
Abstract
Purpose
Primary immunodeficiencies (PID) are a group of heterogeneous diseases. Epidemiological studies from databases worldwide show geographical variation. In this study the objective is to determine the spectrum of PID in Saudi Arabia by analyzing the database in a referral tertiary hospital.
Methods
This is a prospective data collection by interviews and medical chart review for all PID patients followed at the King Faisal Specialist Hospital & Research Center (KFSH&RC) from May 2010 to April 2013.
Results
A total of 502 patients presented (53 % male and 47 % female). Combined immunodeficiencies were the most common (59.7 %), followed by predominantly antibody deficiencies (12.3 %), congenital defects of phagocyte (9.4 %), combined immunodeficiencies with associated or syndromic features (6.2 %), disease of immune dysregulation (6 %), complement deficiencies (5.8), and defects in innate immunity (0.6 %). The most common combined immunodeficiencies phenotype was T-B-SCID (17 %). The patients’ ages ranged from less than 1 year old to 78 years, and 394 patients (78.2 %) are in the paediatrics age group (<14 years). The overall mean age of symptoms onset was 17 months and the overall mean delay in diagnosis was 21.6 months. Recurrent infections were the most common occurring clinical presentation (66 %), followed by family history (26 %). Consanguinity was found in 75 % of the patients. A total of 308 (61 %) patients had undergone stem cell transplantation (SCT).
Conclusion
The study revealed that combined immunodeficiencies are not uncommon and are the most frequent occurring diagnosis in our patient population. This study is a prerequisite to establish a national registry of primary immunodeficiency in Saudi Arabia.
Similar content being viewed by others

Introduction
Primary immunodeficiency diseases (PID) are considered to be rare disorders worldwide. Therefore, establishing a database is important to determine the magnitude, types and spectrum of PID disease encountered in a certain population. Similar databases worldwide have shown geographical and racial variation in the spectrum of PIDs [1–14]. Registry data from Saudi Arabia are limited to two studies. Both are from a homogeneous population and from only one region of the country, and hence likely does not represent the whole nation [15, 16]. Nevertheless, they are important preliminary results, and highlight the need for ongoing, systematic data collection to learn more about PIDs in Saudi Arabia. Furthermore; consanguineous marriages (first cousin marriages) in Saudi Arabia are high, present up to 60 % [17]. This has provided a background for genetic diseases to be abundant in the Saudi population. The elevated rate of consanguineous marriages was shown in the studies of a relatively large number of patients diagnosed with an autosomal recessive inherited form of PIDs as in severe combined immunodeficiency (SCID) [18], hyper-IgE syndrome [19], and hyper-IgM syndrome [20]. Therefore a registry data from Saudi Arabia is important in particular to autosomal recessive inherited PIDs. Moreover the overall incidence of combined immunodeficiencies (CID) is estimated to be 1 in 75,000 to 100,000 live births [21] and since most forms of CID are inherited as autosomal recessive traits, therefore anecdotal experience suggests that the incidence of CID is higher in our region than in western countries, probably exceeding 1 in 10,000 live births [22]. However accurate epidemiologic data remain scarce.
In order to determine the spectrum of PIDs in a Saudi tertiary hospital, we established a database and registered all presenting patients diagnosed with PIDs. Thus we report here the results for the past 3 years. Our goal is for this database to be the foundation for a national registry.
Methods
The King Faisal Specialist Hospital and Research Center (KFSHRC) is a tertiary level hospital for both pediatric and adult patients in the Kingdom of Saudi Arabia. The hospital has received referrals from all around the country since its opening in 1975. A pediatric clinical immunology service was established in 1984 and since then the hospital began to be a major referral for PIDs patients. In May of 2010 a PID database was established as a prospective ongoing hospital-based registry of all patients diagnosed with any of the PID diseases that meet the criteria for diagnosis by the World Health Organization. Data capture forms were developed. Permission was sought and granted by the local Institutional Review Board. Consent was obtained by the patients or their parents. The variables identified in the data capture forms are basic demographic, diagnosis details and treatment procedures. The medical diagnoses are coded with the International Classification of Diseases 10 (ICD-10 AM). Microsoft SQL Server was used to develop and administer the database, which itself is accessed using a standard web browser. All data are confidential and access is restricted to persons with a valid username and password.
