
Abstract
Ag–bentonite was prepared by ion exchange process to sorb iodide and chloride ions in batch experiments. The modified bentonite was examined with XRF and XRD. 75% of the cation exchange capacity was exchanged by silver ions. It was found that the sorption of chloride ions is an exothermic precipitation process because the solubility decreases with increasing temperature. In the case of iodide sorption, the dissolution of AgI was observed under high concentration of non-radioactive iodide ions, which is well known in analytical chemistry. The phenomenon occurs not only in the bulk aqueous phase but also in the interlayer space of montmorillonite.
Similar content being viewed by others


Introduction
The water-soluble species can be sorbed on solid surfaces by adsorption, precipitation or ion-exchange [1], or by the combination of these processes. Under geological conditions, because of their negative layer charge, and negative pH-dependent (often called edge) charge, the sorption of cations on clay minerals is the characteristic process. However, there are some long-lived anionic radioactive species, such as 36Cl, 129I, 79Se, and 14C and 99Tc, which are highly soluble in water and do not sorb on the materials of engineering barriers or natural clays. Therefore, they can cause some potential risk in the environment of nuclear waste disposals.
In the environment, 36Cl can be found naturally as a result of cosmic-ray spallation of 40Ar in the atmosphere. Moreover, it occurs by interactions between K, Ca and Cl and cosmic radiation in near-surface rocks and soils, and it can be found in the environment due to the activation of stable chlorine-35 by naturally generated thermal neutrons in the subsurface [2,3,4]. In the stratosphere, approximately two-thirds of natural 36C1 production can be found. Spontaneous fission of 238U will also induce alpha particle irradiation of elements such as O, Na, Mg, Al, Si and neutron release, which can lead to 36C1 production [5]. In surroundings of natural U deposits, 36C1 in groundwater is produced due to cosmogenic radiation [6]. The thermal neutrons of the nuclear reactors also produce 36Cl from 35Cl in the structural material introduced with the chloride content in the water used for concreting [7]. 36C1 may have a significant longterm effect on human health, because of its long half-life and potential mobility in geosphere. 36Cl is a weak β-emitter, with half-life 3.01 × 105 years. Moreover, mobility of this radionuclide has not yet been extensively studied.
Radioactive iodine arises as a fission product in the nuclear fission of 235U, by nuclear fuel reprocessing or nuclear accidents (Fukushima, Chernobyl), and can be generated during medical applications [8]. Radioactive iodine isotopes have different half-lives, the shortest (131I) is 8.04 days and the longest (129I) half-life of a radioactive iodine isotope is 1.57 × 107 years. Some iodine species are mobile in natural water entering aquatic environments (oceans, groundwater and rivers) without treatment [9]. The accumulation of radioactive iodine can cause thyroid cancer, leukemia and metabolic disorders in the human body [10]. Usually, radioactive iodine is present as inorganic iodine (I−, IO3−) and organic iodine in aqueous media. At positive redox potentials and in acidic and neutral pH values, I− is the dominant species.
In the literature, different methods have been investigated for the removal of radioactive I− from aqueous media. These are, for example, membrane separation [11], adsorption [12,13,14,15], precipitation [16], or ion exchange [17, 18].
Several organic and inorganic, natural and artifcial sorbents were found to remove radioactive I− from the environment, for example: activated carbon [19]; metal oxides [20]; natural organic materials [21]; composite absorbents [22]; zeolite [23]; inorganic anion exchanger [24]. Lettinga et al. [25] reported that activated carbon removes 131I with low efficiency. Inorganic ion exchangers are difficult to produce and are not suitable for large scale applications. In addition, there are inorganic anion exchangers that are toxic. Several sorbents use compounds containing Bi3+, Hg2+, Cu +, and Pb2+ cations that precipitate with iodide ions [26,27,28]. According to Lefevre et al. [26], Cu+ ion is the most promising cation for I− removal due to its relatively low toxicity and lower cost. Liu et al. [16] used CuCl to remove I− in the form of a precipitate from aqueous solutions. The disadvantage is that dissolved oxygen easily oxidizes copper. Recently, silver and its compounds have received more attention. Silver chloride [22], argentite (AgS) [29], and Ag-modified activated carbon, zeolites, and porous silica are effective for iodine removal, because iodide forms insoluble AgI with silver ion. However, due to their small specific surface area, these compounds had low sorption capacity and the process proceeded slowly [27, 30]. To solve this problem, natural clay minerals can be modified with silver ions. The use of different species of silver as bactericidal agent such as Ag+, Ag0, or Ag nanoparticles [31, 32], Ag2O [33], AgBr [34], and also Ag+ exchanged montmorillonite (MMT-Ag) [35] has been largely reported.
