
Abstract
The aim of this work was to prepare and characterize the novel antibacterial nanocomposites based on chitosan, grafted acrylic acid and silver nanoparticles (AgNPs). Chitosan has been functionalised by chemical reaction with acrylic acid, and then mixed with silver nanoparticles (AgNPs). AgNPs have been obtained formerly by chemical reduction of silver nitrate in the presence of mercaptosuccinic acid as a nanoparticles stabilizer. Structure and properties of obtained nanocomposite have been characterised by UV–vis, FTIR and 13C-NMR spectroscopy. The morphology of silver chitosan-g-acrylic acid nanocomposite has been studied by electron microscopy (SEM/EDX and HR-TEM). Thermal stability has been investigated by thermogravimetric analysis. The grafted chitosan contained 70.9 % wt acrylic acid, as determined by gravimetric method. It was found that modified chitosan forms water-insoluble gel with lower thermal resistance than pure chitosan. However, the difference between thermal properties of grafted chitosan and its nanocomposite with AgNPs is negligible. The grafting reaction mechanism has been discussed in detail on the basis of the obtained results and current literature data.
Similar content being viewed by others
Introduction
Chitosan (CS) - abundant biopolymer, a derivative of chitin produced in nature by living organisms (e.g. by crabs, lobsters and shrimps), has excellent properties such as biocompatibility, biodegradability, non toxicity, moisture uptake, capability to complex formation and absorption of various chemical elements or compounds [1–5]. As CS is composed of D-glucosamine and N-acetyl-D-glucosamine units, its properties depend not only on the molecular weight but also on the deacetylation degree. Owing to the presence of side groups (OH, NH2, NH-CO-CH3) in polysaccharide chain, the structure of CS can be easily modified into various chemical reactions, leading to new properties and broader applications. At present, CS is used as a material for tissue engineering (scaffolds), in biotechnology (delivery of genes, antigens, proteins), pharmacy (drug delivery systems), medicine (cancer diagnosis, wound healing, component of fibres and fabrics [6, 7]. Recently, CS has been proposed as valuable material for nanotechnology and biotechnology. Some of the new review papers discussed the preparation process of CS-based nanocomposites or hybrid materials with biological activity, for example, by introducing silver nanoparticles (AgNPs) to the matrix of this polysaccharide [8–11]. AgNPs are known antibacterial and antifungicidal agents and their implementation of different polymers is often reported in the literatures [12–14]. The final properties of composites containing nanoparticles of noble metals depend not only on their content but mainly on their size, aggregation degree and distribution in the polymer matrix. Moreover, the proper choice of NPs stabilizer, protecting from agglomeration, is also very important.
The aim of present work was to prepare and characterize the bioactive chitosan nanocomposite. In the first step, the polysaccharide has been functionalised through grafting reaction with acrylic acid (AA). Such modification leads to amphiphilic properties due to both amine and carboxylic groups present in macrochains. This polymer is expected to be sensitive to pH, because in acidic solutions NH2 groups are protonated (−NH2 + H3O+ → -NH3 + + H2O), while in basic environment carboxylic groups are dissociated (−COOH + OH¯ → -COO¯ + H2O). Moreover, taking account the behaviour of CS origin and poly(acrylic acid) homopolymer, the strong water sorption and the capability of gel formation in CSAA is expected. In order to obtain antibacterial properties, AgNPs (prepared by chemical reduction of silver salt) were added to graft chitosan.
Spectroscopic and microscopic methods have been used to investigate the formed CSAA/Ag/MSA nanocomposite. The thermogravimetric analysis has been also carried out to evaluate its potential application.
The described synthesis is a promising method of obtaining a novel biocidal material, which can be proposed to usage in biomedicine, pharmacy, textile and cosmetic industry as well as in production of antibacterial absorbers or filters (destined to purification processes). The important issue is that the supposed advantage of this novel bioactive nanocomposite is that its both components: chitosan derivative and silver nanoparticles are safe in contact with human body. This assumption is based on the literature data [15].
