
Abstract
Purpose
Glioblastoma is the most common brain tumor in adults and is virtually incurable. Therefore, new therapeutic strategies are urgently needed. Over the last decade, multiple growth-promoting functions have been attributed to CD95, a prototypic death receptor well characterized as an apoptosis mediator upon CD95L engagement. Strategic targeting of non-apoptotic or apoptotic CD95 signaling may hold anti-glioblastoma potential. Due to its antithetic nature, understanding the constitutive role of CD95 signaling in glioblastoma is indispensable.
Methods
We abrogated constitutive Cd95 and Cd95l gene expression by CRISPR/Cas9 in murine glioma models and characterized the consequences of gene deletion in vitro and in vivo.
Results
Expression of canonical CD95 but not CD95L was identified in mouse glioma cells in vitro. Instead, a soluble isoform-encoding non-canonical Cd95l transcript variant was detected. In vivo, an upregulation of the membrane-bound canonical CD95L form was revealed. Cd95 or Cd95l gene deletion decreased cell growth in vitro. The growth-supporting role of constitutive CD95 signaling was validated by Cd95 re-transfection, which rescued growth. In vivo, Cd95 or Cd95l gene deletion prolonged survival involving tumor-intrinsic and immunological mechanisms in the SMA-497 model. In the GL-261 model, that expresses no CD95, only CD95L gene deletion prolonged survival, involving a tumor-intrinsic mechanism.
Conclusion
Non-canonical CD95L/CD95 interactions are growth-promoting in murine glioma models, and glioma growth and immunosuppression may be simultaneously counteracted by Cd95l gene silencing.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
CD95, also referred to as Fas or APO-1, is a pleiotropic cytokine receptor that, upon cognate stimulation with CD95 ligand (CD95L), may lead either to tumor-suppressing or tumor-promoting signaling. Initially, CD95L/CD95 interactions were described to result in the apoptotic cell death of CD95-expressing cells. When induced in cancer cells, CD95 signaling was therefore considered tumor-suppressing. Accordingly, agonistic CD95 targeting to induce apoptosis in CD95-expressing cancer cells was investigated upon the discovery of CD95 [1, 2], including in glioblastoma [3, 4]. However, therapeutic stimulation of apoptotic CD95 signaling in cancer cells is generally considered clinically impracticable due to major associated side effects [5, 6]. Furthermore, cumulative evidence suggests that cognate interactions between CD95L and CD95 may likewise prompt non-apoptotic tumor-promoting features such as proliferation, invasiveness and stemness in CD95-expressing cancer cells [7,8,9,10,11,12] in various malignancies including glioblastoma [13,14,15]. The notion of such tumor-promoting CD95 signaling entailed a paradigm shift in therapeutic CD95 targeting. Specifically, CD95 signaling inhibition using CD95L scavenging strategies rather than stimulation was considered for the treatment of glioblastoma [16].
Therapeutic CD95L inhibition has been generally oriented at blocking non-apoptotic, tumor-promoting signal transduction in cancer cells [17, 18]. However, there is also a scenario where apoptotic CD95 signaling may be tumorigenic, that is, when the apoptotic cell is not a cancer cell but a CD95-expressing effector immune cell. CD95 expression in immune cells renders them susceptible to apoptosis induced by CD95L expressed in the tumor. Interactions between CD95L expressed by glioma cells [19,20,21] or the tumor microenvironment with CD95 expressed in leukocytes have been proposed to induce immune cell apoptosis and subsequent immunosuppression [22,23,24,25]. However, this notion remains controversial since T cell killing assays have failed to reproduce this tumor counterattack [26], and studies where tumor counterattack was shown in vitro but not in vivo have been published, too [27].
Nevertheless, strategic inhibition of CD95 signaling to simultaneously block CD95-mediated cancer cell growth and the apoptotic death of cells in the tumor microenvironment with antitumor potential, including CD95-expressing immune effector cells, may represent a suitable alternative therapeutic strategy targeting the CD95-CD95L system. In glioblastoma, a virtually incurable cancer to date, such strategies may be of high interest since they may theoretically affect two of the major factors contributing to glioblastoma aggressiveness and resistance to therapy: the rapid and infiltrative glioblastoma cell growth and the highly immunosuppressive tumor milieu. Therefore, in this study, we explored comprehensively the role of cancer cell-intrinsic and tumor microenvironment-mediated CD95L/CD95 interactions using syngeneic mouse glioma models. To ensure the study of constitutive CD95 signaling without artifacts, specific gene deletion of Cd95 and Cd95l was utilized here to elucidate the role of CD95 signaling in glioblastoma models.