By the chemical modification of clays, sorbents precipitating anionic species, including radioactive ones can be prepared. The sorption of phosphate ion by rare earth cation exchanged bentonites used efficiently against the eutrophication of lakes is mentioned as an example [36]. In our previous three articles, we have successfully used rare earth cation exchanged bentonites for removing phosphate [37] and arsenite ions [38]; and Sn(II), Cr(III)- modified bentonites for removing pertechnetate ions [39].
In case of halogenides, namely Cl and I studied in this work, the interlayer cations of montmorillonite of bentonite clay are exchanged to cations, namely Ag, which precipitate with halogenide ions. The reactions and their stability (solubility) constants are well known (see “Speciation modelling of the proportion of chemical forms in the silver ion-iodide ion system” section). For this reason Ag–bentonite was prepared from Ca–bentonite. The sorption experiments of halogenide ions on Ag–bentonite were carried out. This supposed mechanism is shown in Fig. 1. The aim of this work is to test the sorption capacity of Cl and I ions on Ag–bentonite as well as to reveal the sorption mechanism.

The scheme of interaction between iodide/chloride ions and silver in the interlayer space, where Na+ and I− ions can be substituted by K+ or Cl−, respectively
Experimental
Preparation of Ag–bentonite
Ag–bentonite was prepared by an ion exchange process using Ca–bentonite (Istenmezeje, Hungary) and AgNO3 solution. Elemental analysis of the original Ca–bentonite (measured with EDAX) resulted in the following composition: 73.29% SiO2, 18.71% Al2O3, 4.23% MgO, 2.29% CaO and 1.48% Fe-oxide [40]. X-ray diffraction and thermoanalytical analysis show 71 m/m % montmorillonite, 12 m/m % cristobalite/opal CT, 8 m/m % illite, 4 m/m % quartz, 3 m/m % kaolinite, and 2 m/m % calcite in it. The cation exchange capacity (CEC) of Ca–bentonite determined by ammonium acetate method [41] is 81 meqv/100 g.
Ca -bentonite (40 g) was suspended in AgNO3 (Reanal,p.a.) solution (125 cm3, 1.08 mol dm−3). The suspension was shaken for 5 h. It was filtered through a 0.45 μm pore size membrane filter. The exchange has been done only one, because previous experiments have shown that after 3 exchanges, the exchange was not more effective. The pH of the filtrate was 6.5. Then the Ag–bentonite was washed with distilled water three times and dried at room temperature in a desiccator over silica gel for 6 weeks and powdered in agate mortar. The silica gel was ignited every week. The preparation was made in dark. As light can reduce silver ions, sorption of iodide ions on Ag–bentonite was carried out on Ag–bentonite stored in light and in dark too.
The Ag concentration of bentonite was determined by X-ray fluorescence analysis (XRF) using the Kα-lines of silver (22.1 keV). The parts of XRF: 185 MBq 241Am excitation source, Si(Li) detector with 20 mm2 surface and 3.5 mm evaporated layer (Atomki, Debrecen, Hungary), Canberra DSA 1000 digital spectrum analyzer and Canberra Genie 2000 3.0 spectroscopy software (Canberra Industries, Meriden, CT 06450, USA).
The Ag concentration measured with XRF was 6 × 10−4 mol g−1. This means that about 75% of the cation exchange capacity was exchanged by silver ions in one ion exchange.
Kinetic studies of iodide sorption on Ag modified bentonite
The NaI, KI solutions were prepared by dissolving NaI (Reanal, puriss), KI (Sigma-Aldrich, puriss) in water. The kinetic of iodide sorption labeled with Na131I isotope (Institute of Isotopes Co., Ltd.) was studied in batch experiments. The desired mass of Ag–bentonite (10 mg) was added to 19.5 cm3 distilled water. All samples were placed in a Bühler KS15A shaking bath at 25 °C for 30 min to reach the equilibrium between the liquid and the solid phase. After this time, 0.5 cm3 carrier free 131I was added to the samples. All samples were shaken for 5–120 min then filtered through a 0.45 μm pore size cellulose nitrate filter and the radioactive intensity of the solution (ce) and the solid phase was measured with a multi-channel gamma spectrometer equipped with (NaI)Tl scintillation detector. pH of the solution was measured with a Clean pH200 pH meter and a Clean Cs1030 combined glass electrode. It was determined that the 0.45 μm pore size cellulose nitrate filter does not react with the ions in the solution.