Materials and methods
Materials
High molecular weighed chitosan, CS (viscosity-average molecular weight was 7.5 × 105, and its N-deacetylation degree was 85 %) and acrylic acid, CH2 = CHCOOH, AA, were purchased from Sigma Aldrich (Saint-Quentin Fallavier, France). Cerium (IV) ammonium nitrate, (NH4)2Ce(NO3)6, CAN; mercaptosuccinic acid, HOOC-CH(SH)-CH2COOH, MSA, and other reagents (acetic acid, sodium hydroxide, sodium borohydride and silver nitrate) were supplied by Sigma Aldrich (Saint Louis, MO). Before using, the AA was distilled under the reduced pressure for purification and inhibitor removing. The other reagents were high grade purity and were used without further purification.
Synthesis of chitosan-g-acrylic acid copolymer – CSAA
CSAA was synthesized according to the modified previously reported method [16] . 0.9973 g of chitosan was first dissolved in 50 ml 5 % aqueous solution of acetic acid. Monomer - AA (5 ml) and CAN (1.0197 g in 10 ml H2O) as the initiators were added to a solution of chitosan. The mixture was refluxed and stirred under an inert (helium) atmosphere for 4 h at 60 °C. After this time, the mixture became solid. The obtained viscous gel was rinsed in hot water and methanol for 2 h in Soxhlet extractor to remove the monomer residue and water soluble homopolymer of AA. The procedure was repeated in hot acetic acid to extract the soluble fraction of CSAA copolymer. Subsequently, precipitate has been filtered. The both white solid products (soluble and insoluble fractions of CSAA) were dried at 40 °C under vacuum to a constant weight.
CS sample has been weighed before and after grafting reaction for evaluation of the grafting degree (GD).
where mg – mass of grafted polymer and mo – is initial mass of polymer (before reaction).
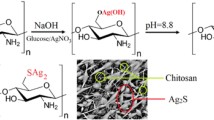
Synthesis of stabilised silver nanoparticles – Ag/MSA
The details of Ag/MSA synthesis was described in the previous work [17]. The preparation of Ag/MSA was based on the reduction of AgNO3 by NaBH4 in the presence of mercaptosuccinic acid (MSA) as a stabilizer.
AgNO3 was dissolved in methanol and added dropwise to the solution of MSA. The mixture was stirred at the room temperature until obtaining the yellow colour. Thereafter, NaBH4 was added to the solution and mixed. The obtained solution of a brown colour was being cooled in the refrigerator for 3 h. The product was filtered and washed by methanol. Finally, nanoparticles were dried at room temperature.
Preparation of nanosilver nanocomposites based on chitosan-g-acrylic acid copolymer – CSAA/Ag/MSA
The obtained soluble fraction of CSAA was dissolved in 0.1 M acetic acid at a concentration of 5 % (m/m). Ag/MSA/NPs solution was added to acidic solution of CSAA and intensively mixed at room temperature overnight. The content of the introduced silver nanoparticles in the nanocomposite was 3 % wt. Thin solid films have been obtained by casting of mixed solution on levelled polystyrene plates or spectroscopic windows. After solvent evaporation, the samples were carefully dried in dark place in vacuum at room temperature.
Methods of characterisation
The 13C NMR spectra in a solid state were recorded at room temperature with Bruker Avance III 700 MHz spectrometer.
The Fourier Transform Infrared (FTIR) spectra (range of 700–4000 cm−1) were recorded with a Mattson Genesis II spectrophotometer at room temperature in hexachloro-1,3-butadiene (HCBD) suspension.
UV–vis spectra of samples in the form of thin film or solution were obtained by means of UV-PC1600 Shimadzu spectrophotometer.
The thickness of polymeric films was measured by Sylvac 229 type digital indicator (Sylvac, Switzerland). The films were 6 μm thick.
The TG, DTG and DTA curves were recorded using TA Instrument SDT 2920 Simultaneous DSC-TGA. The following conditions were applied: temperature range - 20–600 °C, heating rate – 10 °C/min, and nitrogen atmosphere. Typical thermal degradation parameters were determined from the thermogravimetric curves for all stages of degradation: onset temperature (To), temperature at the maximum process rate (Tmax), temperature of process end (Tend), weight loss (% ∆m) and carbonaceous residue at 550 °C.
