
Abstract
Iron-based magnetic nanoparticles (MNPs) have attracted considerable attention as promising draw solutes in forward osmosis processes due to various advantages. In the present study, magnetite (Fe3O4) nanoparticles (MNPs) with diameters in the range of 12 ± 2 nm were synthesized by applying the coprecipitation method and stabilized using (3-aminopropyl)triethoxysilane (APTES). The amino groups of APTES were further functionalized with various amounts of citric acid (CA) and poly(acrylic acid) (PAA) to achieve higher osmotic pressure. The as-prepared and functionalized nanoparticles were characterized using different methods: Fourier-transform infrared spectroscopy (FTIR) confirmed the binding of APTES and CA/PAA on the surface of the MNPs, while the size of the particles was determined by transmission electron microscopy (TEM) and X-ray powder diffraction. The isoelectric point, zeta potential, and the particle size in suspension were determined by dynamic light scattering (DLS)/zeta potential measuring system. The amount of free –COOH groups was determined by conductometric titrations. The amount of organic matter (APTES, CA, and PAA) bound on the surface was determined by thermogravimetric analysis (TG), giving results between 7.6% for samples stabilized with APTES only and 17.5% for samples functionalized with APTES + PAA. TG provides a simple and efficient method for determining the amount of organic compounds bound to the surface of MNPs. The osmotic pressure measurements of double-layer coated MNPs provided encouraging results of up to 28 bar, enabling using the samples as draw solutions in an experimental forward osmosis system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Water scarcity is becoming one of the major worldwide environmental issues due to industrial and agricultural pollution as well as enormous increase in global population, a major challenge that requires an adequate solution in the near future. It is reported that by 2025, two thirds of the world’s population may face water shortages. The problem could be overcome by developing an efficient method of converting salt or polluted water into potable water with low energy demand [1]. The traditional method of water desalination using multi-stage flash distillation still produces more than 50% of all desalinated water in the world; however, the energy demand of the process is still high and can be regarded as a major drawback in times of high energy prices. Reverse osmosis (RO) is often regarded as a second generation of desalination processes. In this method, pressure is applied across a semipermeable membrane for water treatment. The method is used in most of the desalination plants installed nowadays. The two main issues with RO methods are energy consumption and membrane fouling due to the high hydraulic pressures used to run the process [1, 2]. On the other hand, forward osmosis (FO) technologies exploit the natural osmotic pressure gradient between two sides of a selective permeation membrane to achieve spontaneous water transfer from the solution of lower osmotic pressure feed solution (FS) to that of higher osmotic pressure draw solution (DS). The method, based on the natural phenomenon of a solvent passing from an area with low osmotic pressure through a semipermeable membrane to an area with high osmotic pressure, has gained significant interest in recent years as an alternative to conventional flash distillation and RO processes in wastewater treatment and desalination. The diffusing water dilutes the DS by concentrating the FS simultaneously, so no physical pressure is needed. Any energy required would only be for pumping the FS and DS toward the membrane [3]. In the FO process, the membrane plays a crucial role and must be designed to specific requirements to align with the intended application [4]. Although there are many references on the preparation of FO membranes in the literature, currently there are only a few global FO membrane suppliers, and the products of most companies are still at the very basic research or development stage [5]. To maximize the efficiency of the FO process, it is necessary to identify a low-cost DS solute that can generate high osmotic pressures, high water flux, and minimal reverse flux, moreover, is non-toxic, and is economically reconcentrated after use [6]. Furthermore, the DS solute must meet stringent criteria for limiting diffusion through the porous membrane support. This restriction is essential to prevent undesired effects such as osmotic pressure build-up and subsequent decline in both flux and overall membrane performance [7]. Unlike RO, the end product of FO is not purified water, but diluted draw solution. Unless the diluted DS is of use of itself, a second separation step is necessary to reconcentrate the DS and obtain purified water [8]. Multiple suitable draw solutions were investigated though decades, including volatile solutes such as SO2, ammonium bicarbonate, fertilizers, simple carbohydrates, polymers [2, 8].
Iron-based magnetic nanoparticles (MNPs) like magnetite and maghemite could provide ideal candidates for DS for a multitude of reasons: They are simple to prepare, inexpensive, exhibit good biocompatibility, and good magnetic properties, allowing an easy recovery by using an external magnetic field. Iron-based MNPs have been successfully used in environmental applications such as removal of copper [9] and lead [10] from industrial effluents and are recently under investigation for treating cancer via the process of magnetic hyperthermia [11]. Two predominant methods for the preparation of iron-based MNPs are reported in recent the literature, one being the thermal decomposition of an appropriate precursor, mostly iron(III) acetylacetonate, Fe(acac)3 [12,13,14,15]. The most commonly used method seems to be coprecipitation, where Fe3O4 precipitates from a mixture of Fe2+ and Fe3+ ions in alkaline solution [16,17,18,19,20]. The coprecipitation method offers various advantages when compared to other reported synthesis pathways, being simple, energy-efficient, avoiding the use of toxic precursors and complex equipment, and enabling a narrow size distribution of as-prepared MNPs [21].
To induce a high osmotic pressure in the FO process, the size of MNPs should be as small as possible, since osmotic pressure is a colligative property. On the other hand, small MNPs tend to aggregate in aqueous solutions [12], thus they must be functionalized with molecules that generate sufficient osmotic pressure and prevent agglomeration. Numerous coating molecules have been investigated and reported in the literature, including poly(acrylic acid), PAA [12, 13], poly(N-isopropylacrylamide) [12], 1,6-hexanediamine [9], polyvinyl alcohol (PVA) [11], 2-pyrrolidone [13], triethylene glycol [13], poly(sodium acrylate) [14, 18], siloxanes [15], and trisodium citrate salt [16, 17]. Examples of other possible coatings, synthetic approaches, as well as a review of the current status in the area are provided, among others, in the review paper by Hafiz and coworkers [21] and in the book chapter by Roach et al. [22]. Among the various coatings, double-layer coatings, consisting of an inner silica layer, functionalized with an outer layer, have attracted considerable attention recently. The silica layer shields the iron-based MNPs from reaction, especially in acidic media, increases the surface area, and can be used for linkage with the outer layer [10]. Those kind of MNPs, coated with tetraethyl orthosilicate (TEOS) and modified with a polyethyleneimine outer layer, were investigated as nanoadsorbents [10]. Among various silica precursors, tetraethyl orthosilicate (TEOS) seems to be the most popular choice for coating MNPs for different applications, e.g., in biomedical applications like hyperthermia and targeted drug delivery [23, 24].
In the area of FO, MNPs stabilized with TEOS and functionalized with sodium citrate [19] and MNPs covered with TEOS and 3-(trimethoxysilyl) propyl methacrylate [20] were studied. When compared to TEOS, (3-aminopropyl)triethoxysilane (APTES) has the comparative advantage due to the presence of –NH2 groups which can act as bonding sites for further functionalization of silica-covered MNPs with an acidic outer layer, e.g., citric or poly(acrylic acid). Encouraging results in FO methods utilizing iron-based MNPs functionalized with different organic coatings have been reported recently. Ban et al. [25] have reported a good osmotic pressure of ≈ 30 bar for magnetite MNPs, coated with poly(sodium acrylate) (PSA); however, they observed that the amount of PSA attached to the MNPs decreased during recycle steps. Promising FO performance, with osmotic pressures in the range of 18 bar, achieved by citric acid (CA)-coated magnetite MNPs, was reported by Petrinić et al. [26], while lower values of 4.1 bar are reported by Vohl et al. [27] for PAA-coated maghemite (γ-Fe2O3).
Thermal analysis has long been proven to be a powerful characterization technique in the study of various inorganic, organic, and hybrid materials, as well as in applied life sciences [28, 29]. In the particular field of iron-based MNPs, the use of TG analysis for the investigation of thermal degradation of magnetite NPs with polysiloxane coatings, under investigation as drug carriers for targeted drug delivery, was reported [30]. The conversion of magnetite NPs to maghemite was studied by TG/DSC measurements [31], and the thermal stability of PVA-coated MNPs was evaluated using TG [11]. Further recent publications, reporting on successful applications of thermoanalytical methods in characterization of iron-based MNPs, include the assessment of thermal stability of silanol/alkoxysilane-coated maghemite/magnetite [32], investigations of maghemite MNPs used in photolytic degradation of pollutants in wastewater [33], experimental studies of magnetite-based dielectric fluids for subsea cooling applications [34], and investigations of iron/carbon nanotubes by TG/DTG methods [35].
Previous studies have illustrated that iron-based MNPs show a lot of promise as draw solutions for the FO process, but additional research of appropriate surface coatings is needed in order to obtain stable and robust draw solutions. In all the previously mentioned studies, a single-layer coating was used during the experiments. The primary aim of this work was to study the effectiveness of double-layered coated magnetite MNPs, consisting of an inner APTES layer which enhances the stability of MNPs and enables the covalent bonding of an outer CA/PAA layer with a high number of hydrophilic groups to improve solubility and generate an adequate osmotic pressure. To the best of our knowledge, the present study is the first one reporting on the application of double-layer coated iron-based MNPs, stabilized with APTES and functionalized with CA, in the FO process.
One of the main challenges remains the choice of suitable characterization techniques to determine the amount of respective coatings and the stability of bonding. Thermogravimetric analysis (TG) has been shown to be a simple, yet reliable method able to provide a good assessment of the mass fraction of APTES, CA, and PAA bound to the surface of Fe3O4 MNPs. The present paper seems to be the first one to report on a systematic thermogravimetric approach, revealing increasing mass losses in the series bare MNPs < MNP/APTES < MNP/APTES_CA < MNP/APTES_PAA.
Experimental
Materials
All chemicals were obtained from commercial sources and used as purchased, without further purification: Iron(II) chloride tetrahydrate (FeCl2·4H2O), iron(III) chloride hexahydrate (FeCl3·6H2O), and ethanol (99.9%) were obtained from Carlo Erba. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), citric acid, glycerol (≥ 99.5%), N-hydroxysuccinimide (NHS), and poly(acrylic acid) (PAA, M ≈ 1800 g mol−1) were purchased from Sigma-Aldrich; (3-aminopropyl)triethoxysilane (APTES) was bought from Alfa Aesar. Ammonia solution (25%) and methanol (99.8 + %) were obtained from Lab Expert. Hydrochloric acid (≥ 37%) and sodium hydroxide (≥ 98%) were purchased from Honeywell.
Preparation and functionalization of magnetite nanoparticles
The preparation procedure for bare Fe3O4 MNPs followed the method reported by Mashhadizadeh and Amoli-Diva [36] with some minor modifications. Calculated amounts of FeCl2·4H2O (4.3 g, 0.00216 mol) and FeCl3·6H2O (11.68 g, 0.00432 mol) were dissolved in 200 mL of deionized water (DW) by magnetic stirring inside a 250-mL three-neck round bottom flask. The solution was refluxed on an oil bath in nitrogen atmosphere. When the temperature reached 80 °C, 45 mL of 25% NH3(aq) was added, causing the solution to change the color from orange to black. The solution was kept for further 60 min at 80 °C under magnetic stirring. After cooling, the as-prepared magnetic nanoparticles (MNPs) were separated magnetically and washed three times with 100 mL of DW and twice with 100 mL of 0.02 M NaCl solution, using ultrasonic bath (US) for 5 min after each washing.
For the fabrication of magnetite nanoparticles functionalized with APTES (MNP/APTES), the entire batch of freshly prepared MNPs was dispersed in 80 mL of a 10% (v/v) APTES solution and 40 mL of glycerol and heated in an oil bath for 120 min at 90 °C under constant magnetic stirring in nitrogen atmosphere. After cooling, the functionalized MNPs were separated magnetically, washed in turn three times with 200 mL of DW, five times with 70 mL of methanol, and three times with 200 mL of DW. The particles were kept in a suspension inside a 100-mL glass vial. The mass concentration of MNP/APTES in the suspension was determined gravimetrically by drying an aliquot of the suspension to constant mass.
For the preparation of magnetite nanoparticles functionalized with APTES and citric acid (MNP/APTES_CA), 8.9 mL of the MNP/APTES suspension, corresponding to 0.5 g of MNP/APTES nanoparticles, was diluted with DW to a volume of 50 mL. Different amounts of CA were added, and the suspension was magnetically stirred for 10 min. Calculated amounts of EDC, acting as crosslinking agent, were added, and the pH was increased to 3.5 with 1-M NaOH. The solution was stirred for another 60 min, magnetically separated, and the solid product washed in turn five times with 80 mL of DW and once with 80 mL of ethanol. After each washing, the suspension was sonicated for 5 min in an ultrasonic bath. After the last washing, ethanol was carefully removed by decantation, DW was added to the MNPs, the suspension was heated for 60 min at 95 °C to remove the residual ethanol, and finally filled up to a volume of 40 mL with DW. The mass concentration of MNP/APTES_CA in the suspension was determined gravimetrically by drying an aliquot of the suspension to constant mass. Different masses of CA (0.1, 0.2, 0.5, 0.8, and 1.0 g) were used in the experiments. The amounts of EDC needed for crosslinking were calculated based on the assumption that at pH = 3.5, one –COOH group per CA molecule is deprotonated [37, 38], resulting in the molar ratio CA: EDC = 1: 1. The amounts of CA and EDC used in the synthesis are given in Table 1, and the preparation is summarized in Scheme 1.