Iodide ions were sorbed on Ag–bentonite so fast that the kinetic rate constant could not be determined by the batch technique. This fast reaction suggests the precipitation of bentonite-silver ion-iodide ion complex.
Sorption isotherms of chloride and iodide sorption on Ag modified bentonite
The sorption of chloride and iodide ions was studied similarly to kinetic investigations by batch technique (“Kinetic studies of iodide sorption on Ag modified bentonite” section). The sorption experiments were carried out on bentonite stored in light and in dark too, because light can reduce silver ions.
The mass of Ag–bentonite was 10 mg; the volume of the solution was 20 cm3. The initial chloride concentrations varied from 7.75 × 10−5 to 1.5 × 10−3 mol dm−3. NaCl solution was labeled with 36Cl tracer (Izinta, 4.8 M Bq/g Cl).
The iodide concentration varied from carrier-free (10−10 mol dm−3) to 2 × 10−3 mol dm−3 initial concentrations. For the higher concentrations, inactive potassium and sodium iodide solutions were added to the carrier-free 131I solutions. Suspensions were shaken at the desired temperature (25, 30, 40 °C) for 120 min, so sorption equilibrium is reached. The additional steps were carried out as in “Kinetic studies of iodide sorption on Ag modified bentonite” section. The radioactive intensity of the solution (ce) and the solid phase for iodide ion was measured with a multi-channel gamma spectrometer equipped with (NaI)Tl scintillation detector (“Kinetic studies of iodide sorption on Ag modified bentonite” section), while the radioactive intensity of chloride just in the solution (ce) was measured using its beta radiation with liquid scintillation spectrometer (Wallac 1409). 1 cm3 samples were mixed with 4 cm3 scintillation cocktail. The scintillator cocktail consist of: 4.0 g 1,5-diphyenil-oxazol (PPO), 257 cm3 Triton X-100,37 cm3 ethylene glycol, 106 cm3 ethanol, diluted to 1000 cm3 with xylene.
Determination of d(001) basal spacing of montmorillonite in Ag–bentonite with XRD
The d(001) basal spacing of Ag-montmorillonite before and after the sorption of iodide ion was determined with X-ray diffraction (XRD). The additional steps were carried out as in “Sorption isotherms of chloride and iodide sorption on Ag modified bentonite” section . The concentration of inactive KI and NaI solutions were 5 × 10−4 mol dm−3. On the measured solid phase the sorbed amount of iodide ions from KI solution was approx. 2.3 × 10−4 mol g−1 and from NaI solution was approx. 3.2 × 10−4 mol g−1. After the sorption and filtration, the solid phase was dried at room temperature and powdered, then the solid phase was measured with a Philips PW1710 powder diffractometer equipped with a CuKα source and a graphite monochromator (operating at 30 mA, 40 kV). The scan rate was 2°2Θ/min.
Results and discussion
The effect of light and aging on Ag–bentonite
The light could reduce silver ions, so it was necessary to determine how it affects the sorption of the halogenide ions. Therefore, sorption of iodide ions on Ag–bentonite was investigated on Ag–bentonite stored in light and in dark, as shown in Fig. 2, which was obtained by plotting (a mol g) versus ce (mol dm−3), where a is the amount sorbed at equilibrium (mol g), ce is the equilibrium solution concentration. The effect of light was investigated on a freshly made Ag–bentonite. The data series refer to 35–74 days old Ag–bentonites. Figure 2 shows that the effect of light is negligible.

The effect of light for the sorption of iodide ions on Ag–bentonite (bentonite stored in light is marked with red and bentonite stored in dark is shown with blue); 10 mg Ag–bentonite, 20 cm3 solution, pH = 6, T = 25 °C
We were interested if the sorption capacity of the bentonite is affected by its ageing. Therefore, the sorption of iodide ions was investigated on 36, 191, 198 and 600 days old Ag–bentonite stored in dark, as shown in Fig. 3. It can be concluded that Ag–bentonite ages with time. After 600 days the iodide ion sorption capacity decreased to the half. The duration of the use of the prepared Ag–bentonite requires further investigation.