The SEM images of the dried samples were gained by a microscope LEO Electron Microscopy Ltd, England and SEM/FIB (scanning electron microscope / focused ion beam) Quanta 3D FEG.
The EDX microanalysis was performed with a spectrometer Quantax 200 with XFlash 4010 (Bruker AXS) coupled to the scanning electron microscope. SEM was also equipped with secondary electron detector (SE). The images of the high resolution transmission electron microscopy (HR-TEM) were recorded with a FEI Europe; model Tecnai F20 X-Twin (2011) at high vacuum. The samples were placed directly on standard Cu grids.
Results
Preparation of CSAA/Ag/MSA nanocomposites
The schematic diagram of the preparation process of CSAA/Ag/MSA nanocomposite is illustrated in Fig. 1. The first step was the CS reaction with a monomer - AA in the presence of CAN according to the procedure published previously [16]. During this reaction, carboxylic groups were introduced into polymer chain. Probably main substitution points were amine side groups (where N-H was transformed into N-C bond) and chain ends. The degree of grafting, determined by gravimetry was 70.9 % wt.

Schematic representation of silver chitosan-g-acrylic acid nanocomposite (CSAA/Ag/MSA) preparation
The second important step was to introduce the previously prepared Ag nanoparticles (stabilised by MSA) to chitosan grafted by AA. The nanocomposite contained 3 % wt. of silver nanoparticles.
Both specimens – CSAA copolymer and CSAA/Ag/MSA nanocomposite were obtained in the form of insoluble in neutral aqueous solution gel.
Discussion
Characteristics of obtained materials by UV–vis, FTIR and NMR spectroscopy
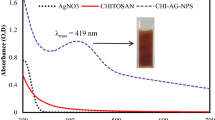
For the spectral properties of obtained CS, soluble fraction of CSAA and CSPAA/Ag/MSA composite have been studied by UV–vis spectroscopy (Fig. 2). Pure, unmodified chitosan does not absorb above 220 nm, only low intensive absorption branch is observed at about 250 nm. It is probably caused by the internal impurities. However, after CS modification, strong absorption appears at 270–300 nm range due to the presence of carboxylic groups in macrochains.

UV–vis spectra of CS, CSAA, Ag/MSA and CSAA/Ag/MSA
The spectrum of the Ag nanoparticles stabilized by MSA shows the absorption in the range of 250–500 nm with a clear maximum at 395 nm, which is typical for the Ag plasmon [14, 18]. The spectrum of obtained CSAA/Ag/MSA nanocomposite also exhibits plasmon band of AgNPs but its maximum appears at 420 nm. The shift of this band to longer wavelengths indicates the changes in the structure of Ag/MSA and suggests that NPs are attached to CSAA.
For confirmation of expected structures of studied polymers, FTIR and 13C NMR spectra in a solid state have been done.
CS
13C NMR, (700 MHz), δ (ppm), 24.8 (CH3), 58.3 (C2), 60.6 (C6), 75.5 (C3,5), 83.4 (C4), 105.8 (C1), 175.2 (very weak) (C = O).
IR: ν (cm−1); 3000–3700 (N-H stretching), 1652 (amide I, C = O stretching), 1563 (amide II, NH2 bending), 1415 (C-H stretching of the CH2 group of the C6), 1379 (C-H stretching of the CH3 group), 1318 (amide III, C-N stretching), 1153 (anti-symmetric stretching of the C-O-C bridge), 1077 and 1034 (skeletal vibration C-O).
PAA
13C NMR, (700 MHz), δ (ppm), 39.1 (CH2), 45.8 (CH), 184.2 (C = O).
IR: ν (cm−1); 2500–3500 (O-H stretching), 1710 (C = O stretching), 1400–1460 (C-H stretching of the CH2 and CH3groups), 1150–1250 (skeletal vibration C-O).
CSAA
13C NMR, (700 MHz), δ (ppm), 22.9 (CH3), 41.8 (CH2), 56.6 (C2), 60.1 (C6), 71.5–74.9 (C3,5), 97.7 (CH2 od C4), 179.4 (C = O).