Material and methods
Cell lines
SMA-497, SMA-540 and SMA-560 cells were kindly provided by Dr. D. D. Bigner (Durham, NC) and GL-261 cells were obtained from ATCC (Rockville, MD). Human embryonic kidney (HEK) 293T cells were purchased from Open BioSystems (Huntsville, AL). Cells were grown as adherent monolayers in Dulbecco´s modified Eagle´s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 2 mM L-glutamine (Gibco, Waltham, MA). Cells were regularly tested for mycoplasma contamination using the enzymatic MycoAlert™ PLUS Mycoplasma Detection Kit (Lonza, Basel, Switzerland).
Animal studies
Details for animal study methods are provided in Supplementary Material and Methods. Briefly, 500 − 100’000 glioma cells were stereotactically implanted into the right striatum of 4 to 16 weeks old mice. The onset of neurological symptoms defined end-stage survival.
Statistical analysis
Data are expressed as mean and standard deviation (SD) or standard error of the mean (SEM) and are representative of several independent experiments. Statistical analyses were performed with GraphPad Prism version 8 (GraphPad Software, San Diego, CA, http://www.graphpad.com). One- or two-way ANOVA with Bonferroni post-hoc tests were applied for multiple comparison significance assessments. Survival curve estimation was performed by Kaplan-Meier survival analysis and statistical significance in survival experiments was assessed by means of log-rank test. Significance levels for all analyses are *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Results
CD95 and CD95L expression in murine glioma cell lines
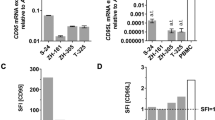
Cd95 mRNA expression was confirmed in four murine glioma cell lines (SMA-497, SMA-540, SMA-560, GL-261) (Fig. 1a). CD95 protein was detected on the surface of SMA-497 and SMA-540, but only at low levels (SFI < 1.5) in SMA-560 and not at all in GL-261 cells (Fig. 1b, S1a) (Note S1). Two murine CD95L protein isoforms have been described: the full-length canonical isoform, containing an extracellular domain, a transmembrane domain and a cytoplasmic domain; and a shorter non-canonical isoform, CD95Ls, which lacks the cytoplasmic and the transmembrane domains [28]. Full-length CD95L is encoded by the Cd95l transcript variant 1, consisting of four exons, and CD95Ls is encoded by the Cd95l transcript variant 2, consisting of part of the first and the fourth exons of the canonical Cd95l sequence [29]. Canonical Cd95l was expressed in none of the glioma cell lines in vitro, as assessed by RT-qPCR using a primer pair targeting a region of the first Cd95l exon uniquely present in transcript variant 1 (Fig. 1c, left). However, Cd95l transcript expression was detected in all cell lines using primers targeting a region of the fourth exon of Cd95l, common to transcripts 1 and 2 (Fig. 1c, right). Since Cd95l transcript was detected using primers targeting both variants but not with primers targeting variant 1 exclusively, we inferred that murine glioma cells express only the transcript variant 2, encoding the soluble CD95L isoform CD95Ls. Accordingly, CD95L protein was not detected on the surface of either glioma cell line, but also not on splenocytes (Fig. 1d, S1b) (Note S2). Exposure to temozolomide has been suggested to upregulate CD95L in human glioblastoma cells [13]. Cell surface CD95L expression was, however, not induced by exposure to temozolomide at 10 µM or 100 µM for 24 to 120 h in SMA-497 cells in vitro (Fig. S2).

CD95 and CD95L expression in mouse glioma cells. a. SMA-497, SMA-540, SMA-560 or GL-261 cells were assessed for expression of Cd95 by PCR using Hprt1 as endogenous control. b. Protein levels were assessed by flow cytometry. c. To assess Cd95l mRNA expression, two primer pairs, one targeting the first exon of the complete Cd95l coding sequence and one targeting the fourth exon of the complete Cd95l coding sequence, were used. d. CD95L protein levels were assessed by flow cytometry. Activated splenocytes (2 µg/ml concanavalin A) were included as positive control for CD95 protein and Cd95 mRNA and for CD95l mRNA. Data in a,c are presented as mean and SD. In c, a.t. indicates CT values above reliability threshold, CT>32.
CD95L is upregulated in murine gliomas in vivo
The analysis of Cd95l transcript levels in the tumors of SMA-497, SMA-540, SMA-560 and GL-261 glioma cell-bearing mice revealed an upregulation of canonical Cd95l mRNA in vivo (Fig. 2b,c). Cd95l transcript levels were studied using primers targeting part of the first Cd95l exon or the third-to-fourth exon junction, exclusive of the canonical transcript variant 1 (Fig. 2a,b) and primers targeting part of the fourth Cd95l exon common to both the canonical transcript variant 1 and the CD95Ls-encoding variant 2 (Fig. 2a,c). CD95L was also detected on the surface of iRFP720-labelled GL-261 cells isolated from end-stage tumor-bearing mice, confirming not only protein expression but also glioma cell origin (Fig. 2d).

