
Abstract
Background
The Yellowfin tuna (Thunnus albacares) is a large tuna exploited by major fisheries in tropical and subtropical waters of all oceans except the Mediterranean Sea. Genomic studies of population structure, adaptive variation or of the genetic basis of phenotypic traits are needed to inform fisheries management but are currently limited by the lack of a reference genome for this species. Here we report a draft genome assembly and a linkage map for use in genomic studies of T. albacares.
Methods and results
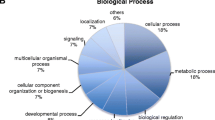
Illumina and PacBio SMRT sequencing were used in combination to generate a hybrid assembly that comprises 743,073,847 base pairs contained in 2,661 scaffolds. The assembly has a N50 of 351,587 and complete and partial BUSCO scores of 86.47% and 3.63%, respectively. Double-digest restriction associated DNA (ddRAD) was used to genotype the 2 parents and 164 of their F1 offspring resulting from a controlled breeding cross, retaining 19,469 biallelic single nucleotide polymorphism (SNP) loci. The SNP loci were used to construct a linkage map that features 24 linkage groups that represent the 24 chromosomes of yellowfin tuna. The male and female maps span 1,243.8 cM and 1,222.9 cM, respectively. The map was used to anchor the assembly in 24 super-scaffolds that contain 79% of the yellowfin tuna genome. Gene prediction identified 46,992 putative genes 20,203 of which could be annotated via gene ontology.
Conclusions
The draft reference will be valuable to interpret studies of genome wide variation in T. albacares and other Scombroid species.



Similar content being viewed by others


Data availability
The sequencing data obtained during this project (PacBio and Illumina sequences used in assembly, dd-RAD illumine sequencing reads) were uploaded in genbank (Bioproject PRJNA504596). Supplementary Materials (Supplementary Methods and Materials, Supplementary Figs. 1–3) are included as supplementary Materials published with this manuscript. Supplementary File 2 (GO annotations) is uploaded in the Aquila Repository of the University of Southern Mississippi ((https://aquila.usm.edu/datasets/7/).
References
Collette BB, Nauen CE (1983) FAO species catalogue. Scombrids of the world. An annotated and illustrated catalogue of tunas, mackerels, bonitos and related species known to date. Food and Agriculture Organization of the United Nations, Rome
ISSF (2022) Status of the World Fisheriesfor Tuna: March 2022. International Seafood Sustainability Foundation, Washington, D.C.
Sibert J, Hampton J (2003) Mobility of tropical tunas and the implications for fisheries management. Mar Policy 27:87–95. https://doi.org/10.1016/S0308-597X(02)00057-X
Margulies D, Scholey VP, Wexler JB, Stein MS (2016) Research on the reproductive biology and early life history of yellowfin tuna Thunnus albacares in Panama. Advances in tuna aquaculture. Elsevier, pp 77–114.
Carvalho GR, Hauser L (1994) Molecular genetics and the stock concept in fisheries. Rev Fish Biol Fish 4:326–350. https://doi.org/10.1007/BF00042908
Schmid M, Frei D, Patrignani A et al (2018) Pushing the limits of de novo genome assembly for complex prokaryotic genomes harboring very long, near identical repeats. Nucleic Acids Res 46:8953–8965. https://doi.org/10.1093/nar/gky726
Ye C, Hill CM, Wu S et al (2016) DBG2OLC: efficient assembly of large genomes using long erroneous reads of the third generation sequencing technologies. Sci Rep 6:31900. https://doi.org/10.1038/srep31900
Ma ZS, Li L, Ye C et al (2019) Hybrid assembly of ultra-long nanopore reads augmented with 10x-Genomics contigs: demonstrated with a human genome. Genomics 111:1896–1901. https://doi.org/10.1016/j.ygeno.2018.12.013
Haghshenas E, Asghari H, Stoye J et al (2020) HASLR: fast hybrid assembly of long reads. iScience 23:101389. https://doi.org/10.1016/j.isci.2020.101389
McWilliam S, Grewe PM, Bunch RJ, Barendse W (2016) A draft genome assembly of southern bluefin tuna Thunnus maccoyii. arXiv. Doi: 10.48550/arxiv.1607.03955.