IR: ν (cm−1); 2500–3700 (N-H and O-H stretching), 1715 (C = O stretching), 1642 (amide I, C = O stretching), 1546 (amide II, NH2 bending), 1400–1460 (C-H stretching of the CH2 and CH3groups), 1324 (amide III, C-N stretching), 1159 (anti-symmetric stretching of the C-O-C bridge), 1079 and 1037 (skeletal vibration C-O).
The infrared spectra of CS and soluble fraction of CSAA are shown in Fig. 3. The characteristic wide band with maximum at 3379 cm−1, assigned to the N-H stretching vibrations of amine group, can be found in CS spectrum. The amide I, amide II and amide III bands have been observed, respectively, at 1652 cm−1, 1563 cm−1 and 1318 cm−1. Moreover, the typical bands for saccharides appeared (at 1153 cm−1, 1077 and 1034 cm−1). The presence of band at 1379 cm−1 attributed to the CH3 groups of the acetamide group indicates that chitosan is not completely deacetylated [19]. After grafting, the new band with maximum at 1715 cm−1 appears in carbonyl region in CSAA spectrum. It corresponds to C = O stretching vibrations of carboxyl groups.

The FTIR spectra of CS, PAA and its copolymer - CSAA (marked the band confirming the grafting process)
The hydroxyl band in CSAA spectrum is broader and shifted to the lower frequency comparing to the initial unmodified CS. This is the result of the presence of OH in carboxylic groups of CSAA in addition to OH saccharide moieties. The significant broadening of hydroxyl band in CSAA indicates the formation of hydrogen bonds between carboxyl and hydroxyl groups. For comparison, the spectrum of PAA, being by-product, was presented (Fig. 3).
Certainly, these observations confirmed the introduction of AA to the chitosan macromolecules during the grafting process. As expected, there are no significant differences between the IR spectra of CSAA and CSAA/Ag.
For more precise determination of structures of synthesized CSAA, the 13C NMR spectroscopy has been used. The CS spectrum shows characteristic for saccharide structure peaks at around 58.3, 60.6, 75.5, 83.4, and 105.8 ppm. The very weak peaks at 24.8 (CH3) and 175.2 (C = O) groups confirmed the presence of acetamide groups in an incompletely deacetylated chitosan (Fig. 4).

Solid state 13C NMR spectra of CS and CSAA
In the 13C NMR spectrum of both soluble and insoluble fractions of CSAA, the similar peaks at 56.6 and 71.5–74.9 ppm were also observed. However, the signals at 105.8 and 83.4 ppm are lost while new signal at 97.7 ppm appears. These changes indicate oxidation of chitosan by Ce4+ ion at C1 and C4 positions. Furthermore, the peaks at 41.8 and 179.4 ppm (attributed to methyl and carbonyl groups, respectively) from grafted AA were detected. The 13C NMR spectrum of PAA homopolymer is shown for comparison (Fig. 4).
The details of the mechanism involved in the chitosan-acrylic acid copolymer formation have not been understood well yet. Several proposals of copolymerization mechanism initiated using cerium ions have been reported. Therefore, the very disputable issue is the site in macrochains where the grafting takes place.
On the one hand, it is indicated that the copolymerization of chitosan in the presence of Ce4+ ions could directly occur onto amino groups [20, 21], which is presented in a structure in Fig. 5a. This thesis has been confirmed experimentally by: weight analysis, conductometric and potentiometric titrations [22], or FTIR spectroscopy [23].

Reaction scheme for grafting AA onto chitosan
Contrary to this view, it is assumed that grafting using CAN as an initiator could lead to radical active centre on C6 atoms - Fig. 5b. The evidence has been proved by the usage of NMR and IR spectroscopy [24].
However, it is pointed out that mechanism may be similar to those observed in cellulose [25, 26]. According to this point of view, the most probable position of cleavage in glucosidic ring is C2–C3 bond. Doba et al. [27] suggested, considering the large number of repeating diol groups along the polysaccharide chain, that both Cl-C2 (end groups) and C2-C3 glycol groups are predominant sites for initiation of graft copolymerization. Thus, the anhydroglucose units of chitosan interacting with the Ce4+ ions are precisely oxidized in this position.