CD95L expression in murine gliomas in vivo. a. Diagram of the two known murine Cd95l transcript variants and their sequence correspondence with the canonical Cd95l coding sequence (Cd95l CDS) and the putative murine CD95L and CD95Ls protein domains (created with BioRender.com). Three primer pairs (PP) used for the analysis of Cd95l transcript expression in vivo and their approximate amplifying regions in the transcript variants they are predicted to amplify are depicted. The CDS sequence, transcript elements, protein sequences and domain correspondence depiction were based on the data annotated in the Ensembl and UniProt databases [29, 35]. The 3D protein structures are AlfaFold predictions. The CD95L structure was extracted from the AlphaFold database [36] and the CD95Ls 3D structure was generated using the AlphaFold Colab notebook [37]. Structure coloring indicates model confidence (dark blue, pLDDT > 90; light blue, pLDDT 90 − 70; yellow, pLDDT 70 − 50; orange, pLDDT < 50). CD, death domain; TMD, transmembrane domain; ECD, extracellular domain; PRD, proline-rich domain; SA, self-assembly domain; THD, TNF homology domain. b,c. Cells were maintained in vitro or implanted in syngeneic mice and tumors from end-stage glioma-bearing mice were isolated. Expression of Cd95l mRNA in tumors and the surrounding tumor-bearing brain tissue (C57BL/6 brain and VM/Dk brain) was analyzed by RT-qPCR using primer pairs targeting the first exon of Cd95l or the sequence spanning the third and four exons of the complete Cd95l coding sequence (b) or a primer pair targeting the fourth exon of Cd95l (c). Hprt1 was used as internal control. Three tumor-bearing mice (in vivo 1, in vivo 2, in vivo 3) per model were studied. Data are represented as mean and SD (a.t., CT values above reliability threshold, CT>32). d. iRFP720-labelled GL-261 cells (GL-261 iRFP720+) were implanted in syngeneic mice. Tumors from end-stage mice were isolated and analyzed by flow cytometry. Flow cytometry histograms showing cell surface CD95L protein levels in GL-261 cells gated based on iRFP720 positivity are depicted. SFI, specific fluorescence index (median fluorescence intensity of anti-CD95L antibody, black ÷ median fluorescence intensity of isotype control, grey).
CD95 and CD95L gene disruption in mouse glioma cells
To investigate the role of constitutive CD95 signaling in murine glioma cells, CD95 or CD95L were knocked out by means of CRISPR/Cas9. CD95 depletion was mediated by two sgRNA directing the disruption of the second Cd95 exon, which is shared by all Cd95 transcript variants and encodes the N-terminus region of the extracellular CD95 domain, essential for CD95L binding [30]. CD95L depletion was mediated by a sgRNA directing a double-strand DNA break on the first Cd95l exon, which encodes the transmembrane CD95L domain, only present in canonical CD95L; and a sgRNA directing a double-strand DNA break in a region of the fourth Cd95l exon, which encodes part of the extracellular CD95L domain, common to both CD95L isoforms.
CD95 knockout clonal sublines were selected based on the absence of the mRNA sequence situated between the two predicted sgRNA-directed double-strand DNA break sites (Fig. 3a; left y axis). CD95 knockout was confirmed on protein level based on the absence of cell surface protein (Fig. 3a; right y axis, S3). Because of the lack of cell surface CD95 detection in naïve SMA-560 and GL-261 cells, CD95 knockout confirmation in these cell lines relied uniquely on the absence of the target mRNA sequence.

CD95 and CD95L CRISPR knockout (KO) in mouse glioma cells. Cd95 or Cd95l genes were knocked out via CRISPR/Cas9. a. Clonal CD95 KO cells were assessed for expression of the Cd95 transcript sequence spanning two predicted double-strand DNA break sites by RT-qPCR using Hprt1 as endogenous control and of protein by cell surface flow cytometry. RT-qPCR data are expressed as mean and SD. SFI, specific fluorescence index (median fluorescence intensity of experimental antibody ÷ median fluorescence intensity of isotype control). b. Naïve, CRISPR control or CD95 KO cells were stimulated with 10 or 1000 ng/ml exogenous CD95L (Mega-Fas-Ligand, MFL) in the absence or presence of cycloheximide (CHX, 10 µg/ml) for 6 h and assessed for Ac-DEVD-amc-cleaving (DEVDase) activity. As a positive control, cells were treated with 1 µM staurosporine (stauro). As a negative control, cells were treated with 10 µM zVAD-fmk in combination with staurosporine. A representative independent experiment is shown for each cell line. Data are expressed as mean and SD of six replicates. Statistical significances were assessed by two-way ANOVA followed by Bonferroni’s post hoc test (**p < 0.01, ****p < 0.0001 versus vehicle; ####p < 0.0001 versus CHX; +p < 0.05, ++++p < 0.0001 versus stauro). A.F.U., arbitrary fluorescence units. c. Clonal CD95L KO cells were assessed for expression of the Cd95l transcript sequence targeted by a sgRNA directing a Cas9-mediated double-strand DNA break in the fourth exon of Cd95l by RT-qPCR using Hprt1 as endogenous control. Data are expressed as mean and SD. Naïve refers to non-transfected cells, and CRISPR control to cells transfected with non-targeting sgRNA/pSpCas9(BB)-2 A-GFP plasmids.