The effect of aging on Ag–bentonite, the sorption of iodide ions was investigated on 36, 191, 198 and 600 days old Ag–bentonite stored in dark, 10 mg Ag–bentonite, 20 cm3 solution, pH = 6, T = 25 °C
Sorption isotherms of chloride and iodide ion sorption on Ag ion modified bentonites
When the sorption equilibrium was reached, the sorbed quantity of chloride ion vs equilibrium concentration showed saturation (Fig. 4). This means that the halogenide sorption on Ag–bentonite can be formally described by a Langmuir isotherm equation. It is interesting to mention that the Langmuir isotherm model and equation was originally derived for gas adsorption with four assumptions. These are:

36Cl sorption on Ag–bentonite at different temperatures (10 mg 84 days old Ag–bentonite stored in dark, 20 cm3 solution, 25 °C, pH = 6)
-
1.
The adsorption can take place on the free sites of the surface;
-
2.
The adsorption is monomolecular;
-
3.
The adsorption sites are energetically homogeneous and there is no interaction among the adsorbed species;
-
4.
Adsorption equilibrium of the phases exists.
For the chloride sorption on Ag -bentonite, all these assumptions are fulfilled, as follows:
The sorption of halogenide ions takes place on free sites produced by the silver cations in the interlayer space of montmorillonite. As a result of the equivalency of monovalent halogenide and silver ions, the silver-halogenide sorption species are neutral, thus no electrostatic interaction occurs between the particles.
Equation (1) used is equivalent to Langmuir representation with a reciprocal Langmuir constant and can be expressed mathematically as follows:
where a is the amount sorbed at equilibrium (mol g−1), ce is the equilibrium solution concentration (mol dm−3), z is the maximum sorption capacity (mol g−1) and K is the solubility of the sorption complex (mol dm−3) [1].
The maximum sorption capacity and the solubility of the sorption complex (K) (Eq. 1) were calculated using the software called Scientist (Micromath Scientific Software, Salt Lake City, UT). The Scientist program uses the least squares method.
Figure 4 shows the sorbed amount of chloride ions (a) as a function of equilibrium concentration (ce) on silver bentonite at various temperatures. The experiments were carried out at 25, 30 and 40 °C on 84 days Ag–bentonite stored in dark.
Figure 4 shows that as the quantity of sorbed chloride ions decreases as the temperature increases; the sorption is the highest at 25 °C. The experimental data were fitted by Eq. 1.
The maximum sorption capacity and the solubility of the sorption complex (K) are listed in Table 1. Despite the trend of solubility decrease with increasing temperature is not statistically significant, the exothermic reaction may be supposed (Table 1). The solubility of the sorption complex obtained is not equals with the solubility of AgI. The solubility of the bentonite-silver ion-iodide ion sorption complex is 7.9 × 10−6 mol dm−3. The concentration of the saturated silver iodide solution is 1.2 × 10−8 mol dm−3. This means that the solubility of the bentonite-silver ion-iodide ion sorption complex is about three orders of magnitude higher than that of the silver iodide.
Figure 5 shows the sorbed amount of iodide ions (a) versus equilibrium concentration (ce,). The figure shows the 131I sorption on silver bentonite with NaI carrier at 25, 30 and 40 °C. The a/ce isotherm (Fig. 5) shows that the sorbed amount of iodide ion behaves differently at different concentrations. For natural materials, these uncertainties are considered completely acceptable in the literature.

Na131I sorption on Ag–bentonite at different temperatures, 10 mg 195 days old Ag–bentonite stored in dark, 20 cm3 solution, Ag content: 6 × 10−4 mol g−1, pH = 6
In Fig. 5 two phenomena can be observed. The first phenomenon is observed at low concentrations, where the sorbed amount of iodide ions are increasing until approximately 5 × 10−5 mol dm−3 equilibrium concentration and the maximum sorbed amount of iodide ions is 3.4 × 10−4 mol g−1. At high concentrations a decrease is observed. These data were not fitted by Scientist because the sorption was influenced by iodide carrier solution.
Experiments were carried out with KI carrier solution as well. Figure 6 shows the sorbed amount of iodide ions (a) as a function of equilibrium concentration (ce). Differences can be observed between the sorption of iodide depending on whether the inactive carrier used to increase the concentration of I in NaI or KI solution.

