. The same happens with the planarity of benzene with respect to the hydrogen atoms that is also lost at 2.2 Å. Thus, the most important geometrical changes happen around the HEP. Finally, Fig. S1 in the Supporting Information encloses the orbital correlation diagram for the attempted face-to-face dimerization. The [6+6] pathway with D6h symmetry is the least-motion reaction for the cycloaddition of two benzene molecules, and is forbidden by orbital symmetry. There are three occupied–unoccupied orbital crossings in the correlation diagram.

Potential energy curve for the [6+6] cycloaddition of two benzene molecules. Energy differences with respect to two benzene molecules separated by 4.2 Å are depicted

Evolution of the C–C bond length and the HCCC dihedral angle along the cycloaddition reaction of two benzene molecules
The trends of the HOMA, FLU, and PDI aromaticity profiles depicted in Fig. 9 indicate that the aromaticity of the two approaching 6-MRs remains almost constant until 2.2 Å, it reduces somewhat in the region around the HEP, and decreases drastically at 1.8 Å when the new C–C bonds can be considered already formed. Unlike HOMA and FLU, PDI shows a minor increase of aromaticity from 1.8 Å to hexaprismane. Thus, Fig. 9 perfectly shows the transition from an aromatic 6-MR in benzene to nonaromatic 6-MRs in hexaprismane. This trend is additionally supported by the evolution of the different DIs along the path given in Fig. 10. δ(C,C)ortho reduces from 1.4 to 0.9 e, thus indicating the evolution from aromatic C–C bonds in benzene to single C–C bonds in the product. It is also noticeable that until R = 1.8 Å, δ(C,C)para is greater than δ(C,C)meta, a characteristic associated to aromatic 6-MRs [59, 95]. Therefore, according to these DIs, the corresponding HEP is aromatic. Finally, δ(C···C) shows the formation of new bonds between the two benzene molecules at about 1.8 Å.

Evolution of HOMA, FLU, and PDI along the cycloaddition reaction of two benzene molecules

Evolution of relevant delocalization indices (in electrons) along the cycloaddition reaction between two benzene molecules
Just to conclude this aromaticity analysis, NICS measures have also been calculated along the reaction path (see Fig. 11). NICS(0), NICS(1)in, and NICS(1)out experience abrupt changes in the region around the HEP, with a sudden increase first and a subsequent decrease in this zone. However, like in the case above, the out-of-plane component of NICS, NICSzz, provides a similar trend to other aromatic criteria, thus better reflecting the contributions to the π-system than NICS itself, which is largely affected by the in-plane components. As before the aromaticity profiles given by NICS(0) and NICS(1)in deviate substantially from the rest, again proving the effects to the magnetic couplings between superimposed rings that largely affect the NICS(0) and NICS(1)in measures of local aromaticity, but apparently have only minor effects on the NICSzz values. It is noticeable that NICSzz(1)in values near the HEP point out slight aromatic character, whereas NICSzz(0) values attribute an antiaromatic behavior.