SMA-497, SMA-540 and SMA-560 CD95 knockout sublines, contrary to naïve and CRISPR control cells, did not respond to stimulation with exogenous CD95L by increasing DEVD-amc cleaving activity, which characterizes the activity of the apoptotic effector caspase 3 as part of canonical CD95 signaling. In these sublines, DEVD-amc cleaving activity was also abrogated during stimulation with CD95L in combination with the protein synthesis inhibitor cycloheximide, which sensitizes glioma cells to CD95-mediated apoptosis induction [4]. The increase of DEVD-amc cleaving activity upon CD95L stimulation in SMA-560 naïve and control cells and its abrogation upon CD95 knockout suggested the expression of functional CD95, although undetectable by flow cytometry. An increase in the DEVD-amc cleaving activity upon stimulation with CD95L was not observed in GL-261 cells, indicating either insufficient CD95 protein levels or resistance to CD95-mediated apoptosis (Fig. 3b). CD95L knockout clonal sublines were selected based on the absence of the Cd95l mRNA target sequence of the sgRNA targeting exon 4 (Fig. 3c).
Phenotypic characterization of CD95 and CD95L knockout in murine gliomas in vitro
CD95 and CD95L knockout SMA-497 sublines exhibited reduced cell growth under conventional adherent culture conditions (Fig. 4a). Under suspension spheroid stem cell culture conditions, the growth reduction upon CD95 knockout was accentuated (Fig. 4b). The cell growth decrease upon knockout was reproduced in CD95L knockout SMA-540, SMA-560 and GL-261 cells (Fig. S4a-c) but not in CD95 knockout SMA-540, SMA-560 and GL-261 sublines, which exhibited major clonal variation (data not shown). To confirm that the observed cell growth reduction in various CD95 knockout clones was a specific consequence of CD95 depletion, CD95 knockout SMA-497 cells were re-transfected with Cd95. Indeed, cell growth was rescued in CD95 knockout cells re-expressing CD95 (Fig. 4c, S5). However, CD95 overexpression in non-transfected, constitutively CD95 and CD95L-expressing naïve and CRISPR control cells did not promote cell growth (Fig. S6), suggesting that CD95 signaling stably maintains cell growth, but does not accelerate growth with a linear dose response relationship.

Effect of CD95 and CD95L knockout (KO) in SMA-497 cell growth and T cell killing in vitro. a,b. The growth of CRISPR control, CD95 KO or CD95L KO SMA-497 cells was estimated by crystal violet staining in limiting dilution assays of cells grown in adherence conditions in the presence of fetal calf serum (a) or by MTT assay in cells cultured as spheres in suspension in the absence of fetal calf serum, but with EGF and FGF supplementation (b). c,d. Cell growth of adherent CD95 KO (clone 6) or CD95L KO (clone 3) SMA-497 cells transfected with Cd95 or Cd95l, respectively, was estimated in limiting dilution assays by crystal violet staining. Data in a-d are expressed as mean and SEM of representative experiments. Statistical significances (main column effect) were determined by means of a two-way ANOVA test followed by Bonferroni’s post hoc test. Data were reproduced in three independent experiments. e. Specific killing of activated splenocytes after 24 h co-culture with CRISPR control, CD95L KO (clone 3) or Cd95l-transfected CD95L KO (clone 3) SMA-497 cells. Data in e are expressed as mean and SD. Statistical significances were calculated by one-way ANOVA followed by Bonferroni’s post hoc test. A.U., arbitrate units; ns, not significant; ****, p < 0.0001.
Functional difference of CD95Ls and canonical full-length CD95L
Transfection of CD95L knockout cells with the complete coding sequence of Cd95l resulted in reduced cell growth (Fig. 4d) likely due to augmented CD95L-induced apoptosis, since growth reduction was not observed upon transfection of Cd95l into CD95 knockout cells (data not shown). Furthermore, contrary to naïve murine glioma cells, which have been observed to exclusively express the CD95Ls-encoding Cd95l transcript in vitro, canonical Cd95l transfectants were capable of killing activated splenocytes, presumably via CD95L-CD95-mediated apoptosis (Fig. 4e). A decrease in specific killing of splenocytes was observed upon CD95L knockout neither in SMA-497 nor in SMA-540, SMA-560 or GL-261 cells (Fig. 4e, data not shown).