Sorption isotherms of 131I ions on 195 days old Ag–bentonite in the case of NaI and KI (10 mg Ag–bentonite, 20 cm3 solution, 25 °C, Ag content: 6 × 10−4 mol g−1, pH = 6)
The sorbed amount of iodide ions increases to a concentration of 10−6 mol dm−3 and then decreases. The maximum sorbed amount of iodide ions is higher in the case of NaI, it was 3.35 × 10−4 mol g−1 and in the case of KI it was 2.29 × 10−4 mol g−1. Thus, potassium ions decreases the sorption of iodide, but at high concentrations the sorbed amount of iodide is approximately the same in NaI and KI solutions. Therefore, further experiments were carried out with NaI carrier.
The maximum sorption capacities listed in this study are in mol g−1. At 25 °C, these values were calculated to mg g−1, too, in order to collate our results with literature data. Accordingly, the maximum sorption capacities of Ag–bentonite for iodide at 25 °C was 55.8 mg g−1. In literature lower results were found, for example, the maximum sorption capacity of silver-impregnated activated carbon was 38.1 mg g−1 [42]. Moreover, the maximum sorption capacity of the nano Cu2O/Cu modified activated carbon, which synthesized by a simple and efficient hydrothermal method and used to adsorb iodide ion (127I), was also lower, 41.2 mg g−1 [12]. For chloride sorption no literature data was found.
Speciation modelling of the proportion of chemical forms in the silver ion-iodide ion system
To understand the sorption results the proportion of chemical forms in the silver ion-iodide ion system was calculated using the Visual MINTEQ chemical equilibrium model [43]. Calculations refer to 25 °C. The concentrations of the silver and iodide ions were input into the program as components. The thermodynamic equilibrium data (solubility, stability constants) were provided by the database of the Visual MINTEQ chemical equilibrium program (Eqs. 2 and 3).
The silver concentration was the silver content in modified bentonite (6 × 10−4mol dm−3) converted to the volume of the system (20 cm3), 4 × 10−4 mol dm−3 was obtained. The total concentration of iodide was changed from 3 × 10−8 to 5 × 10−3 mol dm−3. Using these data, the relative ratios of the chemical species, namely Ag+, AgI and AgI2− were calculated. Figure 7 shows the ratios of these species expressed as a percentage of silver ion as a function of the total equilibrium concentration of the iodide ion. At the same time, the sorbed amount of iodide ions on Ag–bentonite (a, mol g−1) were plotted. Figure 7 shows that the distribution of the species is in a good agreement with the sorption data, namely the decrease in the sorbed quantity begins when the formation of the soluble AgI2− species starts. As seen in Fig. 5 at high concentrations a decrease is observed due to the formation of a soluble silver diiodide complex at high concentration of iodide (Eq. 3). This means that by increasing the concentration of the inactive iodide ion carrier, a portion of the sorbed radioactive iodide can be dissolved by the higher iodide amount as a silver ion-diiodide-complex known from analytical chemistry. Equations 2 and 3 occurs simultaneously [44]:

Relative ratios of Ag+, AgI and AgI2− as a percentage of silver ion and the sorbed amount of iodide ions (a, mol g−1) at 25 °C, co is the concentration of iodide solution (mol dm−3)
Although the dissolution was only slightly observed with chloride (Fig. 4), in the case of chloride ion sorption on Ag–bentonite AgCl speciation was also calculated with Visual MINTEQ chemical equilibrium model as in the case of iodide ion. The total concentration of chloride solution was changed from 10−5 to 5 × 10−3 mol dm−3. Figure 8 shows the species received per total silver ion in %. The total concentration of chloride solution was changed from 10−5 to 5 × 10−3 mol dm−3.

Relative ratio of Ag+, AgCl and AgCl2− expressed as a percentage of silver ions and the sorbed amount of chloride ions (a, mol g−1) at 25 °C; co is the concentration of chloride solution (mol dm−3)
Figure 8 shows the species received per total silver ion in %. As the amount of AgCl2− increases in the solution, the sorbed amount begins to decrease. Here, the same phenomenon can be observed as with iodide ion, that is by increasing the concentration of the inactive chloride ion carrier, a portion of the radioactive chloride can form soluble silver ion-dichloride complex, but this phenomenon is much less present in the case of chloride sorption, then for iodide ion sorption, for which the difference in the stability constant of the dihalogenide complexes is responsible.