In the literature, two methods of initiation were proposed. First method presents the direct detachment of hydrogen atom from the carbon having a vicinal hydroxyl group. This process yields free radical on the carbon [28–30]. The second type of the process shows a reversible reaction of Ce4+ ions with nucleophilic group leading to a complex formation [25, 26]. It results in breakdown the C2-C3 bonds between the functional end groups. In the case of chitosan, the NH2 and reactive adjacent OH groups may form a complex with cerium ion, capable to dissociation, causing an increase in the content of free-radicals in the polysaccharide backbone. For that reason, a general scheme of grafting may occur as shown in Fig. 5c. Experimental evidences of this mechanism are given in the cited publication. Authors also show the possibility of ring opening reaction [31, 32].
In addition, the location of the radical on the C2 carbon atom has been suggested. In this case, an aldehyde group should be formed on carbon C3 after the breaking of C2-C3 bond caused by CAN (Fig. 5d). Such a mechanism can be found in Pourjavadi work [33] on grafting of polyacrylonitrile onto chitosan as well as in a publication by Jung [34] concerning grafting of the eugenol monomer on the amino groups of chitosan.
The direct acid–base reaction between COOH and NH2 groups leading to ionic bonds is also very probable [35].
Yilmaz et al. [36] suggested that the CAN reaction on chitosan results in products consisting of pyrone and pyranose rings. In the presence of a monomer (triethylene glycol dimethacrylate), the grafting at the oxidation site takes place. Nevertheless, at higher grafting yields, amide and amine functionalities of chitosan also serve as active sites (Fig. 5e). This mechanism of grafting AA onto chitosan is very probable also in the reaction described in this paper, which is confirmed by 13C NMR and FTIR spectroscopy analysis. The intensity of signals in NMR spectra indicated that AA underwent substitution at most radical points of chitosan and caused elongation of side chains of the chitosan backbone.
It should be emphasized, a wide variety of possible reactions at the conditions of usage makes impossible to specify the exact structure of the received product.
The lack of total solubility of the CSAA indicates the high crosslinking degree. There may appear covalent, ionic and hydrogen bonds between different groups.
Thermogravimetric analysis (TA)
TG and DTG curves of all received materials: CS, CSAA, CSAA/Ag/MSA are shown in Fig. 6. The thermal parameters obtained from TGA curves are listed in Table 1.

TG and DTG curves of: a) CS, b) CSAA and c) CSAA/Ag/MSA
The initial weight loss (below 150 °C), observed in all chitosan films, can be attributed to the loss of moisture. Although, all samples were dried and stored in a vacuum dryer, the strongly bonded water is still present due to the high sorption capacity of chitosan and its derivative. The content of water is the highest in CSAA (14 %) among studied specimens.
The heated chitosan decomposes in two steps. Despite the fact that water evaporation is connected to 7 % weight loss, the main decomposition (57 %) occurs at 250–400 °C range (II step). It is due to the chain scission and dehydration of glycoside units of chitosan. The carbonaceous residue is about 36 % at 550 °C, which is in accord with the previous report [19].
However, CSAA and CSAA/Ag/MSA composite undergo a thermal degradation in three steps. It is clearly visible that the II step of the degradation process starts earlier in the modified chitosan (CSAA) and in CSAA/Ag/MSA nanocomposite than in CS. This is because carboxyl or carboxyethyl groups are more susceptible to abstraction comparing to amine groups. There are no differences in To and Tmax for CSAA and CSAA/Ag/MSA, only weight loss is 6 % larger in nanocomposite in this step. However, the weight loss in the II step in both specimens containing modified chitosan (i.e. in CSAA and CSAA/Ag/MSA) is significantly lower (19–25 %) than in unmodified CS (57 %) but the maximum rate in copolymer appears at a temperature of about 50 of degrees lower. It indicates that only part of CSAA undergoes faster decomposition than corresponding fraction in CS. The second larger part of CSAA decomposes later, at higher temperatures (above 400 °C), suggesting the degradation of more stable, probably crosslinked structures.