Despite evident mRNA expression upon Cd95l transfection (Fig. S5c, S7a), cell surface CD95L was not observed (Fig. S5d, S7b). However, CD95L was detected in the lysates and supernatants of Cd95l transfectants, using an antibody with affinity for the canonical CD95L extracellular domain (Fig. S8). In cell lysates, a ~40 kDa band, corresponding to the CD95L predicted mass, was observed while in cell culture supernatants a band corresponding to a ~25 kDa protein was additionally detected. Since the expected mass of CD95Ls is ~8–16 kDa [31], this band likely resulted from degradation and cleavage upon the artificial condition of overexpression (Note S3).
Cd95 or Cd95l gene deletion delay growth in syngeneic murine glioma models
Syngeneic VM/Dk mice orthotopically implanted with CD95 or CD95L knockout SMA-497 cells (Fig. 5a) developed smaller tumors than mice implanted with control cells (Fig. 5b). Control tumors appeared to exhibit less regular borders than CD95L knockout tumors (Fig. 5b, S9a). However, significant differences in the number of tumor satellites, quantified to infer invasion, were not revealed between groups, despite a trend towards reduced satellite number in CD95L knockout tumors potentially driven by tumor size differences (Fig. S9a). Immunocompetent syngeneic mice implanted with CD95 or CD95L knockout SMA-497 cells survived significantly longer than mice implanted with control cells (Fig. 5c). The median survivals of control, CD95 knockout and CD95L knockout tumor-bearing mice were 15, 26 and 33 days, respectively.

Effect of CD95 and CD95L knockout (KO) in the SMA-497 model in vivo. a. Syngeneic (immunocompetent) VM/Dk or athymic (immunocompromised) Foxn1nu mice were orthotopically implanted with control (naïve and CRISPR control), CD95 KO (clones 5 and 6) or CD95L KO (clones 3 and 5) SMA-497 cells (illustration created with BioRender.com). b. Tumor volumes from animals sacrificed at the first onset of symptoms were determined by hematoxylin/eosin staining (scale bar = 1 mm). Data are expressed as mean and SD of n = 4 VM/Dk mice. Statistical significances between CD95 KO or CD95L KO and control tumor-bearing brains were determined by means of a one-way ANOVA test followed by Bonferroni’s post hoc test. c,d. End-stage survival was recorded. Statistical significances between CD95 KO or CD95L KO glioma-bearing mice and control glioma-bearing mice within the same mouse strain were determined by means of a log-rank test. Median survival in days provided in brackets (n = 7 mice per group). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Cancer cell-intrinsic and immunological mechanisms mediate increased survival upon CD95L knockout in the SMA-497 model
The increase in median survival observed in fully immunocompetent mice implanted with CD95 or CD95L knockout cells was reproduced in mature T cell-deficient Foxn1nu mice (Fig. 5d). However, we noted differences by mouse strain for overall outcome: while 40% of immunocompetent mice bearing CD95L knockout tumors achieved long-term survival (Fig. 5c) and did not develop neurologic symptoms after tumor rechallenge (data not shown), none of the CD95L knockout cell-bearing immunodeficient mice survived for more than 60 days after glioma cell implantation (Fig. 5d). These data suggest a glioma CD95L-mediated T cell suppression that is abrogated in CD95L-depleted glioma cells.
Mean CD3+ T cell numbers in the tumors at an early time point did not differ overall between groups. However, while only 50% of CD95L-expressing control or CD95 knockout tumors exhibited CD3+ T cell infiltration, CD3+ T cells were observed in 100% of CD95L knockout tumors (Fig. S9b). Significant inter-group differences in the numbers of CD11b+ microglia/macrophages and CD31+ endothelial cells were not revealed either (Fig. S9c,d) (Note S4).
Conversely, in syngeneic GL-261 gliomas, tumor intrinsic effects only mediated increased survival upon CD95L knockout (Fig. S10a) and immune cell-dependency was not observed (Fig. S10b-d) (Note S5).
Discussion
The role of CD95 signaling in glioblastoma and in many other cancers has remained obscure and controversial, rendering efforts at targeting these molecules therapeutically challenging. Careful consideration needs to be given to the fact that CD95L/CD95 interactions are pleiotropic and subsequent signaling may entail a complex network of not yet completely elucidated signaling pathways leading to distinct outcomes, contributing to either tumor suppression or progression in a context-dependent manner.
Here we sought to gain insight into the function of CD95 and CD95L in murine glioma models using CRISPR/Cas9-mediated gene deletion as the key experimental strategy. We demonstrate that the spontaneous (SMA-497, SMA-540, SMA-560) and chemically induced (GL-261) murine glioma models express canonical Cd95 and a non-canonical soluble Cd95l splice variant, encoding a soluble isoform of CD95L (CD95Ls) in vitro (Fig. 1). Full-length canonical Cd95l is expressed only when glioma cells are implanted in syngeneic mouse brains (Fig. 2). To disrupt CD95/CD95L interactions, we introduced genetic deletions in Cd95 and Cd95l by CRISPR/Cas9-mediated knockout.