Results of d(001) basal spacing of montmorillonite of Ag–bentonite with XRD
Since it was previously assumed that Ag- ions in the interlayer space of montmorillonite sorb the halogenide ions, it was examined how this can affect the structure, a good indicator of which is the d(001) basal spacing determined by XRD. Table 2 shows the results of XRD used to determine the d(001) basal spacing of Ag- montmorillonite before and after sorption of iodide from KI and NaI solution. The initial concentration of KI and NaI solutions was 5 × 10−4 mol dm−3. The sorbed amount of iodide ions from KI solution was approx. 2.3 × 10−4 mol g−1 and from NaI solution was approx. 3.2 × 10−4 mol g−1. In NaI solution, higher sorption capacity was observed, which can be due to the incorporation of potassium ion into the crystal lattice, which decreases the layer charge. Another reason can be that the sodium ion swells the montmorillonite, providing additional binding sites for the iodide ion.
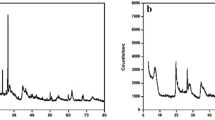
The d(001) basal spacing of the original Ca-montmorillonite is 1.5092 nm showing that mostly bivalent calcium ions are in the interlayer space. After the Ag+ exchange, a decrease in d(001) basal spacing of montmorillonite (1.4888 nm) is observed proving the ion exchange of monovalent Ag cation, because the d(001) basal spacing of monovalent montmorillonite is less than that of bivalent montmorillonite. This effect can explain the decrease of the basal spacing after ion exchange. As a result of the iodide sorption from potassium iodide solution, the d(001) basal spacing of montmorillonite decreased to 1.412 nm; and d(001) basal spacing increased to 1.5235 nm after sorption of iodide ions from NaI solution. The diffractograms (Fig. 9) show the presence of iodargyte (AgI).

XRD diffractrogram of Ag- montmorillonite before and after sorption of iodide from KI and NaI solution., the unit of y axis is arbitrary unit (a.u.) (sm-smectite, cr-cristobalite, q- quartz, i- iodargyrite)
Conclusions
The preparation of Ag–bentonite (silver content is 6 × 10−4 mol g−1) from Ca–bentonite was successful, the XRD measurments showing the changes in the d(001) basal spacing, and the formation of the new phase iodargyte (AgI).
The silver ions are present in the interlayer space of montmorillonite and can precipitate with chloride and iodide anions from sodium chloride, potassium and sodium iodide solutions. The uptake of iodide ions on Ag–bentonite is rapid and the equilibrium is reached within a few minutes so the rate constant could not be determined by batch technique.
In the case of iodide ion, the uptake was influenced by iodide carrier solution. Increasing the concentration of the inactive iodide ion reduces the uptake due to the formation of a soluble silver diiodide complex. The proportion of chemical forms in the silver-iodide/silver-chloride system was calculated using the Visual MINTEQ chemical equilibrium model. The distribution of species can interpret the sorption data, the decrease in sorbed amount begins when soluble Agl −2 complex formation begins. In the case of chloride sorption, the phenomenon is the same as in the case of the iodide sorption. However, the sorption quantity differs, for which the difference in the stability constant of the dihalogenide complexes is responsible. These results are consistent with analytical chemistry. The phenomenon occurs not only in the bulk aqueous phase but also in the interlayer space of montmorillonite. The equilibrium relationship between Ag–bentonite and dissolved chloride ion concentration was described with Langmuir isotherm. Both the amount of sorbed chloride ions and the solubility of the soprtion complex decreases as the temperature increases, thus the sorption of chloride ions is an exothermic surface precipitation reaction.
The effect of light and aging on Ag–bentonite was examined. It was found that the effect of light is negligible, but Ag–bentonite ages with time. In 600 days the sorption of iodide ions decreases to the half. The duration of the use of the prepared Ag–bentonite requires further investigation. These results show that Ag- modified bentonite could play a role in the treatment of radioactive waste as an engineering barrier to sorb halogenide ions.