Evolution of different measures of NICS along the cycloaddition reaction between two benzene molecules
Conclusions
The present work analyzes the evolution of aromaticity along the thermally forbidden [4+4] and [6+6] dimerizations of cyclobutadiene and benzene to give cubane and hexaprismane, respectively. Even though these processes do not correspond to experimentally observed reaction paths, their analyses provide new clues for a better comprehension of the connection between aromaticity and reactivity. Dimerization of cyclobutadiene to yield cubane is found to be a quite exothermic process that can take place with a relatively low energy barrier. On the contrary, the dimerization of benzene to give hexaprismane is a highly endothermic process that goes through a high energy barrier. These two cycloadditions are also quite different from the point of view of the evolution of aromaticity from reactants to product. Thus, whereas in the first case cyclobutadiene dimer is antiaromatic, goes through an intermediate aromatic region, and ends in an antiaromatic cubane; in the second case benzene dimer presents the highest aromaticity, which slightly decreases at the region of highest energy, and reaches the nonaromatic 6-MRs of hexaprismane. HOMA, FLU, PDI (for hexaprismane), and NICSzz indexes have proven to be appropriate aromaticity measures to evaluate this property along both reactions. Different aromatic indexes have been applied because of the multidimensional character of this phenomenon, and the convergence of their conclusions makes us confident about the trends presented for both reactions.
References
Mehta G, Padma S (1991) Tetrahedron 47:7783
Dailey WP (1987) Tetrahedron Lett 28:5787
Golobish TD, Dailey WP (1996) Tetrahedron Lett 37:3239
Eaton PE, Cole TW (1964) J Am Chem Soc 86:3157
Katz TJ, Acton N (1973) J Am Chem Soc 95:2738
Eaton PE, Or YS, Branca SJ (1981) J Am Chem Soc 103:2134
Chou TC, Lin GH, Yeh YL, Lin KJ (1997) J Chin Chem Soc 44:477
Fleischer EB (1964) J Am Chem Soc 86:3889
Almenningen A, Jonvik T, Martin HD, Urbanek T (1985) J Mol Struct 128:239
Cole TW Jr, Perkins J, Putman S, Pakes PW, Strauss HL (1981) J Phys Chem 85:2185
Wiberg KB (1984) J Comput Chem 5:197
Li Y, Houk KN (1996) J Am Chem Soc 118:880
Sinnokrot MO, Sherrill CD (2006) J Phys Chem A 110:10656
Tauer TP, Sherrill CD (2005) J Phys Chem A 109:10475
Sinnokrot MO, Sherrill CD (2004) J Phys Chem A 108:10200
Ringer AL, Sinnokrot MO, Lively RP, Sherrill CD (2006) Chem Eur J 12:3821
Poater J, Bofill JM, Alemany P, Solà M (2006) J Org Chem 71:1700
Moran D, Manoharan M, Heine T, Schleyer PvR (2003) Org Lett 5:23
Schleyer PvR, Maerker C, Dransfeld A, Jiao HJ, Hommes N (1996) J Am Chem Soc 118:6317
Chen Z, Wannere CS, Corminboeuf C, Puchta R, Schleyer PvR (2005) Chem Rev 105:3842
Shiina I, Uchimaru T, Shoji M, Kakeyam H, Osada H, Harashy Y (2006) Org Lett 8:1041
Barata JFB, Silva AMG, Faustino MAF, Neves MGPMS, Tome AC, Silva AMS, Cavaleiro JAS (2004) Synlett 1291
Leung M, Trahanovsky WS (1995) J Am Chem Soc 117:841
Chou T, Chen H-C, Tsai C-Y (1994) J Org Chem 59:2241
Halevi EA (1992) Orbital symmetry and reaction mechanim. The OCAMS view. Springer-Verlag, Berlin
Woodward RB, Hoffmann R (1969) Angew Chem Int Ed 8:781
Borden WT, Davidson ER (1996) Acc Chem Res 29:67
Bernardi F, Bottoni A, Robb MA, Schlegel HB, Tonachini G (1985) J Am Chem Soc 107:2260
Head-Gordon M, Pople JA, Frisch MJ (1988) Chem Phys Lett 153:503
Ioffe A, Shaik S (1992) J Chem Soc, Perkin Trans 2:2101
Dinadayalane TC, Vijaya R, Smitha A, Sastry GN (2002) J Phys Chem A 106:1627
Apeloig Y, Matzner E (1995) J Am Chem Soc 117:5375
Jiao H, Schleyer PvR (1998) J Phys Org Chem 11:655
Bernardi F, Celani P, Olivucci M, Robb MA, Suzzi-Valli G (1995) J Am Chem Soc 117:10531
Sakai S (2006) J Phys Chem A 110:6339
Santos JC, Andres J, Aizman A, Fuentealba P (2005) J Chem Theor Comput 1:83
Poater J, García-Cruz I, Illas F, Solà M (2004) Phys Chem Chem Phys 6:314
Poater J, Solà M, Viglione RG, Zanasi R (2004) J Org Chem 69:7537
Poater J, Duran M, Solà M, Silvi B (2005) Chem Rev 105:3911
Jug K, Koester AM (1990) J Am Chem Soc 112:6772
Merino G, Vela A, Heine T (2005) Chem Rev 105:3812
Katritzky AR, Jug K, Oniciu DC (2001) Chem Rev 101:1421
Katritzky AR, Karelson M, Sild S, Krygowski TM, Jug K (1998) J Org Chem 63:5228
Krygowski TM, Cyrański MK (2001) Chem Rev 101:1385
Cyrański MK, Krygowski TM, Katritzky AR, Schleyer PvR (2002) J Org Chem 67:1333
Hehre WJ, Ditchfield R, Pople JA (1972) J Chem Phys 56:2257
Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, Defrees DJ, Pople JA (1982) J Chem Phys 77:3654
Hariharan PC, Pople JA (1973) Theor Chim Acta 28:213