Synthesis and functionalization of MNP/APTES_CA
Finally, for the preparation of magnetite nanoparticles functionalized with APTES and poly(acrylic acid) (MNP/APTES_PAA), calculated amounts of PAA, EDC, and NHS were dissolved in 5 mL of DW. Then, 5 mL of the MNP_APTES suspension and another 5 mL of DW were added, and the pH was increased to 4.5 with 1-M NaOH. The suspension was stirred for 180 min at room temperature. The nanoparticles were magnetically separated and washed five times with 30 mL of DW and once with 30 mL of ethanol. After the final washing and decantation of ethanol, DW was added to the MNPs, the suspension was sonicated for 10 min in an US bath, and subsequently heated for 60 min at 95 °C inside a drying chamber to remove the residual ethanol. The suspension was filled up to a volume of 25 mL with DW, and the mass concentration of MNP/APTES_PAA in the suspension was determined gravimetrically as described above. Masses of all the precursors were calculated as follows: The amount of nitrogen in MNP/APTES was determined by CHNS analyzer, giving the result of 0.75% N. Since the mass of MNP/APTES in the suspension was determined gravimetrically, it is possible to calculate the amount of –NH2 groups in the sample and the needed amount of PAA. It is known from the literature [39] that at pH = 4.5, the degree of dissociation for PAA equals to 0.5 and it can be calculated that PAA with M ≈ 1800 g mol−1 contains on average 25 –COOH groups, from which 12–13 are deprotonated. We tried to activate on average 3 –COOH groups per PAA molecule for binding with EDC, which combined with using 50% excess amount results in a molar ratio n(EDC) = 4.5 × n(PAA). The amount of NHS was calculated as n(NHS) = 3 × n(PAA). The amounts of PAA, EDC, and NHS used in the synthesis are given in Table 2, and the synthesis is summarized in Scheme 2.