A growth reduction was evidenced upon CD95 knockout (Fig. 4), although major clonal variation was observed. CD95 KO clones showing no phenotype may have undergone genetic compensation, a phenomenon suggested in various studies involving genetic editing [32]. In fact, we showed a growth rescue upon Cd95 retransfection, confirming the specificity of CD95 deletion in the growth reduction observed in the CD95 knockout sublines that exhibited a phenotype (Fig. 4). Unlike CD95 re-expression in CD95 knockout cells, CD95 overexpression in parental cells did not increase growth, suggesting that a threshold level of CD95 maintains glioma cell growth whereas supraphysiological levels exert no additional biological effect.
All glioma cell lines were sensitive to CD95-mediated apoptosis (Fig. 3). Therefore, in contrast to previous reports which suggested resistance to CD95-mediated apoptosis as a requisite for growth-promoting CD95 signaling [7, 14], we report growth-promoting constitutive CD95 signaling in CD95-mediated apoptosis-sensitive glioma cells.
Despite the impossibility of detecting CD95Ls protein due to the lack of isoform-specific antibodies, all CD95L knockout cells exhibited a marked growth reduction in vitro (Fig. 4, S4). At the time of its discovery, CD95Ls was described to negatively regulate apoptosis [31]. Our data raise the possibility that CD95Ls may, in fact, induce non-apoptotic tumor-promoting signaling.
In vivo, knockout of CD95 or CD95L in the SMA-497 and GL-261 models had both shared and model-specific effects: Cd95l gene depletion uniformly prolonged survival in both models, whereas Cd95 deletion prolonged survival in the SMA-497 model only (Fig. 5, S10). This may be explained by the low-to-null constitutive level of CD95 in GL-261 cells. Although tumor invasiveness has been reported to be inhibited in the murine SMA-560 glioma upon pharmacologic CD95L blockade [14, 17], significant differences in the number of tumor satellites were not observed in our SMA-497 model. Tumors formed by CD95 and CD95L knockout SMA-497 cells were significantly smaller than control tumors, suggesting that the decreased tumorigenicity of CD95 and CD95L knockout cells is mainly driven by growth inhibition. Reduced tumor incidence and size have also been described upon CD95 depletion in ovarian and liver cancer [12].
While a similar median survival gain was observed in mature T cell-competent and deficient mice, long-term surviving mice after inoculation of CD95L knockout SMA-497 cells were exclusively observed in immunocompetent mice. Furthermore, CD3+ T cells infiltrated all CD95L KO tumors but not all CD95L-expressing tumors (Fig. S9), suggesting that CD95L halts CD3+ T cells in some mice, presumably due to apoptosis induction. Therefore, we propose that the abrogation of tumor CD95L acting on CD95 expressed on immune cells may allow immunocompetent mice to achieve long-term survival more frequently. Of note, recent observations of CD95-dependent natural killer cell suppression in triple negative breast cancer models [33, 34] indicate that the growth delay of CD95-depleted tumors may not only depend on slowed growth properties as documented in vitro (Fig. 4), but also on altered natural killer cell activity, which is retained in the immunocompromised mouse models used in the present study. Overall, based on our results, we hypothesize that cancer cell-intrinsic growth-promoting CD95 signaling may be mediated by CD95Ls while the potential CD95L-mediated T cell killing is likely induced by canonical CD95L. This is because we did not observe apoptosis-inducing capacity in cells expressing only CD95Ls, but only growth inhibition upon CD95L knockout (Fig. 4a,b, S4). In contrast, ectopic expression of full length CD95L, which is expressed in vivo but not in vitro (Fig. 2), resulted in specific splenocyte killing (Fig. S8). The survival gain mediated by Cd95l gene deletion was maintained in GL-261 cell-bearing Rag-/- mice lacking T and B cells as well as in Faslpr mice lacking functional CD95, suggesting a tumor-intrinsic effect of CD95L disruption only that might involve interactions with other death ligands or even backward signaling.
Limitations of our study include the risk of confounding factors derived from the use of clonal sublines and potential off-target Cas9-mediated DNA double-strand breaks, but multiple clones and cell lines were used, and rescue experiments were performed, to maximize reliability (data partly not shown). Moreover, we noted that multiple commercially available antibodies are not specific as evidenced by unaltered staining patterns in confirmed knockout sublines (Supplementary notes). This may explain apparent contradictions between the findings reported here and those of previous studies on CD95L in murine cancer models in vitro and in vivo.
Anyhow, we report here that constitutive CD95 signaling in glioblastoma may be tumor-promoting via both intrinsic growth regulation and immunosuppression. However, CD95 signaling remains complex and the biological context may determine the outcome of modulating this pathway.