Statistical analysis was done using the SAS® software package, version 9.4 (Statistical Analysis System, SAS Institute Inc., Cary, NC, USA). Descriptive statistics were reported for all variables in the study. All categorical variables were reported as counts and percentages. Continuous variables were reported as means, medians, standard deviations, minima, maxima and ranges.
Results
Patient’s Distribution
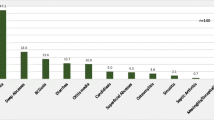
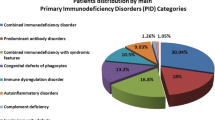
A total of 502 patient encounters were registered in the database from May 2010 to April 2013. The patients were diagnosed with 34 different primary immunodeficiency diseases which belong to 7 main PID categories. None of the patients were identified in the autoinflammatory disorders category or the phenocopies of PID. Combined immunodeficiencies it’s the predominant category, forming 59.7 % of the patients (300 patients), followed by predominantly antibodies deficiency in 12.3 % (62 patients), congenital defect of phagocyte in number and/or function in 9.4 % (47 patients), combined immunodeficiencies with associated or syndromic features in 6.2 % (31 patients), diseases of immune dysregulation in 6 % (30 patients), complement deficiencies in 5.8 % (29 patients), and innate immunity in 0.6 % (4 patients) (Fig. 1 and Table 1). Among the combined immunodeficiencies, SCID T−B− was the most common type in 17 % (85 patients). SCID T−B+ was found in 5 % (25 patients), followed by Omenn syndrome in 3.6 % (18 patients). SCID patients who did not match any phenotype of the combined immunodeficiencies by the IUIS classification was diagnosed as SCID- not otherwise specified (SCID- NOS) and this was in 3.2 % (16 patients). ADA deficiency and combined immunodeficiency (CID) (CID are defined as patients with partial T&B cell abnormalities who survived after the age of 2 years without immunologic reconstitution) were in 2.8 % (14 patients) each. The least common diagnoses were PNP and CD8 deficiency in 0.8 % (4 patients each) and reticular dysgenesis in 0.6 % (3 patients). Major Histocompatibility Class II deficiency (MHCII deficiency) was the second most common diagnosis in 12.4 % of the patients (62 patients) (Table 1). Common variable immunodeficiency (CVID), chronic granulomatous disease (CGD), Wiskott-Aldrich syndrome, and Griscelli syndrome were the most commonly diagnosed PID diseases in their respective categories, and they were diagnosed in 7.6, 6.2, 3.8, and 3.8 % respectively among the total number of patients (Table 1). In the Complement Deficiencies category, Hereditary Angioedema due to C1-estrease deficiency was diagnosed in 5 % (25 patients). The Innate Immunity category had the least number of patients. The regional distribution of patients was determined on the basis of the patient’s home address (Fig. 2).

Patients distribution by main primary immunodeficiencies classification

Regional distribution of PID patients
Age, Gender and Race
The patients’ ages ranged from 0 to 78 years, and 394 patients (78.2 %) were in the pediatrics age group (<14 years) (Table 2 and Fig. 3). The male to female (M: F) ratio was 1.1:1.0 overall. This (M: F) ratio remains the same in the combined immunodeficiencies and in the diseases of immune dysregulation category. In contrast the ratio (M:F) was 2.1:1.0 for well-defined syndrome with immunodeficiency, predominantly antibodies deficiency (0.7:1), congenital defect of phagocyte in number and/or function category (1.6:1), Complement Deficiencies (0.7:1) and in The Innate Immunity category (3:1) (Table 2). 97 % of the patients were from a Saudi Arabian ethnic background and 3 % were from other Arabian or Asian ethnic background.