References
Nagy MN, Kónya J (2009) Interfacial chemistry of rocks and soils. CRC Press, Taylor and Francis Group, New York
Bentley HW, Phillips FM, Davis SN (1986) Chlorine-36 in the terrestrial environment. In: Fritz P, Fontes JC (eds) Handbook of environmental isotope geochemistry. Elsevier, Amsterdam, pp 427–480
Phillips FM, Zreda MG, Smith SS, Elmore D, Kubik PW, Sharma P (1990) Cosmogenic chlorine-36 chronology for glacial deposits at Bloody Canyon, Eastern Sierra-Nevada. Science 248:1529–1532
Davis RF, Higinbotham N (1976) Electrochemical gradients and K+ and Cl− fluxes in excised corn roots. Plant Physiol 57:129–136
Bird JR, Davie RF, Chivas AR, Fifield LK, Ophel TR (1991) 36C1 production and distribution in Australia. Palaeogeogr Palaeoclimatol Palaeoecol 84:299–307
Jiang SS, Jiang S, Guo H, Du SB, Chen ZR, Guo OF, Zhao Y (1994) Determination of 36C1 in the groundwaters and ores around a uranium deposit. Nucl Instrum Methods Phys Res B 92:385–388
Sheppard SC, Johnson LH, Godwin BW, Tait JC, Wuschke DM, Davison CC (1996) Chlorine-36 in nuclear waste disposal Assessment results for used fuel with comparison to I-129 and C- 14. Waste Manag 16:607–614
Buesseler KO, Jayne SR, Fisher NS, Rypina II, Baumann H, Baumann Z, Breier CF, Douglass EM, George J, Macdonald AM (2012) Fukushima-derived radionuclides in the ocean and biota off Japan. PNAS 109:5984–5988
MacLean LC, Martinez RE, Fowle DA (2004) Experimental studies of bacteria– iodide adsorption interactions. Chem Geol 212:229–238
Kinly III D (2005) Chernobyl’s legacy: Health, environmental and socio-economic impacts and recommendations to the Governments of Belarus, the Russian Federation and Ukraine The Chernobyl Forum
Sancho M, Arnal J, Verdú G, Lora J, Villaescusa J (2006) Ultrafiltration and reverse osmosis performance in the treatment of radioimmunoassay liquid wastes. Desalination 201:207–215
Zhang X, Gu P, Li X, Zhang G (2017) Efficient adsorption of radioactive iodide ion from simulated wastewater by nano Cu2O/Cu modified activated carbon. Chem Eng J 322:129–139
Choung S, Um W, Kim M, Kim MG (2013) Uptake mechanism for iodine species to black carbon. Environ Sci Tech 47:10349–10355
Nenoff T, Krumhansl J, Gao H, Rajan A, McMahon K (2007) Iodine waste form summary report SAND 2007-6202 Sandia National Laboratory Albuquerque NM
Theiss FL, Ayoko GA, Frost RL (2016) Iodide removal using LDH technology. Chem Eng J 296:300–309
LiuY GuP, Jia L, Zhang G (2016) An investigation into the use of cuprous chloride for the removal of radioactive iodide from aqueous solutions. J Hazard Mater 302:82–89
Decamp C, Happel S (2013) Utilization of a mixed-bed column for the removal of iodine from radioactive process waste solutions. J Radioanal Nucl Chem 298:763–767
Tachibana Y, Nogami M, Nomura M, Suzuki T (2015) Simultaneous removal of various iodine species in aqueous solutions of high salt concentrations using novel functional adsorbents. J Radioanal Nucl Chem 307:1911–1918
Sinha PK, Lal KB, Ahmed J (1997) Removal of radioiodine from liquid effluents. Waste Manag 17(1):33–37
Zhang X, Stewart S, Shoesmith DW, Wren JC (2007) Interaction of aqueous iodine species with Ag2O∕Ag surfaces. J Electrochem Soc 154(4):F70
Xu C, Zhang S, Sugiyama Y, Ohte N, Ho YF, Fujitake N, Kaplan DI, Yeager CM, Schwehr K, Santschi PH (2016) Role of natural organic matter on iodine and 239,240 Pu distribution and mobility in environmental samples from the northwestern Fukushima Prefecture, Japan. J Environ Radioact 153:156–166
Zhang H, Gao X, Guo T, Li Q, Liu H, Ye X, Guo M, Wu Z (2011) Adsorption of iodide ions on a calcium alginate–silver chloride composite adsorbent. Colloids Surf A Physicochem Eng Asp 386(1–3):166–171