The value of To and Tmax from the third step is lower about 15–16 °C in CSAA/Ag/MSA in comparison to CSAA. The detailed analysis of DTG curve for CSAA leads to a conclusion, that this step is complex and consists of at least two overlapping processes.
Carbon residue at 550 °C is lowest in CSAA (20 %) and largest in CS (36 %), while intermediate value (30 %) was recorded for CSAA/Ag/MSA. It should be added that silver is also present in residue of nanocomposite.
Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray spectroscopy (EDX)
SEM analysis shows that the surface of CSAA is not flat and has irregular scattered conglomerates of different sizes and shapes, typical for powder systems (Fig. 7a). The cross section of CSAA exhibits many of dimples and small voids, indicating on the sample porosity (Fig. 7b). In the case of CSAA/Ag/MSA, SEM does not show any other particular surface structures and defects. Surface of this sample is not smooth and random nodules, probably caused by nanoparticles aggregation, appear.

SEM image of CSAA: a – surface view, b – cross section
The elemental composition of CSAA/Ag/MSA was investigated by EDX analysis (Fig. 8a).

SEM/EDX image of CSAA/Ag/MSA film: a – distribution of sulfur and silver in nanocomposite, b – topography
It can be noticed that silver nanoparticles distribution in thin CSAA films is uniform, thus the sample is relatively homogeneous. In particular, the content of silver and other expected elements (carbon, nitrogen, oxygen and sulphur) at the surface of CSAA/Ag/MSA film is given in Table 2. The values obtained by EDX can be somewhat different to predicted ones for the whole bulk.
High Resolution Transmission Electron Microscopy (HR-TEM)
More information on the structure of silver chitosan-g-acrylic acid nanocomposite supplies HR-TEM. The silver particles are almost spheroidal in shape and have diameter in range of 40–80 nm. An example of the typical Ag/MSA nanoparticles embedded in CSAA matrix is presented in Fig. 9. The structure visible in Fig. 9a is an aggregate composed of few smaller oval particles. Higher magnification clearly shows the order inside the silver nanoparticles (i.e. the parallel arrangement of planes in Ag crystals).

HR-TEM images of CSAA/Ag film: Ag nanoparticle at different magnification (a, b); marked square at 8b indicates the area selected for calculation of d-spacing, shown at left corner
The calculated d-spacing (Fig. 8c) equals 0.239 nm, which corresponds well to literature value for metallic silver (0.2343 nm) [37]. It confirms Ag presence in the modified chitosan.
Summary and conclusions
This studies report the preparation of biocidal nanocomposite based on chitosan containing carboxylic group and silver particles.
The functionalisation of chitosan by grafting of acrylic acid leads to obtaining CSAA copolymer, which was applied as matrix for silver nanoparticles. The structure of prepared material has been confirmed by spectroscopic (UV–vis, FTIR, NMR) and microscopic (SEM/EDX, HR-TEM) analysis. Mechanism of grafting reaction has been discussed in details.
It was found that the modified chitosan is characterised by the lower thermal stability than the unmodified one but there are no significant differences between thermal behaviour of CSAA and CSAA/Ag/MSA.
The preparation method leading to silver chitosan-g-acrylic acid nanocomposite is relatively simple and can be recommended for application in practice. Certainly, the obtained material can be used in the various fields where the particular sterile conditions are needed, for example, in medicine and pharmacy. Furthermore, the preparation of this nanocomposite from renewable biomaterial like chitosan is the additional benefit.