Patients distribution in each age group
Symptoms Onset, Diagnosis and Delay of Diagnosis
The overall mean age of symptoms onset was 17 months, where combined immunodeficiencies had the earliest onset of symptoms (7.3 months) while the complement deficiencies had the latest onset of symptoms (103 months) (Table 2). The mean age of diagnosis was 39 months for all PID categories. Majority of the patients (470 patients 93 %) were diagnosed during their childhood (<14 years); from among these (280 patients 56 %) were diagnosed before 1 year of age. Combined immunodeficiencies were diagnosed at a mean age of 21 months, in contrast to predominantly antibodies deficiencies which were diagnosed at a mean age of 76.4 months (Table 2). The overall mean for delay in diagnosis was 21.6 months (ranging from less than 1 month to 38 years). The delay in diagnosis in combined immunodeficiencies was 13.5 months and that for those with predominantly antibodies deficiencies were 46 months (Table 2). There were no antenatal diagnoses among patients registered in the database.
Clinical Presentation
Recurrent infections were the most common occurring clinical presentation (66 %), followed by family history (26 %), atypical infection (22 %), failure to thrive (11 %), recognized clinical syndrome (8 %). Atypical infections among the registered patients were disseminated Mycobacterium bovis secondary to Bacillus Calmette- Guerin (BCG) vaccination, Cytomegalovirus (CMV), Pneumocystis jiroveci pneumonia (PJP), Epstein–Barr virus (EBV), and aspergillus in 54, 18, 6, 4 and 2 patients respectively. Disseminated Mycobacterium bovis was found in 49 SCID patients out of 114 SCID receiving the BCG vaccine at birth (43 %). Overall the most common type of recurrent infection at presentation was lower respiratory tract infection (50 %), skin Infections (cellulitis, abscesses) (25 %), chronic diarrhea (24 %), upper respiratory tract infection (21 %), oral thrush (13 %), and deep abscesses (6 %).
Consanguinity and Family History
Consanguinity is defined in this study as patients for whom their parents are first degree cousins. The overall rate for consanguinity was 75 % of all patients. The rate of consanguinity varied among the groups of PID (Table 1). Immune dysregulation had the highest rate of consanguinity at 83 %, followed by combined immunodeficiencies at 78 %, while in the other groups the rates were - predominantly antibodies deficiencies (74 %), phagocytic defects (72 %), well-defined immunodeficiency syndrome (61 %), and complement deficiencies (45 %). The lowest rate of consanguinity was recorded for Innate Immunity at 25 %. Family history of PID in other members of the patient’s family was found in 306 patients (61 %) (Table 1). 132 patients (26 %) were diagnosed at birth through targeted newborn screening because of family history of previous siblings affected (Table 1).
Treatment
Treatment modalities were captured in the database. Intravenous immunoglobulin (IVIG) replacement was documented for 386 (76 %) of the patients. The mean duration for IVIG replacement was 19.2 months (ranging from 0 to 199 months). In 9 patients IVIG was discontinued because of side effects. Prophylaxis antibiotics were used in 330 patients (65 %). A total of 308 (61 %) patients had undergone stem cell transplantation (SCT). SCT sources were bone marrow in 255 patients (82.8 %) and cord blood in 53 patients (17.2 %). The bone marrow transplant donors were match related donor (MRD) in 218 patients (sibling in 162, parents in 37, and other family related donor in 19 patients), mismatch related donor (MMRD) in 18 patients, haploidentical donor in 17 patients, and matched unrelated donor (MUD) in 2 patients The mean age of transplant was 21.2 months (ranging from 0.5 to 197 months).
Mortality
Out of the 502 patients in the database, 52 patients (31 males and 21 females) died (10.3 %) at some point following registration. The age range of death was 4 to 403 months with a mean age of death of 52 months. 45 patients (88 %) died during childhood, which includes 20 patients before 1 year of age, 32 patients before 2 years, and 41 patient before 5 years of age (38, 63 and 79 %, respectively). The diagnosis of these patients was SCID (27 patients), MHCII deficiency (8 patients), CGD (4 patients), Griscelli Syndrome (3 patients), Hyper-IgE syndrome (2 patients), EBV related lymphoproliferative disorder (2 patients), and dyskeratosis congenital, CVID, ataxia telangiectasia, Wisckott Aldrich syndrome, hyper-IgM syndrome, and leukocyte adhesion deficiency 1 (1 patient each). 34 of these patients (67 %) underwent stem cell transplantation. The donor sources were HLA-matched related in 15 patients, unrelated cord blood in 13 patients, and mismatched related in 6 patients. The most common immediate cause of death was severe pneumonia presenting or progressing to acute respiratory distress syndrome / air leak syndrome with or without pulmonary hemorrhage. The other causes were septic shock, multiorgan failure and refractory cytopenia.