Tsuzuki S, Uchimaru T, Matsumura K, Mikami M, Tanabe K (2000) Chem Phys Lett 319:547
Tsuzuki S, Honda K, Uchimaru T, Mikami M, Tanabe K (2002) J Am Chem Soc 124:104
Lee EC, Kim D, Jurecka P, Tarakeshwar P, Hobza P, Kim KS (2007) J Phys Chem A 111:3446
Sinnokrot MO, Valeev EF, Sherrill CD (2002) J Am Chem Soc 124:10887
Park YC, Lee JS (2006) J Phys Chem A 110:5091
van Mourik T, Gdanitz RJ (2002) J Chem Phys 116:9620
Ye X, Li Z-H, Wang W, Fan K, Xu W, Hua Z (2004) Chem Phys Lett 397:56
Godfrey-Kittle A, Cafiero M (2006) Int J Quantum Chem 106:2035
Cerny J, Hobza P (2005) Phys Chem Chem Phys 7:1624
Tsuzuki S, Luthi HP (2001) J Chem Phys 114:3949
Poater J, Fradera X, Duran M, Solà M (2003) Chem Eur J 9:400
Matito E, Duran M, Solà M (2005) J Chem Phys 122:014109. Erratum, íbid (2006) 125:059901
Kruszewski J, Krygowski TM (1972) Tetrahedron Lett 13:3839
Krygowski TM (1993) J Chem Inf Comp Sci 33:70
Wolinski K, Hilton JF, Pulay P (1990) J Am Chem Soc 112:8251
Corminboeuf C, Heine T, Seifert G, Schleyer PvR, Weber J (2004) Phys Chem Chem Phys 6:273
Schleyer PvR, Manoharan M, Jiao HJ, Stahl F (2001) Org Lett 3:3643
Lazzeretti P (2004) Phys Chem Chem Phys 6:217
Fradera X, Austen MA, Bader RFW (1999) J Phys Chem A 103:304
Bader RFW (1990) Atoms in molecules: a quantum theory. Clarendon, Oxford
Poater J, Solà M, Duran M, Fradera X (2002) Theor Chem Acc 107:362
Matito E, Solà M, Salvador P, Duran M (2007) Faraday Discussions 135:325
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski G, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 03. Gaussian Inc., Pittsburgh
Biegler-König FW, Bader RFW, Tang T-H (1982) J Comput Chem 3:317 (http://www.chemistry.mcmaster.ca/aimpac/)
Levchenko SV, Krylov AI (2004) J Chem Phys 120:175
Carsky P, Bartlett RJ, Fitzgerald G, Noga J, Spirko V (1988) J Chem Phys 89:3008
Eckert-Maksic M, Vazdar M, Barbatti M, Lischka H, Maksic ZB (2006) J Chem Phys 125:064310
Hedberg L, Hedberg K, Eaton PE, Nodari N, Robiette AG (1991) J Am Chem Soc 113:1514
Hirota E, Endo Y, Fujitake M, Della EW, Pigou PE, Chickos JS (1988) J Mol Struct 190:235
Hückel E (1931) Z Physik 70:104
Hückel E (1931) Z Physik 72:310
Hückel E (1932) Z Physik 76:628
Hückel E (1937) Z Elektrochemie 43:752
Shaik SS, Shurki A, Danovich D, Hiberty PC (2001) Chem Rev 101:1501
Alajarín M, Sánchez-Andrada P, Cossío FP, Arrieta A (2001) J Org Chem 66:8470
Portella G, Poater J, Bofill JM, Alemany P, Solà M (2005) J Org Chem 70:2509. Erratum, íbid (2005) 70:4560
Steiner E, Fowler PW, Jenneskens LW (2001) Angew Chem Int Ed 40:362
Fallah-Bagher-Shaidaei H, Wannere CS, Corminboeuf C, Puchta R, Schleyer PvR (2006) Org Lett 8:863
Merino G, Heine T, Seifert G (2004) Chem Eur J 10:4367
Schleyer PvR, Manoharan M, Wang ZX, Kiran B, Jiao HJ, Puchta R, van Eikema Hommes NJR (2001) Org Lett 3:2465
Heine T, Islas R, Merino G (2007) J Comput Chem 28:302
Jeffrey GA, Ruble JR, McMullan RK, Pople JA (1987) Proc R Soc Lond A 414:47
Christiansen O, Stanton JF, Gauss J (1998) J Chem Phys 108:3987
Moran D, Simmonett AC, Leach FE III, Allen WD, Schleyer PvR, Schaefer HF III (2006) J Am Chem Soc 128:9342
Jaffe RL, Smith GD (1996) J Chem Phys 105:2780
Puzder A, Dion M, Langreth DC (2006) J Chem Phys 124:164105
Poater J, Fradera X, Duran M, Solà M (2003) Chem Eur J 9:1113
Acknowledgments
Financial help has been furnished by the Spanish MEC project No. CTQ2005-08797-C02-01/BQU and by the Catalan Departament d’Universitats, Recerca i Societat de la Informació (DURSI) of the Generalitat de Catalunya project No. 2005SGR-00238. M. A. thanks the MEC for the predoctoral fellowship AP20034882. J. P. also acknowledges the European Union for a Marie Curie fellowship. We also thank the Centre de Supercomputació de Catalunya (CESCA) for partial funding of computer time. We are deeply indebted to Prof. Josep Maria Bofill for very valuable discussions.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Dedicated to Prof. Tadeusz Marek Krygowski in recognition of his distinguished and pionnering works on electron delocalization and aromaticity.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11224_2007_9240_MOESM1_ESM.doc
Figure S1 with the orbital correlation diagram for the [6+6] cycloaddition of two benzene molecules, and Cartesian coordinates of the most relevant intermediate points along the PES (DOC 127 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Alonso, M., Poater, J. & Solà, M. Aromaticity changes along the reaction coordinate connecting the cyclobutadiene dimer to cubane and the benzene dimer to hexaprismane. Struct Chem 18, 773–783 (2007). https://doi.org/10.1007/s11224-007-9240-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-007-9240-4