Synthesis and functionalization of MNP/APTES_PAA
Characterization
Thermogravimetric (TG) measurements of the as-prepared and functionalized magnetite nanoparticles were taken with a METTLER TGA/SDTA 851e system using the following parameters: temperature range of 25–900 °C, heating range 10 °C min−1, nitrogen flow 100 mL min−1, 70-μL Al2O3 crucibles, sample masses of approximately 15 mg. Particle size and zeta potential were measured using a Zetasizer Nano ZS, Malvern, UK, operated with a 4-mW He–Ne laser at 633 nm. Analyses were performed on dilute suspensions, so multiple scattering was considered negligible. The isoelectric points were determined by titration and measurement of the respective zeta potentials of the samples, using an MPT-2 titrator. ATR-IR spectra were recorded at a scan range of 4000–650 cm−1 (IRAffinity-1S FTIR, Shimadzu). The morphological properties of selected samples were analyzed by TEM imaging, area electron diffraction (SAED), and energy-dispersive X-ray analysis (EDX) with a transmission electron microscope JEOL 2100J, Japan. The particle size and size distribution were determined using the particle imaging software DigitalMicrograph 2.11 (Gantan, USA). Elemental analyses were carried out on a PerkinElmer CHNS/O 2400 Series II elemental analyzer. X-ray diffraction patterns were collected using an AXS-Bruker/Siemens D5005 X-ray powder diffractometer in the range of 10° < 2θ < 70°, utilizing graphite monochromated CuKα radiation (λ = 1.5418 Å) with a step size of 0.033° and step time of 1 s. The average nanoparticle size was estimated using the Scherrer equation.
The osmolality of the suspensions prepared from functionalized nanoparticles was determined using a freezing point osmometer, Gonotec Osmomat 3000, Berlin, Germany. The device operates using the principle of freezing point depression, measuring the difference between the freezing point of the measured suspension and the freezing point of DW. The osmolality is then calculated using the equation:
where bosm is the osmolality of the suspension (osmol/kg), ΔTf is the difference between the freezing points of the suspension and pure DW (°C), and Kk is the cryoscopic constant of water (= 1.858 °C kg/osmol).
From the osmolality, osmotic pressure can be calculated using Van’t Hoffs law:
Here, π corresponds to the osmotic pressure (bar), c is the molar concentration, R is the gas constant (expressed as 0.0831 bar L mol−1 K−1), and T is the temperature (K). Considering T = 298.15 K and bosm ≈ cosm (due to the density of the aqueous medium which being ≈ 1 kg L−1), π can easily be calculated as π = 24.7 × bosm.
Results and discussion
Thermogravimetric analysis
The curves obtained by thermogravimetric measurements (TG) of as-prepared (bare) MNPs and two independent batches of APTES-coated MNPs are presented in Fig. 1. Magnetite in bulk form is known to be stable under inert atmosphere up to temperatures exceeding 1000 °C; hence, all the observed mass losses can be attributed either to the loss of water or to the decomposition of APTES. The small mass losses in the range of ≈ 3%, observed for uncoated MNPs, were ascribed to desorption of adsorbed (up to 140 °C) and chemically bound (140–570 °C) water. For both MNP/APTES samples, the mass losses in the same temperature range are significantly higher, ranging between 7.6 and 7.7%. By a simple subtraction of both values, the approximate mass fraction of APTES on the coated MNPs can be estimated, as shown in Table 3. Samples coated with CA were prepared from the batch of APTES-coated MNPs labeled as MNP/APTES(1), while those coated with PAA were prepared from the batch MNP/APTES(2).