Discussion
Studies of PID databases worldwide are important contributors to our knowledge of the epidemiology, characteristics and features of PIDs in different regions of the world. This study analysed the PID database from the KFSH&RC - a tertiary care hospital in Saudi Arabia with 800 beds, and a major referral centre for stem cell and organ transplants, cancer therapy and inherited diseases. KFSH&RC is the only centre in the country which provides HSCT for PID patients.
The Kingdom of Saudi Arabia’s population census (up to 2013) has reported a population 19,838,448 million and 27.6 % are children [23]. The PID prevalence has been calculated for the Saudi pediatrics age groups (0–14 years) since the majority at the time of the study are in this age group (394 patients). Furthermore, the total number of registered predominant antibodies deficiency (the most frequent in adult group), does not reflect the actual number of patients in the population, and therefore the prevalence in the whole population will be underestimated. Accordingly, a minimal estimate for the prevalence of all PIDs is 7.2 in 100,000 Saudi children population. The prevalence and incidence for CID, SCID and MHCII is reported in Table 3. Since the hospital is the major referral centre for the Kingdom, this may present an almost complete number of cases in the country. CID estimated prevalence from our data is 4.82 / 100 000 Saudi children population (Table 3) this high prevalence was also shown in previous publications from regional studies to be at 7 and 2.6 / 100 000 from the eastern province of Saudi Arabia [16] and Kuwait [6] respectively. This in compared to CID prevalence reported from other registries in the UK [1], France [2] and Germany [11] at 0.35, 0.52 and 0.05 respectively. As well as SCID prevalence and incidence (Table 3) from our data showed to be high and up to 10 times that published in studies from other countries as UK [1], France [4], Australia and New Zealand [3], Latin America [4], and Japan [5]. Moreover MHC class II deficiency registered in our database in 62 patients, this is another rare autosomal recessive disease where only about 150 patients reported in the literature, the majority are from a similar highly inbred tribes in north Africa [24] and the Arabian peninsula [25]. This could be attributed to the high rate of consanguineous marriages and autosomal recessive diseases in our population. The overall consanguinity (first degree cousin marriages) rate in our registry was 75 %; this high rate has also been reported in other regional PID registries as in Kuwait [6], Oman [7], Egypt [8], Tunisia [9] and Iran [10] at a rate of 77, 81, 63, 46 and 63.1 % respectively. This in contrast to the rate of consanguinity in registries from other countries as, UK [1], France [2], Germany [11] South Africa [12], at 3, 15, 8.6, and 1.2 %, respectively.
The distribution pattern of PID in our center shows a higher frequency of combined immunodeficiencies (59.7 %) (Table 1 and Fig. 1), This estimate from our center could be biased because of the fact that our center is the referral center for stem cell transplantation (SCT) in the country as well as diagnostic and therapeutic modalities for other form of PIDs, especially for antibodies deficiency patients are available in other regional hospital cross the country. However, registries from other Arab populations have showed that the rate of combined immunodeficiency is higher than that in the rest of the world; the eastern province of Saudi Arabia (47 %) [16], Egypt (29 %) [8], Tunisia (24 %) [9], and Kuwait (21 %) [6], as compared to data from other worldwide registries; the European Society for Immunodeficiencies registry (7.78 %) [13], Australian / New Zealand (6.2 %) [3], USA (8.8 %) [12], Japan (7 %) [5] and Latin America (10.1 %) [4].
Autosomal Recessive (AR) SCID is the most common type of SCID in our population (90 % of SCID patients). This is consistent with previous regional studies. For example, AR-SCID for the eastern province of Saudi Arabia [16], Kuwait [6], and Oman [5], were 83, 87, and 96 % respectively. Moreover AR-SCID was also found to be the most common type in other registries, as in the Australian/New Zealand [3] and the ESID [13] (88 and 82 % respectively). X-linked SCID has been widely recognized as the most common type of SCID. This is probably true in only certain parts of the world as in North America [26] and Japan [5]. This emphasizes the racial and geographical variation of PID in different countries. This can also be explained by the higher rate of consanguineous marriages (75 %) in our PID population which in turn leads to a higher prevalence of Autosomal Recessive inherited diseases.