Sato I, Kudo H, Tsuda S (2011) Removal efficiency of water purifier and adsorbent for iodine cesium, strontium, barium and zirconium in drinking water. J Toxicol Sci 36:829–834
Kodama H (1998) Bismuth lead compound. US Patent 5720883
Lettinga G (1972) Radioactivity and water supplies. Diss Technische Hogeschool Delft. 281–304
Lefevre G, Bessiere J, Ehrhardt JJ, Walcarius (2003) A Immobilization of iodide on copper(I) sulfide minerals. J Environ Radioact 70:73–83
Chapman KW, Chupas PJ, Nenoff TM (2010) Radioactive iodine capture in silver-containing mordenites through nanoscale silver iodide formation. J Am Chem Soc 132(26):8897–8899
Sava DF, Rodriguez MA, Chapman KW, Chupas PJ, Greathouse JA, Crozier PS, Nenoff TM (2011) Capture of volatile iodine, a gaseous fission product, by zeolitic imidazolate framework-8. J Am Chem Soc 133(32):12398–12401
Asmussen RM, Neeway JJ, Lawter AR, Wilson A, Qafoku NP (2016) Silver-based getters for 129I removal from low-activity waste. Radiochim Acta 104(12):905
Lefèvre G, Walcarius A, Ehrhardt JJ, Bessière J (2000) Sorption of iodide on cuprite (Cu2O). Langmuir 16(10):4519–4527
Alexander JW (2009) History of the Medical Use of Silver. Surg Infect 10(3):289–292
Chernousova S, Epple M (2012) Silver as antibacterial agent: ion, nanoparticle, and metal. Angew Chem Int Ed 52(6):1636–1653
Shaffiey SR, Shaffiey SF, Ahmadi M (2015) Synthesis and evaluation of bactericidal properties of Ag2O nanoparticles against Aeromonashydrophila. Int J Nano Dimens 6(3):263–269
Suchomel P, Kvitek L, Panacek A, Prucek R, Hrbac J, Vecerova R, Zboril R (2015) Comparative study of antimicrobial activity of AgBr and Ag nanoparticles (NPs). PLoS ONE 10(3):e0119202
Malachová K, Praus P, Rybková Z, Kozák O (2011) Antibacterial and antifungal activities of silver, copper and zinc montmorillonites. Appl Clay Sci 53(4):642–645
http://www.phoslock.com.au/IRM/content/default.aspx. Accessed 06 June 2017
Buzetzky D, Nagy N, Kónya J (2017) Use of La-, Ce-, Y-, Fe- bentonites for removing phosphate ions from aqueous media. Period Polytech Chem Eng 61(1):27–32
Buzetzky D, Tóth N, Nagy N, Kónya J (2019) Application of modified bentonites for arsenite(III) removal from drinking water. Period Polytech Chem Eng 63(1):113–121
Buzetzky D, Kovács EM, Nagy MN, Kónya J (2019) Sorption of pertechnetate anion by cation modified bentonites. J Radioanal Nucl Chem 322:1771–1776
Kuzmann E, Singh LH, Garg VK, de Oliveira AC, Kovács EM, Molnár ÁM, Homonnay Z, Kónya P, Nagy MN, Kónya J (2016) Mössbauer study of the effect of rare earth substitution into montmorillonite. Hyperfine Interact 237:1–8
Richards LA (1957) Diagnosis and Improvement of Saline and Alkaline Soils. U.S. Dept Agr Handbook 60
Hoskins JS, Tanju K, Serkiz SM (2002) Removal and sequestration of iodide using silver-impregnated activated carbon. Environ Sci Technol 36:784–789
https://vminteq.lwr.kth.se/download/. Accessed 12 Jan 2019
Martell EA, Smith MR (1976) Critical stability constants Springer Science Business Media LLC
Acknowledgements
The authors thank Peter Konya for the X-ray diffraction measurements. The project presented in this article is supported by the EU and co-financed by the European Regional Development Fund under the project GINOP-2.3.2-15-2016-00008. The work was supported by the Hungarian National Research, Development, and Innovation Office (NKFIH K 120265).
Funding
Open access funding provided by University of Debrecen.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Buzetzky, D., Nagy, N.M. & Kónya, J. Use of silver–bentonite in sorption of chloride and iodide ions. J Radioanal Nucl Chem 326, 1795–1804 (2020). https://doi.org/10.1007/s10967-020-07457-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-020-07457-2