References
Jayakumar R, Prabaharan M, Nair SV et al (2010) Novel carboxymethyl derivatives of chitin and chitosan materials and their biomedical applications. Prog Mater Sci 55:675–709. doi:10.1016/j.pmatsci.2010.03.001
Pillai CKS, Paul W, Sharma CP (2009) Chitin and chitosan polymers: chemistry, solubility and fiber formation. Prog Polym Sci 34:641–678. doi:10.1016/j.progpolymsci.2009.04.001
Kaczmarek H, Zawadzki J (2010) Chitosan pyrolysis and adsorption properties of chitosan and its carbonizate. Carbohydr Res 345:941–947. doi:10.1016/j.carres.2010.02.024
Chang H-S, Lin Y-S, Tsai Y-D, Tsai M-L (2013) Effects of chitosan characteristics on the physicochemical properties, antibacterial activity, and cytotoxicity of chitosan/2‐glycerophosphate/nanosilver hydrogels. J Appl Polym Sci 127:169–176. doi:10.1002/app.37855
Singh J, Dutta PK (2009) Spectroscopic and conformational study of chitosan acid salts. J Polym Res 16:231–238. doi:10.1007/s10965-008-9221-3
Sailaja GS, Velayudhan S, Sunny MC et al (2003) Hydroxyapatite filled chitosan-polyacrylic acid polyelectrolyte complexes. J Mater Sci 38:3653–3662. doi:10.1023/A:1025689701309
Kumar MNVR (2000) A review of chitin and chitosan applications. React Funct Polym 46:1–27. doi:10.1016/S1381-5148(00)00038-9
Białas S, Mucha M (2013) Influence of nanosilver on thermal stability of chitosan. Prog Chem Appl Chitin Deriv 18:85–92
Chang H-W, Lin Y-S, Tsai Y-D, Tsai M-L (2013) Effects of chitosan characteristics on the physicochemical properties, antibacterial activity, and cytotoxicity of chitosan/2-glycerophosphate/nanosilver hydrogels. J Appl Polym Sci 127:169–176. doi:10.1002/app.37855
Tylman M, Mucha M (2014) Chitosan scaffolds with nanosilver layer for bone implantation obtained by electrolytic method. Mater Sci Technol 30:582–586. doi:10.1179/1743284713Y.0000000497
Zheng Y, Monty J, Linhardt RJ (2014) Polysaccharide-based nanocomposites and their applications. Carbohydr Res. doi:10.1016/j.carres.2014.07.016
Da Silva Paula MM, Franco CV, Baldin MC et al (2009) Synthesis, characterization and antibacterial activity studies of poly-(styrene-acrylic acid) with silver nanoparticles. Mater Sci Eng C 29:647–650. doi:10.1016/j.msec.2008.11.017
Tyliszczak B, Pielichowski K (2013) Novel hydrogels containing nanosilver for biomedical applications - synthesis and characterization. J Polym Res 20:191. doi:10.1007/s10965-013-0191-8
Radetić M (2013) Functionalization of textile materials with silver nanoparticles. J Mater Sci 48:95–107. doi:10.1007/s10853-012-6677-7
Bae H-S (2010) Functional modification of sanitary nonwoven fabric by chitosan/nanosilver colloid solution and evaluation of applicability. Fibers Polym 11:606–614. doi:10.1007/s12221-010-0607-6
Shantha KI, Bala U, Rao KP (1994) Tailor-made chitosans for drug delivery. Eur Polym J 31:377–382. doi:10.1016/0014-3057(94)00177-4
Ścigalski F, Kaczmarek H, Wolnicka M (2013) Preparation and properties of silver nanoparticles stabilized by mercaptosuccinic acid with a controllable size. Adv Sci Eng Med 5:219–223. doi:10.1166/asem.2013.1256
Kaczmarek H, Metzler M, Ścigalski F (2014) Photochemical stability of poly(acrylic acid)/silver nanocomposite. Mater Lett 135:110–114. doi:10.1016/j.matlet.2014.07.161
Zawadzki J, Kaczmarek H (2010) Thermal treament of chitosan in various conditions. Carbohydr Polym 80:395–401. doi:10.1016/j.carbpol.2009.11.037
Caner H, Yilmaz E, Yilmaz O (2007) Synthesis, characterization and antibacterial activity of poly(N-vinylimidazole) grafted chitosan. Carbohydr Polym 69:318–325. doi:10.1016/j.carbpol.2006.10.008