TG curves and the results of corresponding mass loss calculations for as-prepared magnetite MNPs and coated MNP/APTES
TG and DTG curves of APTES-coated MNPs and a typical MNP functionalized with CA (MNP/APTES_CA0.8) are shown in Fig. 2, while the curves for MNPs functionalized with PAA (MNP/APTES_PAA1:1.25) are shown in Fig. 3. In both cases, curves for samples with the highest mass loss are shown, and curves for MNPs covered with APTES only are provided for comparison. The other corresponding curves show very similar behavior and were omitted for clarity. The thermal decomposition of APTES-coated MNPs shows three characteristic regions: The first one with a DTG peak below 100 °C can be ascribed to the loss of adsorbed water, while the second (DTG peak between 280 and 300 °C) and the third (DTG peak between 620 and 650 °C) can be attributed to decomposition of the corresponding coating molecules. In the case of MNP/APTES_CA samples, the second and the third peaks are shifted to lower temperatures (240 °C and 605 °C, respectively) when compared to MNPs coated with APTES only. The opposite effect can be observed for MNP/APTES_PAA samples, where the corresponding peaks shift to 305 °C and 695 °C. Mass losses for all MNPs functionalized with CA or PAA are larger than those measured for MNPs coated with APTES alone, indicating successful bonding of both acids to APTES-coated MNPs. The mass fraction of acids bound to the MNPs was calculated by subtracting the respective mass losses, resulting in values of 0.7–1.4% for CA and 8.5–9.9 for PAA, as summarized in Table 3. With the exception of the sample MNP/APTES_CA0.1, which is showing significantly lower results, no obvious relation between the mass of added CA and the measured mass losses could be observed, indicating that only a fraction of CA, used in the synthesis, successfully binds to the surface of MNP/APTES. The values obtained for PAA-functionalized MNPs are much higher than those measured for MNPs functionalized with CA, which can be explained with higher molar mass of PAA.

TG/DTG curves and the results of corresponding mass loss calculations for MNP/APTES and functionalized MNP/APTES_CA08

TG/DTG curves and the results of corresponding mass loss calculations for MNP/APTES and functionalized MNP/APTES_PAA1:1.25
In order to further elucidate the thermal behavior of uncoated as well as functionalized MNPs, the obtained results were compared to those reported in the literature. Iron oxide is known to exist in several polymorphic forms, among which only magnetite (Fe3O4) and maghemite (γ-Fe2O3) are ferromagnetic materials suitable for FO applications. However, the saturation magnetization for magnetite is higher than those of maghemite [40]. At the bulk level, the oxidation of magnetite to hematite takes place at temperatures close to 600 °C, while at nanoscale level, this change has been reported to take place at much lower temperatures, even close to room temperature [41]. Ong et al. have studied thermal stability of uncoated magnetite MNPs prepared by coprecipitation using TG in air, reporting mass losses of ≈ 8% up to 900 °C. Combining results of magnetic measurements and XRD, they assumed that at moderate temperatures (275 °C), the mass loss of ≈ 5% is due to desorption of impurities, causing an increase in the saturation magnetization. At 600 °C, minor peaks of hematite started to appear on the XRD patterns, and at 675 °C, magnetite completely transformed to hematite. When compared to those results, bare MNPs prepared in our study exhibit a lower mass loss of ≈ 3.5% at 275 °C and 4.5% at 900 °C. We can conclude that MNPs prepared by our method contain less impurities, possibly due to a different coprecipitation agent, which was NH3 in our study and NaOH in the research reported in [35]. It is generally known that the thermal stability of iron-based MNPs strongly depends on the preparation method and particle size. Kalska-Szostko et al. studied the thermal behavior of magnetite MNPs prepared by two different methods, e.g., coprecipitation and thermal decomposition of Fe(acac)3, reporting a much higher stability in the temperature range up to 500 °C for MNPs prepared by coprecipitation when compared to those obtained by thermal decomposition, which could be explained by a difference in morphology [41].
Another analogous study involving TG–DTA stability analysis of single-layered magnetite-CA MNPs was published by de Sousa et al. [38]. Therein, a two-step mass loss with onsets at 175 and 222 °C is reported and attributed to desorption of CA molecules from the MNP surface. Mass losses in the same temperature range were also observed during our investigation. However, a direct comparison is difficult due to the two-layered design of our MNPs, where peaks related to APTES and CA are likely to overlap. When compared to papers already published by our group for MNPs covered with a single layered of either CA or PAA [26, 27], similar behavior was observed: loss of physically adsorbed water below 160–200 °C, followed by decomposition of bound CA/PAA molecules above 200 °C. A direct comparison of mass losses, especially at higher temperatures, is impractical due to the fact that aforementioned measurements were taken in air [26], causing an additional mass loss due to oxidation of magnetite to hematite.
Particle size, zeta potential, and isoelectric point measurements
The measured values of particle diameter (dH), zeta potential (ζ), and isoelectric point (pHIEP) of as-prepared, APTES, APTES_CA, and APTES_PAA-coated MNPs are summarized in Table 4. The particle diameter is expressed as mean hydrodynamic size (dH), defined by IUPAC as the diameter of a perfect solid sphere that would exhibit the same hydrodynamic friction as the particle of interest. The zeta potential of as-prepared MNPs is slightly negative due to –OH groups on the surface, while the values for APTES-coated MNPs are positive due to basic –NH2 groups of APTES. The zeta potential of APTES-coated MNP suspensions is close to the border of ± 30 mV, with particles having zeta potentials outside of these limits normally considered stable [42, 43], suggesting that the suspensions can be regarded as fairly stable. On the contrary, the suspension of uncoated MNPs is unstable, which can clearly be seen from the much higher dH due to agglomeration. The isoelectric point (pH value at which zeta potential reaches 0 mV) is close to 8 for as-prepared MNPs, while for those coated with APTES, the values are appreciably higher (11.5–11.8) due to basic –NH2 groups of APTES, confirming the successful bonding of APTES to the surface of magnetite nanoparticles.
The measured zeta potential values of functionalized MNP/APTES_CA are in the negative region due to acidic –COOH groups of CA, contributing to negative surface potential. For the sample functionalized with the lowest amount of CA (MNP/APTES_CA0.1), zeta potential is much closer to 0 mV than to the lower stability border of − 30 mV, indicating that the suspension is unstable and prone to agglomeration. All the other suspensions can be regarded as stable. The same can be concluded from dH measurements. The isoelectric point titrations indicate the presence of both basic –NH2 and acidic –COOH groups at the surface of MNPs, resulting in values lower than those obtained for MNPs coated with APTES only.
All zeta potential values of functionalized MNP/APTES_CA samples are negative due to acidic –COOH groups of PAA and below − 30 mV, indicating stable suspensions. The mean hydrodynamic sizes (dH) of all PAA-coated samples are comparable, as well as pH values at the isoelectric point, which are all clearly in the acidic area due to –COOH from PAA groups being in excess of –NH2 from APTES.
Figures 4 and 5 illustrate the fundamentally different behaviors of MNPs coated with APTES only and those additionally functionalized with PAA (molar ratio 1:1) during pH titration. Zeta potential (marked with a green line) strongly depends on pH and exhibits positive values above the isoelectric point and negative below it. As mentioned above, the isoelectric point of MNP/APTES samples (Fig. 4) is decisively in the basic pH range due to basic –NH2 groups of APTES bound to the surface of the MNPs. The average particle size (red line) increases when zeta potential decreases toward 0, due to weakening repulsive forces and consequently increasing agglomeration. The graphs for PAA-functionalized MNP/APTES nanoparticles (Fig. 5) show the isoelectric point clearly in the acidic range, suggesting that acidic –COOH groups from PAA prevail over basic –NH2 groups from APTES. The average particle size reaches its maximum at the isoelectric point, where the particles have no charge, and drops again at higher pH when the zeta potential approaches 30 mV.