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890. https://doi.org/10.1093/bioinformatics/bty560
Marçais G, Kingsford C (2011) A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27:764–770. https://doi.org/10.1093/bioinformatics/btr011
Allam A, Kalnis P, Solovyev V (2015) Karect: accurate correction of substitution, insertion and deletion errors for next-generation sequencing data. Bioinformatics 31:3421–3428. https://doi.org/10.1093/bioinformatics/btv415
Miclotte G, Heydari M, Demeester P et al (2016) Jabba: hybrid error correction for long sequencing reads. Algorithms Mol Biol 11:10. https://doi.org/10.1186/s13015-016-0075-7
Ye C, Ma ZS, Cannon CH et al (2012) Exploiting sparseness in de novo genome assembly. BMC Bioinf. https://doi.org/10.1186/1471-2105-13-S6-S1
Li H (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34:3094–3100. https://doi.org/10.1093/bioinformatics/bty191
Vaser R, Sović I, Nagarajan N, Šikić M (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27:737–746. https://doi.org/10.1101/gr.214270.116
Walker BJ, Abeel T, Shea T et al (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. https://doi.org/10.1371/journal.pone.0112963
Qin M, Wu S, Li A et al (2019) LRScaf: improving draft genomes using long noisy reads. BMC Genomics 20:955. https://doi.org/10.1186/s12864-019-6337-2
Gurevich A, Saveliev V, Vyahhi N, Tesler G (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics 29:1072–1075. https://doi.org/10.1093/bioinformatics/btt086
Simão FA, Waterhouse RM, Ioannidis P et al (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. https://doi.org/10.1093/bioinformatics/btv351
Cusatti S, Margulies D, Scholey V et al (2022) Spawning ecology of captive yellowfin tuna broodstock inferred by the use of mitochondrial DNA sequencing analysis. Aquacult Sci 70(4):331–342
Antoni L, Luque PL, Naghshpour K et al (2014) Development and characterization of microsatellite markers for blackfin tuna (Thunnus atlanticus) with the use of Illumina paired-end sequencing. FB 112:322–325. https://doi.org/10.7755/FB.112.4.8
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Peterson BK, Weber JN, Kay EH et al (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. https://doi.org/10.1371/journal.pone.0037135
Puritz JB, Hollenbeck CM, Gold JR (2014) dDocent: a RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2:e431. https://doi.org/10.7717/peerj.431
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Rastas P (2017) Lep-MAP3: robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics 33:3726–3732. https://doi.org/10.1093/bioinformatics/btx494
Rastas P (2020) Lep-Anchor: automated construction of linkage map anchored haploid genomes. Bioinformatics 36:2359–2364. https://doi.org/10.1093/bioinformatics/btz978
Huang S, Kang M, Xu A (2017) HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 33:2577–2579. https://doi.org/10.1093/bioinformatics/btx220
Flynn JM, Hubley R, Goubert C et al (2020) RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117:9451–9457. https://doi.org/10.1073/pnas.1921046117
Stanke M, Keller O, Gunduz I et al (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34:W435–W439. https://doi.org/10.1093/nar/gkl200
Cantalapiedra CP, Hernández-Plaza A, Letunic I et al (2021) eggNOG-mapper v2: functional annotation, Orthology assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 38:5825–5829. https://doi.org/10.1093/molbev/msab293
Huerta-Cepas J, Szklarczyk D, Heller D et al (2019) eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res 47:D309–D314. https://doi.org/10.1093/nar/gky108534
Marçais G, Delcher AL, Phillippy AM et al (2018) MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol 14:e1005944. https://doi.org/10.1371/journal.pcbi.1005944