Joshi JM, Sinha VK (2006) Graft copolymerization of 2-hydroxyethylmethacrylate onto carboxymethyl chitosan using CAN as an initiator. Polymer 47:2198–2204. doi:10.1016/j.polymer.2005.11.050
Recillas M, Silva LL, Peniche C et al (2009) Thermoresponsive behavior of Chitosan-g-N-isopropylacrylamide copolymer solutions maricarmen recillas. Biomacrom 10:1633–1641. doi:10.1021/bm9002317
Joshi JM, Sinha VK (2007) Ceric ammonium nitrate induced grafting of polyacrylamide onto carboxymethyl chitosan. Carbohydr Polym 67:427–435. doi:10.1016/j.carbpol.2006.06.021
Shantha KL, Harding DRK (2002) Synthesis and characterisation of chemically modified chitosan microspheres. Carbohydr Polym 48:247–253. doi:10.1016/S0144-8617(01)00244-2
Jenkins DW, Hudson SM (2001) Review of vinyl graft copolymerization featuring recent advances toward controlled radical-based reactions and illustrated with chitin/chitosan trunk polymers. Chem Rev 101:3245–3274. doi:10.1021/cr000257f
McDowall DJ, Gupta BS, Stannett VT (1984) Grafting of vinyl monomers to cellulose by ceric ion initiation. Prog Polym Sci 10:1–50. doi:10.1016/0079-6700(84)90005-4
Doba T, Rodehed C, Ranby B (1984) Mechanism of graft co-polymerization onto polysaccharides initiated by metal ion oxidation reactions of model compounds for starch and cellulose. Macromolecules 17:2512–2519. doi:10.1021/ma00142a009
Huang Z, Wu Z, Yang P, Yang W (2014) Chemoselective phototransformation of C-H bonds on a polymer surface through a photoinduced cerium recycling redox reaction. Chem Europ J 20:11421–11427. doi:10.1002/chem.201402786
Graczyk T, Hornof V (1988) Graft copolymerization of cellulose initiated by ceric salts: effect of reaction conditions on the consumption of ceric ion. J Polym Sci Part A: Polym Chem 26:2019–2029. doi:10.1002/pola.1988.080260803
Don T-M, King C-F, Chiu W-Y (2002) Synthesis and properties of chitosan-modified poly(vinyl acetate). J Appl Polym Sci 86:3057–3063. doi:10.1002/app.11329
Zhang J, Yuan Y, Shen J, Lin S (2003) Synthesis and characterization of chitosan grafted poly(N,N-dimethyl-N-methacryloxyethyl-N-(3-sulfopropyl) ammonium) initiated by ceric (IV) ion. Eur Polym J 39:847–850. doi:10.1016/S0014-3057(02)00286-0
Pourjavadi A, Mahdavinia GR, Zohuriaan-Mehr MJ (2003) Modified chitosan. II. H-chitoPAN, a novel pH-responsive superabsorbent hydrogel. J Appl Polym Sci 90:3115–3121. doi:10.1002/app.13054
Pourjavadi A, Mahdavinia GR, Zohuriaan-Mehr MJ, Omidian H (2003) Modified chitosan. I. Optimized cerium ammonium nitrate-induced synthesis of chitosan-graft-polyacrylonitrile. J Appl Polym Sci 88:2048–2054. doi:10.1002/app.11820
Jung B-O, Chung S-J, Lee SB (2006) Preparation and characterization of eugenol-grafted chitosan hydrogels and their antioxidant activities. J Appl Polym Sci 99:3500–3506. doi:10.1002/app.22974
Hu Y, Jiang X, Ding Y, Ge H, Yuan Y, Yang C (2002) Synthesis and characterization of chitosan–poly(acrylic acid) nanoparticles. Biomaterials 23:3193–3201. doi:10.1016/S0142-9612(02)00071-6
Yilmaz E, Adali T, Yilmaz O, Bengisu M (2007) Grafting of poly(triethylene glycol dimethacrylate) onto chitosan by ceric ion initiation. React Funct Polym 67:10–18. doi:10.1016/j.reactfunctpolym.2006.08.003
Satapathy S, Shukla SP, Sandeep KP, Singh AR, Sharma N (2015) Evaluation of the performance of an algal bioreactor for silver nanoparticle production. J Appl Phycol 27:285–291. doi:10.1007/s10811-014-0311-9
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Metzler, M., Chylińska, M. & Kaczmarek, H. Preparation and characteristics of nanosilver composite based on chitosan-graft-acrylic acid copolymer. J Polym Res 22, 146 (2015). https://doi.org/10.1007/s10965-015-0781-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-015-0781-8