pH titration graph for MNP/APTES(2)

pH titration graph for MNP/APTES(2)_PAA1:1
Fourier-transform infrared spectroscopy (FTIR) measurements
The FTIR spectra of the APTES-covered MNPs and those functionalized by CA/PAA (Fig. 6 and Fig. 7) show many common features due to partial overlapping of individual peaks. The spectra recorded for uncoated MNPs are characterized by Fe–O stretching bands at 570–615 cm−1, typical for nanosized Fe3O4 [44, 45]. It is possible to distinguish between spectra of uncoated and APTES-covered MNPs, due to the presence of a weak N–H stretching band at 1600 cm−1 indicating the presence of –NH2 groups on the MNP surface [46]. For CA- and PAA-functionalized MNPs, additional bands between 1600 and 1400 cm−1 can be observed, which can be attributed to C=O stretching and –COO− vibration of the carboxylic groups, the later indicating the bonding of carbonyl groups to the surface of MNPs [26, 27].

FTIR spectra of MNP/APTES and functionalized MNP/APTES_CA nanoparticles

FTIR spectra of MNP/APTES and functionalized MNP/APTES_PAA nanoparticles
TEM analysis
The results of TEM and SAED measurements for bare MNPs as well as for a typical sample of CA-coated MNPs are shown in Figs. 8–10. From Fig. 8a–c, it can clearly be seen that the sample consists of almost perfectly spherical MNPs with diameters in the range of 12 ± 2 nm, which are slightly agglomerated in porous clusters. No significant influence of the coating on the morphology of the nanoparticles could be observed, apart from the formation of a thin amorphous layer on the surface of the coated MNPs. The EDX analysis confirmed the presence of Fe, O, and Si, while nitrogen seems to be present only in a thin layer on the surface, resulting in a concentration which is too low for EDX detection. Nevertheless, the presence of the APTES layer is confirmed by the detection of Si in the samples. The aforementioned amorphous layer, attributed to the presence of APTES on the surface of magnetite particles, can be seen in Fig. 9. The SAED analysis confirms that the MNPs consist mainly of cubic Fe3O4 (Fig. 10).

TEM images of bare MNPs (a) and MNP/APTES_CA0.8 at two different magnifications (b, c)

High-resolution TEM image showing a thin amorphous APTES layer on the surface of MNPs

SAED image of MNP/APTES_CA0.8 (left-hand side) compared with SAED of pure cubic Fe3O4 taken as reference (right-hand side)
XRD analysis
Powder XRD diffraction patterns of bare as well as those of APTES-coated and APTES_CA-functionalized magnetite MNPs are shown in Fig. 11. Distinguishing between magnetite and maghemite by powder XRD can be a challenge, especially in the case of nanosized samples [47]. Nevertheless, after a careful examination of positions and relative intensities of diffraction patterns, measured for our samples, as well as by comparison with data from the literature [44, 45, 47], it can be stated that all diffraction peaks match with reflections of pure cubic magnetite, Fe3O4, with an inverse spinel structure: ICDD No. 00-001-1111. No detectable peaks of other crystalline phases like maghemite or hematite were observed. It should be mentioned that there are no significant differences between patterns of bare MNPs, APTES-covered MNPs (MNP/APTES), as well as those for functionalized MNPs (MNP/APTES_CA). While the presence of organic coatings on the surface of magnetite generally increases the amorphous behavior of nanoparticles, no such effect could be observed in our study, which can be attributed to thin layers of the organic coatings. The average nanoparticle size estimated from Scherrer equation was determined to be in the range between 11 and 14 nm for all the investigated samples, which is a very good agreement with results obtained from TEM images.

X-ray diffraction spectrum of bare magnetite MNPs prepared by coprecipitation (MNP), MNPs coated with APTES (MNP/APTES), and MNPs coated with APTES and functionalized with CA (MNP/APTES_CA). The red lines correspond to the pattern of magnetite, No. 00-001-1111 from the ICDD database
Osmotic pressure measurements
All the prepared samples, functionalized with either CA or PAA, exhibit considerable osmotic pressure. The measured osmolality and osmotic pressure values are summarized in Table 5. The respective results for both samples covered with APTES only are negligible, pointing out the influence of CA/PAA functionalization of MNPs for FO applications. The osmotic pressure for CA-functionalized MNPs shows a general trend of increasing with the rising addition of CA, with a minor decrease for the MNP/APTES_CA0.5 sample. On the other hand, no clear trend can be observed in suspensions of PAA-functionalized MNPs, with a sharp drop measured for the MNP/APTES_PAA1:1.25 sample with the highest amount of added PAA.
For a viable use of the samples as draw solutions in a forward osmosis system, retaining a sufficient osmotic pressure over a period of several weeks is crucial. Consequently, the osmotic pressure measurements were repeated weekly. As shown in Figs. 12 and 13, only small fluctuations occur between the measurements without significant changes over a period of five–six weeks, thus confirming the stability of the suspensions.