References
Yonehara S, Ishii A, Yonehara M (1989) A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med 169:1747–1756. https://doi.org/10.1084/jem.169.5.1747
Trauth BC, Klas C, Peters AMJ et al (1989) Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science (1979) 245:301–305. https://doi.org/10.1126/science.2787530
Weller M, Malipiero U, Rensing-Ehl A et al (1995) Fas/APO-1 Gene Transfer for Human Malignant Glioma. Cancer Res 55:2936–2944
Weller M, Frei K, Groscurth P et al (1994) Anti-Fas/APO-1 antibody-mediated apoptosis of cultured human glioma cells: Induction and modulation of sensitivity by cytokines. J Clin Invest 94:954–964. https://doi.org/10.1172/JCI117462
Ogasawara J, Watanabe-Fukunaga R, Adachi M et al (1993) Lethal effect of the anti-Fas antibody in mice. Nature 364:806–809. https://doi.org/10.1038/364806a0
Rensing-Ehl A, Frei K, Flury R et al (1995) Local Fas/APO‐1 (CD95) ligand‐mediated tumor cell killing in vivo. Eur J Immunol 25:2253–2258. https://doi.org/10.1002/eji.1830250821
Barnhart BC, Legembre P, Pietras E et al (2004) CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J 23:3175–3185. https://doi.org/10.1038/sj.emboj.7600325
Steller EJA, Ritsma L, Raats DAE et al (2011) The death receptor CD95 activates the cofilin pathway to stimulate tumour cell invasion. EMBO Rep 12:931–937. https://doi.org/10.1038/embor.2011.129
Malleter M, Tauzin S, Bessede A et al (2013) CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res 73:6711–6721. https://doi.org/10.1158/0008-5472.CAN-13-1794
Ametller E, García-Recio S, Costamagna D et al (2010) Tumor promoting effects of CD95 signaling in chemoresistant cells. Mol Cancer 9:161. https://doi.org/10.1186/1476-4598-9-161
Ceppi P, Hadji A, Kohlhapp FJ et al (2014) CD95 and CD95L promote and protect cancer stem cells. Nat Commun 5:5238. https://doi.org/10.1038/ncomms6238
Chen L, Park SM, Tumanov A et al (2010) CD95 promotes tumour growth. Nature 465:492–496. https://doi.org/10.1038/nature09075
Drachsler M, Kleber S, Mateos A et al (2016) CD95 maintains stem cell-like and non-classical EMT programs in primary human glioblastoma cells. Cell Death Dis 7:e2209. https://doi.org/10.1038/cddis.2016.102
Kleber S, Sancho-Martinez I, Wiestler B et al (2008) Yes and PI3K Bind CD95 to Signal Invasion of Glioblastoma. Cancer Cell 13:235–248. https://doi.org/10.1016/j.ccr.2008.02.003
Qadir AS, Ceppi P, Brockway S et al (2017) CD95/Fas Increases Stemness in Cancer Cells by Inducing a STAT1-Dependent Type I Interferon Response. Cell Rep 18:2373–2386. https://doi.org/10.1016/j.celrep.2017.02.037
Wick W, Fricke H, Junge K et al (2014) A phase II, randomized, study of weekly APG101 + reirradiation versus reirradiation in progressive glioblastoma. Clin Cancer Res 20:6304–6313. https://doi.org/10.1158/1078-0432.CCR-14-0951-T
Blaes J, Thome CM, Pfenning PN et al (2018) Inhibition of CD95/CD95L (FAS/FASLG) Signaling with APG101 prevents invasion and enhances radiation therapy for glioblastoma. Mol Cancer Res 16:767–776. https://doi.org/10.1158/1541-7786.MCR-17-0563
Merz C, Strecker A, Sykora J et al (2015) Neutralization of the CD95 ligand by APG101 inhibits invasion of glioma cells in vitro. Anticancer Drugs 26:716–727. https://doi.org/10.1097/CAD.0000000000000237
Ichinose M, Masuoka J, Shiraishi T et al (2001) Fas ligand expression and depletion of T-cell infiltration in astrocytic tumors. Brain Tumor Pathol 18:37–42. https://doi.org/10.1007/BF02478923
Weller M, Weinstock C, Will C et al (1997) CD95-Dependent T-Cell Killing by Glioma Cells Expressing CD95 Ligand: More on Tumor Immune Escape, the CD95 Counterattack, and the Immune Privilege of the Brain. Cell Physiol Biochem 7:282–288. https://doi.org/10.1159/000154883
Saas P, Walker PR, Hahne M et al (1997) Fas ligand expression by astrocytoma in vivo: Maintaining immune privilege in the brain? J Clin Invest 99:1173–1178. https://doi.org/10.1172/JCI119273
Yu JS, Lee PK, Ehtesham M et al (2003) Intratumoral T cell subset ratios and Fas ligand expression on brain tumor endothelium. J Neurooncol 64:55–61. https://doi.org/10.1007/BF02700020
Lakins MA, Ghorani E, Munir H et al (2018) Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat Commun 9:948. https://doi.org/10.1038/s41467-018-03347-0
Motz GT, Santoro SP, Wang L-P et al (2014) Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 20:607–615. https://doi.org/10.1038/nm.3541