Suda A, Nishiki I, Iwasaki Y et al (2019) Improvement of the Pacific bluefin tuna (Thunnus orientalis) reference genome and development of male-specific DNA markers. Sci Rep 9:14450. https://doi.org/10.1038/s41598-019-50978-4
Gu Z, Gu L, Eils R et al (2014) Circlize implements and enhances circular visualization in R. Bioinformatics 30:2811–2812. https://doi.org/10.1093/bioinformatics/btu393
Catchen JM, Hohenlohe PA, Bernatchez L et al (2017) Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol Ecol Resour 17:362–365. https://doi.org/10.1111/1755-0998.12669
Jansen HJ, Liem M, Jong-Raadsen SA et al (2017) Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci Rep 7(1):7213
Smith JJ, Timoshevskaya N, Ye C et al (2018) The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat Genet 50:270–277. https://doi.org/10.1038/s41588-017-0036-1
Puncher GN, Cariani A, Maes GE et al (2018) Spatial dynamics and mixing of bluefin tuna in the Atlantic Ocean and Mediterranean Sea revealed using next-generation sequencing. Mol Ecol Resour 18:620–638. https://doi.org/10.1111/1755-0998.12764
Wiley G, Miller MJ (2020) A highly contiguous genome for the Golden-fronted woodpecker (Melanerpes aurifrons) via a hybrid Oxford Nanopore and short read assembly. G3. https://doi.org/10.1101/2020.01.03.894444.
Tan MH, Austin CM, Hammer MP et al (2018) Finding Nemo: hybrid assembly with Oxford Nanopore and Illumina reads greatly improves the clownfish (Amphiprion ocellaris) genome assembly. Gigascience 7:1–6. https://doi.org/10.1093/gigascience/gix137
Tørresen OK, Star B, Mier P et al (2019) Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res 47:10994–11006. https://doi.org/10.1093/nar/gkz841
Lohse, K., Taylor-Cox, E., Darwin Tree of Life Barcoding Collective, et al. (2021). The genome sequence of the speckled wood butterfly pararge aegeria (Linnaeus 1758). Wellcome Open Res. https://doi.org/10.12688/wellcomeopenres.17278.1
Dumschott K, Schmidt MH-W, Chawla HS et al (2020) Oxford Nanopore sequencing: new opportunities for plant genomics? J Exp Bot 71:5313–5322. https://doi.org/10.1093/jxb/eraa263
Lee Y-H, Yen T-B, Chen C-F, Tseng M-C (2018) Variation in the Karyotype, cytochrome b Gene, and 5S rDNA of four Thunnus (Perciformes, Scombridae) Tunas. Zool Stud 57:e34. https://doi.org/10.6620/ZS.2018.57-34
Aird D, Ross MG, Chen W-S et al (2011) Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. https://doi.org/10.1186/gb-2011-12-2-r18
Brekke C, Johnston SE, Knutsen TM, Berg P (2023) Genetic architecture of individual meiotic crossover rate and distribution in Atlantic Salmon. Sci Rep 13(1):20481. https://doi.org/10.1038/s41598-023-47208-3
Santini F, Carnevale G, Sorenson L (2013) First molecular scombrid timetree (Percomorpha: Scombridae) shows recent radiation of tunas following invasion of pelagic habitat. Ital J Zool 80(2):210–221. https://doi.org/10.1080/11250003.2013.775366
Funding
This work was supported by the National Oceanic and Atmospheric Administration Saltonstall-Kennedy program award #NA16NMF4270223.
Views expressed in this paper are those of the authors and do not necessarily reflect those of the.
sponsors.
Author information
Authors and Affiliations
Contributions
Funding acquisition: ES; Study design: ES, PD; Sample acquisition: DM, SC, VS, ES; Data analysis and interpretations: PD, ES; Manuscript drafting: PD, ES; Manuscript reviewing: All authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Protocols employed in this study were approved by the Institutional Animal Care and Use Committee at the University of Southern Mississippi (protocol #1710301).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dimens, P.V., Jones, K.L., Margulies, D. et al. Genomic resources for the Yellowfin tuna Thunnus albacares. Mol Biol Rep 51, 232 (2024). https://doi.org/10.1007/s11033-023-09117-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-023-09117-6