Osmotic pressures during six weeks for MNP/APTES_CA suspensions with different amounts of CA

Osmotic pressures during five weeks for MNP/APTES_PAA suspensions with different amounts of PAA
The suitability of a draw solute is defined by the ability to develop sufficient osmotic pressure to ensure a high water flux while maintaining low (ideally no) diffusion from the DS to the FS. Among recently synthesized MNPs, water-soluble particles (MNP@APTES@PAA) exhibit osmotic pressure of 8.9 bar [46], Fe3O4@CA MNPs (3.7 mass% solutions) had an initial osmotic pressure of 18 bar [26], and synthesized MNP, coated with PAA, yielded an osmotic pressure of ~ 12.8 bar at a 0.7% concentration [27]. From the samples reported in Table 5, the sample MNP/APTES_PAA1:1 exhibited the highest osmotic pressure among the PAA-functionalized MNPs and therefore was selected for further FO experiments. Due to results already reported by Petrinić et al. [26] achieved by CA-coated magnetite MNPs, we did not perform the FO filtrations using MNP/APTES_CA synthesized in this study.
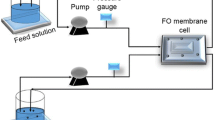
The FO filtration was performed using freshly synthesized MNP/APTES_PAA as a DS. We used 250 mL of DI water as FS and 95 mL of 0.6% freshly prepared suspension of MNP/APTES_PAA as DS. The osmotic pressure of MNP/APTES_PAA suspension was 8.9 bar. During the filtration, the water flux decreased from a maximum value of 4.1–1.8 LMH in 2 h (Fig. 14). During that time, 64.1 mL of water passed through the membrane (water recovery was 25.7%), which corresponds to an increase in the dilution factor of FS from 1 to 1.7. The osmotic pressure of the FS after 2 h increased to 2.1 bar while an increment in conductivity from 10 to 13 μS cm−1 is noticed. The osmotic pressure difference between DS and FS at the end of filtration was 0.4 bar (Fig. 15). DW was used as the FS and the diluted MNP/APTES_PAA1:1 suspension was used as the DS. The water flux (the rate at which water permeates a FO membrane) was measured during 2 h. As expected, the flux at first increases due to the significant difference in the osmotic pressures between the FS and DS, and later, it slowly decreases due to the dilution of the draw solution with DW permeating through the membrane (Fig. 14).