Yu J, Green MD, Li S et al (2021) Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med 27:152–164. https://doi.org/10.1038/s41591-020-1131-x
Favre-Felix N, Fromentin A, Hammann A et al (2000) Cutting Edge: The Tumor Counterattack Hypothesis Revisited: Colon Cancer Cells Do Not Induce T Cell Apoptosis Via the Fas (CD95, APO-1) Pathway. J Immunol 164:5023–5027. https://doi.org/10.4049/jimmunol.164.10.5023
Igney FH, Behrens CK, Krammer PH (2005) CD95L mediates tumor counterattackin vitro but induces neutrophil-independent tumor rejectionin vivo. Int J Cancer 113:78–87. https://doi.org/10.1002/ijc.20538
Bateman A, Martin MJ, Orchard S et al (2021) UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res 49:D480–D489. https://doi.org/10.1093/nar/gkaa1100
Cunningham F, Allen JE, Allen J et al (2022) Ensembl 2022. Nucleic Acids Res 50:D988–D995. https://doi.org/10.1093/nar/gkab1049
Orlinickt JR, Vaishnaw A, Elkon KB, Chao M (1997) Requirement of cysteine,rich repeats of the Fas receptor for binding by the Fas ligand. J Biol Chem 272:28889–28894. https://doi.org/10.1074/jbc.272.46.28889
Ayroldi E, D’Adamio F, Zollo O et al (1999) Cloning and expression of a short Fas ligand: A new alternatively spliced product of the mouse Fas ligand gene. Blood 94:3456–3467. https://doi.org/10.1182/blood.v94.10.3456.422k33_3456_3467
Salanga CM, Salanga MC (2021) Genotype to Phenotype: CRISPR Gene Editing Reveals Genetic Compensation as a Mechanism for Phenotypic Disjunction of Morphants and Mutants. Int J Mol Sci 22:3472. https://doi.org/10.3390/ijms22073472
Qadir AS, Guégan JP, Ginestier C et al (2021) CD95/Fas protects triple negative breast cancer from anti-tumor activity of NK cells. iScience 24:103348. https://doi.org/10.1016/j.isci.2021.103348
Guégan JP, Pollet J, Ginestier C et al (2021) CD95/Fas suppresses NF-κB activation through recruitment of KPC2 in a CD95L/FasL-independent mechanism. https://doi.org/10.1016/j.isci.2021.103538. iScience 24:
Bateman A, Martin M-J, Orchard S et al (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49:D480–D489. https://doi.org/10.1093/nar/gkaa1100
Varadi M, Anyango S, Deshpande M et al (2022) AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50:D439–D444. https://doi.org/10.1093/nar/gkab1061
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Acknowledgements
The authors express their gratitude to J. Friesen for technical assistance and M. Peter (Chicago, IL) and H. Schneider (Zurich, Switzerland) for valuable discussions in the early stages of the project.
Funding
This study was supported by the Oncosuisse grant KLS-3814-02-2016 to M.W.
Open access funding was provided by the University of Zurich.
Author information
Authors and Affiliations
Contributions
Clara Quijano-Rubio and Michael Weller contributed to the study conceptualization and the experimental design. Material preparation, experimental work, data collection and data analysis were performed by Clara Quijano-Rubio. Clara Quijano-Rubio, Manuela Silginer and Michael Weller contributed to data interpretation. The first manuscript draft was written by Clara Quijano-Rubio and revised by Michael Weller. All authors commented on previous versions of the manuscript and revised them critically. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
C.Q.R. and M.S. report no conflicts of interest. M.W. has received research grants from Apogenix, Merck, Sharp & Dohme, Merck (EMD), Philogen and Quercis, and honoraria for lectures or advisory board participation or consulting from Adastra, Bayer, Bristol Meyer Squibb, Medac, Merck, Sharp & Dohme, Merck (EMD), Nerviano Medical Sciences, Novartis, Orbus, Philogen and y-Mabs.
Ethics approval
All animal studies were conducted in accordance with the Swiss federal act on animal protection. All experiments were approved by the cantonal veterinary office under the animal license numbers ZH178/2016 and ZH109/2020.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.










Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Quijano-Rubio, C., Silginer, M. & Weller, M. CRISPR/Cas9-mediated abrogation of CD95L/CD95 signaling-induced glioma cell growth and immunosuppression increases survival in murine glioma models. J Neurooncol 160, 299–310 (2022). https://doi.org/10.1007/s11060-022-04137-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-022-04137-x