Water flux and conductivity increase in FS during 2 h of operation

Osmotic pressures of the FS and DS
Conclusions
Spherical magnetite nanoparticles with diameters in the range of 12 nm have been synthesized by coprecipitation and functionalized with a double APTES_CA or APTES_PAA layer for use as draw solution agents for forward osmosis (FO). Encouraging performances have been achieved in FO experiments, resulting in osmotic pressures up to 28 bar, significant water fluxes, and a high stability of as-prepared draw solutions, retaining a sufficient osmotic pressure over a period of several weeks. While further investigations will be required to provide a definitive answer, it can be concluded that strong peptide bonds between –NH2 groups of APTES and –COOH groups of the particular acid are formed, with EDC acting as a ‘zero-length’ crosslinker.
While the coated MNPs were characterized by several methods, i.e., elemental analysis, FTIR, DLS and zeta potential measurements, powder XRD, TEM including EDX and SAED analysis, it is challenging to determine the amount of the bound APTES, CA, and PAA on the surface of MNPs using only those methods, due to the thinness and amorphous nature of the coatings. Thermogravimetric analysis (TG) has been shown to be a powerful characterization technique able to provide answers to these kinds of concerns. Magnetite is known to be stable up to temperatures exceeding 600 °C, while the thermal degradation of organic molecules used for coating (APTES, CA, and PAA) takes place at significantly lower temperatures. The thermal decomposition of as-prepared MNPs shows three characteristic regions: the first one indicating to the loss of adsorbed water, while the second and the third are ascribed to decomposition of the corresponding coating molecules. The results of TG measurements clearly indicate the difference between mass losses for bare MNPs (2.7% in the temperature range from 30 to 900 °C), MNPs coated only with APTES (7.6–7.7%), MNP/APTES MNPs functionalized with CA (8.4–9.1%), and MNP/APTES MNPs functionalized with PAA (16.1–17.5%). The FO measurements show a strong correlation between the amount of CA/PAA coatings used during the synthesis and the osmotic properties of the MNPs, thus pointing out the necessity of applying a simple and fast, yet effective method for estimating the amount of organic compounds bound to the surface of iron-based magnetic nanoparticles. The results indicate that thermogravimetric analysis can be considered as a reliable method for this task, showing a great potential for future investigations in this increasingly important field of research.
References
Ray SS, Chen S-S, Sangeetha D, Chang H-M, Thanh CND, Le QH, Ku H-M. Developments in forward osmosis and membrane distillation for desalination of waters. Environ Chem Lett. 2018;16:1247–65. https://doi.org/10.1007/s10311-018-0750-7.
Chung T-S, Zhang S, Wang KY, Su J, Ling MM. Forward osmosis processes: yesterday, today and tomorrow. Desalination. 2012;287:78–81. https://doi.org/10.1016/j.desal.2010.12.019.
Haupt A, Lerch A. Forward osmosis application in manufacturing industries: a short review. Membranes. 2018;8:47. https://doi.org/10.3390/membranes8030047.
Kahrizi M, Gonzales RR, Kong L, Matsuyama H, Lu P, Lin J, Zhao S. Significant roles of substrate properties in forward osmosis membrane performance: a review. Desalination. 2022;528: 115615. https://doi.org/10.1016/j.desal.2022.115615.
Li L, Shi W, Yu S. Research on forward osmosis membrane technology still needs improvement in water recovery and wastewater treatment. Water. 2020;12:107. https://doi.org/10.3390/w12010107.
Abounahia N, Ibrar I, Kazwini T, Altaee A, Samal AK, Zaidi SJ, Hawari AH. Desalination by the forward osmosis: advancement and challenges. Sci Total Environ. 2023;886: 163901. https://doi.org/10.1016/j.scitotenv.2023.163901.
Farahbakhsh J, Golgoli M, Khiadani M, Najafi M, Suwaileh W, Razmjou A, Zargar M. Recent advances in surface tailoring of thin film forward osmosis membranes: a review. Chemosphere. 2024;346: 140493. https://doi.org/10.1016/j.chemosphere.2023.140493.
Johnson DJ, Suwaileh WA, Mohammed AW, Hilal N. Osmotic’s potential: an overview of draw solutes for forward osmosis. Desalination. 2018;434:100–20. https://doi.org/10.1016/j.desal.2017.09.017.
Plohl O, Finšgar M, Gyergyek S, Ajdnik U, Ban I, Fras ZL. Efficient copper removal from an aqueous anvironment using a novel and hybrid nanoadsorbent based on derived-polyethyleneimine linked to silica magnetic nanocomposites. Nanomaterials. 2019;9:209. https://doi.org/10.3390/nano9020209.
Tan Y, Chen M, Hao Y. High efficient removal of Pb(II) by amino-functionalized Fe3O4 magnetic nano-particles. Chem Eng J. 2012;191:104–11. https://doi.org/10.1016/j.cej.2012.02.075.
Darvish MSA, Al-Harbi LM, Bakry A. Synthesis of magnetite nanoparticles coated with polyvinyl alcohol for hyperthermia application. J Therm Anal Calorim. 2022;147:11921–30. https://doi.org/10.1007/s10973-022-11393-6.
Ling MM, Chung T-S. Surface-dissociated nanoparticle draw solutions in forward osmosis and the regeneration in an integrated electric field and nanofiltration system. Ind Eng Chem Res. 2012;51:15463–71. https://doi.org/10.1021/ie302331h.
Ling MM, Wang KY, Chung T-S. Highly water-soluble magnetic nanoparticles as novel draw solutes in forward osmosis for water reuse. Ind Eng Chem Res. 2010;49:5869–76. https://doi.org/10.1021/ie100438x.
Dey P, Izake EL. Magnetic nanoparticles boosting the osmotic efficiency of a polymeric FO draw agent: effect of polymer conformation. Desalination. 2015;373:79–85. https://doi.org/10.1016/j.desal.2015.07.010.
Park SY, Ahn HW, Chung JW, Kwak SY. Magnetic core-hydrophilic shell nanosphere as stability-enhanced draw solute for forward osmosis (FO) application. Desalination. 2016;397:22–9. https://doi.org/10.1016/j.desal.2016.06.017.
Kadhim RM, Al-Abodi EE, Al-Alawy AF. Citrate-coated magnetite nanoparticles as osmotic agent in a forward osmosis process. Desalin Water Treat. 2018;115:45–52. https://doi.org/10.5004/dwt.2018.22456.
Na Y, Yang S, Lee S. Evaluation of citrate-coated magnetic nanoparticles as draw solute for forward osmosis. Desalination. 2014;347:34–42. https://doi.org/10.1016/j.desal.2014.04.032.
Zufía-Rivas J, Morales P, Veintemillas-Verdaguer S. Effect of the sodium polyacrylate on the magnetite nanoparticles produced by green chemistry routes: Applicability in forward osmosis. Nanomaterials. 2018;8:470. https://doi.org/10.3390/nano8070470.
Khazaie F, Sheshmani S, Shokrollahzadeh S, Shahvelayati AS. Desalination of saline water via forward osmosis using magnetic nanoparticles covalently functionalized with citrate ions as osmotic agent. Environ Technol. 2022;43:2113–23. https://doi.org/10.1080/09593330.2020.1866087.
Shakeri A, Salehi H, Khankeshipour N, Nakhjiri MT, Ghorbani F. Magnetic nanoparticle-crosslinked ferrohydrogel as a novel class of forward osmosis draw agent. J Nanopart Res. 2018;20:325. https://doi.org/10.1007/s11051-018-4437-6.
Hafiz MA, Hassanein A, Talhami M, Al-Ejji M, Hassan MK, Hawari AH. Magnetic nanoparticles draw solution for forward osmosis: current status and future challenges in wastewater treatment. J Environ Chem Eng. 2022;10: 108955. https://doi.org/10.1016/j.jece.2022.108955.
Roach JD, Bondaruk MM, Burney Z. Nonideal solution behavior in forward osmosis processes using magnetic nanoparticles. In: Du H, Thompsom A, Wang X, editors. Osmotically Driven Membrane Processes. Intech Open, 2018. https://doi.org/10.5772/intechopen.72474
Kristl M, Ban I, Gyergyek S, Maver U, Stergar J. Sol-gel preparation of NixCu1-x/silica nanocomposites using different silica precurors. J Sol-Gel Sci Technol. 2022;101:579–87. https://doi.org/10.1007/s10971-020-05321-z.
Stergar J, Maver U, Bele M, Gradišnik L, Kristl M, Ban I. NiCu-silica nanoparticles as a potential drug delivery system. J Sol Gel Sci Technol. 2022;101:493–504. https://doi.org/10.1007/s10971-020-05280-5.
Ban I, Markuš S, Gyergyek S, Drofenik M, Korenak J, Helix-Nielsen C, Petrinić I. Synthesis of poly-sodium_acrylate (PSA)-coated magnetic nanoparticles for use in forward osmosis draw solutions. Nanomaterials. 2019;9:1238. https://doi.org/10.3390/nano9091238.
Petrinić I, Stergar J, Bukšek H, Drofenik M, Gyergyek S, Hélix-Nielsen C, Ban I. Superparamagnetic Fe3O4@CA nanoparticles and their potential as draw solution agents in forward osmosis. Nanomaterials. 2021;11:2965. https://doi.org/10.3390/nano11112965.
Vohl S, Ban I, Drofenik M, Bukšek H, Gyergyek S, Petrinić I, Hélix-Nielsen C, Stergar J. Microwave synthesis of poly(acrylic) acid-coated magnetic nanoparticles as draw solutes in forward osmosis. Materials. 2023;16:4138. https://doi.org/10.3390/ma16114138.
Kristl M, Muršec M, Šuštar V, Kristl J. Application of thermogravimetric analysis for the evaluation of organic and inorganic carbon contents in agricultural soils. J Therm Anal Calorim. 2016;123:2139–47. https://doi.org/10.1007/s10973-015-4844-1.
Kristl M, Jurak S, Brus M, Sem V, Kristl J. Evaluation of calcium carbonate in eggshells using thermal analysis. J Therm Anal Calorim. 2019;138:2751–8. https://doi.org/10.1007/s10973-019-08678-8.
Varganici C-D, Durdureanu-Angheluta A, Rosu D, Pinteala M, Simionescu BC. Thermal degradation of magnetite nanoparticles with hydrophilic shell. J Anal Appl Pyrol. 2012;96:63–8. https://doi.org/10.1016/j.jaap.2012.03.005.
Stoia M, Istratie R, Pacurariu C. Investigation of magnetite nanoparticles stability in air by thermal analysis and FTIR spectroscopy. J Therm Anal Calorim. 2016;125:1185–98. https://doi.org/10.1007/s10973-016-5393-y.
Vasquez ES, Prehn EM, Walters KB. Assessing magnetic iron nanoparticle properties under different thermal treatments. J Therm Anal Calorim. 2021;143:35–46. https://doi.org/10.1007/s10937-019-09195-4.
Sheikholeslami Z, Kebria DY, Qaderi F. Investigation of photocatalytic degradation of BTEX in produced water using γ-Fe2O3 nanoparticle. J Therm Anal Calorim. 2019;135:1617–27. https://doi.org/10.1007/s10973-018-7381-x.
Dalla Vecchia LC, Rambo CR, Barbosa JR Jr, da Silva AK. An experimental study of a nanoparticle-assisted dielectric fluid in natural convection for subsea cooling applications. J Therm Anal Calorim. 2022;147:8803–14. https://doi.org/10.1007/s10973-021-11143-0.
Alzate M, Gamba O, Daza C, Santamaria A, Gallego J. Iron/multiwalled carbon nanotube (Fe/MWCNT) hybrid materials characterization: thermogravimetric analysis as a powerful characterization technique. J Therm Anal Calorim. 2022;147:12355–63. https://doi.org/10.1007/s10973-022-11446-w.
Mashhadzadeh MH, Amoli-Diva M. Drug-carrying amino silane coated magnetic nanoparticles as potential vehicles for delivery of antibiotics. J Nanomed Nanotechnol. 2012;3:1000139. https://doi.org/10.4172/2157-7439.1000139.
Madruga S, Nedyalkova M, Mas F, Garcés JL. Ionization and conformational equilibria of citric acid: delocalized proton binding in solution. J Phys Chem A. 2017;121:5894–906. https://doi.org/10.1021/acs.jpca.7b05089.
De Sousa ME, Fernández van Raap M, Rivas PC, Zélis PM, Girardin P, Pasquevich GA, Alessandrini JL, Muraca D, Sánchez FH. Stability and relaxation mechanisms of citric acid coated magnetite nanoparticles for magnetic hyperthermia. J Phys Chem C. 2013;117:5436–45. https://doi.org/10.1021/jp311556b.
Wiśniewska M, Urban T, Grządka E, Zarko VI, Gun’ko VM. Comparison of adsorption affinity of polyacrylic acid for surfaces of mixed silica-alumina. Colloid Polym Sci. 2014;292:699–705. https://doi.org/10.1007/s00396-013-3103-x.
Ong BH, Devaraj NK, Matsumoto M, Abdullah MH. Thermal stability of magnetite (Fe3O4) nanoparticles. MRS Online Proc Libr. 2008;1118:309. https://doi.org/10.1557/PROC-1118-K03-09.
Kalska-Szostko B, Wykowska U, Satula D, Nordblad P. Thermal treatment of magnetite nanoparticles. Beilstein J Nanomed Nanotechnol. 2015;6:1385–96.
Kepekçi RA, Ilçe BY, Kanmazalp SD. Plant-derived biomaterials for wound healing. Stud Nat Prod Chem. 2021;70:227–64. https://doi.org/10.1016/B978-0-12-819489-8.00001-6.
Pate K, Safier P. In: Babu SV, editor. Advances in chemical mechanical planarization (CMP). Sawston: Woodhead Publishing, 2016. Chapter 86. https://doi.org/10.1016/C2014-0-01445-1
Shen L, Qiao Y, Guo Y, Meng S, Yang G, Wu M, Zhao J. Facile co-precipitation synthesis of shape-controlled magnetite nanoparticles. Ceram Int. 2014;40:1519–24. https://doi.org/10.1016/j.ceramint.2013.07.037.
Ercuta A, Chirita M. Highly crystalline porous magnetite and vacancy-ordered maghemite microcrystals of rhombohedral habit. J Cryst Growth. 2013;380:182–6. https://doi.org/10.1016/j.jcrysgro.2013.06.003.
Ban I, Drofenik M, Bukšek H, Petrinić I, Helix-Nielsen C, Vohl S, Gyergyek S, Stergar J. Synthesis of magnetic nanoparticles with covalently boned polyacrylic acid for use as forward osmosis draw agents. Environ Sci Water Res Technol. 2023;9:442–53. https://doi.org/10.1039/D2EW00539E.
Kim W, Suh C-Y, Cho S-W, Roh K-M, Kwon H, Song K, Shon I-J. A new method for the identification and quantification of magnetite-maghemite mixture using conventional X-ray diffraction technique. Talanta. 2012;94:348–52. https://doi.org/10.1016/j.talanta.2012.03.001.
Acknowledgements
The authors acknowledge the financial support from the grant number P2-0006 of the Slovenian Research Agency (ARRS). The authors would like to thank Sabina Vohl and Lidija Škodič for help with the laboratory work and prof. dr. Sašo Gyergyek from Jožef Stefan Institute, Ljubljana, for TEM analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kristl, M., Ostroško, U., Ban, I. et al. Thermal study of APTES-functionalized magnetite nanoparticles with citric acid and polyacrylic acid for advanced forward osmosis systems. J Therm Anal Calorim (2024). https://doi.org/10.1007/s10973-024-12983-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10973-024-12983-2