Since they are genetically inherited disorders, the majority of cases manifest in the first year of life. 93 % of our patients were diagnosed during childhood (<14 years). As a result, paediatricians should maintain a high index of suspicion, because early diagnosis and treatment improve the quality of life of patients [27]. Moreover, in severe combined immunodeficiency (SCID), R. Buckley has shown the fact that 36 (97 %) of 37 infants transplanted in her centre in the first 3.5 months of life had survived up to 22 years [28]. This emphasizes the importance of early diagnosis and therapeutic intervention. No new-born screening program for PID is available in our country; however, 26 % (132 patients) were diagnosed at birth by targeted new-born screening for affected families (Table 1). A screening program in our country would be of great benefit in the early case identification of patient with PID, and would lead to an effective enrolment of patients in treatment programs before a life-threatening infection develops. A pilot study is ongoing at our centre using the TREC/KREC assay. The results of this study will help in the developing and adding the TREC/KREC to the national new born screening program.
The mean age at symptoms onset, diagnoses and delay in diagnosis are shown to be variable (Table 2). This reflects the diversity of PIDs. The overall mean for delay in diagnosis for all PIDs was 21.6 months; this is considerably less than what is reported by the EISD registry at 49 months [13]. This finding could be attributed to the low frequency of predominantly antibodies deficiency at 12 % in our registry compare to 55 % in the EISD registry. Patients diagnosed with predominantly antibodies deficiency do have a relatively high delay in their diagnosis [29, 30]; this was also shown in our patients to be at a mean of 44.8 months (Table 2). In addition the mean delay in diagnosis from other regional registries was also less than the EISD registry at 15.4, 18.1 and 28 months in Oman [7], Qatar [31] and Kuwait [6] respectively.
Stem cell transplantation was the mode of treatment in 61 % of patients in the database. The donor commonly was a HLA-genoidentical siblings or phenotypic HLA match, this as a result of the high rate of consanguinity and the number of sibling in the families. For example in 50 % of SCID patients a HLA-genoidentical siblings was the best donor option. The mortality rate in our registry was 10.3 % this in compare to a mortality rate of 7.9 % from the ESID registry [13]; however the mortality rate was variable from other registries around the world and may be related to the severity of the type of PID encountered, the early awareness of PID and/ or the availability of diagnostic and therapeutic modalities as stem cell transplantation. for example the mortality rate were 20, 21.4, 23.4 and 28.8 % in Kuwait [6], Qatar [31], Egypt [8], and Morocco [32] respectively this in contrast to 3.5, 13, and 3.1 % in the UK [1], France [2] and Germany [11] respectively.
BCG vaccine derived from multiple passages of wild-type Mycobacterium bovis is administered routinely at birth to all newborns in Saudi Arabia. BCG vaccine is part of national vaccine program in the developing countries to prevent tuberculosis; however, it is only documented to be protective against meningitis and miliary tuberculosis [33]. Adverse reaction to BCG vaccine is uncommon in immunocompetent individuals and mainly results in local subcutaneous abscess and regional lymphadenopathy, occurring in up to 1 % depending on the vaccine strain [34]. Serious complications in the form of disseminated Mycobacterium bovis disease is common in children with immunodeficiency such as SCID, CGD and IL 12-INF gamma defects and is reported in 30–50 % of SCID patients which compares to our rate of 43 % of SCID patients [35]. BCG-itis in its disseminated form requires an intensive treatment and may result in high morbidity and mortality [36, 37]. Delaying BCG vaccine to at less 6 month of age, where most SCID patients by then are diagnosed, is recommended [38].
Conclusion
This study showed a high incidence and prevalence of PIDs in the country. It is only a single center study and multicenter data collection is needed in order to have a comprehensive analysis of PIDs in the country. Nevertheless, this database is a cornerstone for a PIDs Saudi national registry in the future. Combined immunodeficiencies and SCID, in particular, are not uncommon in the country. Therefore, strategic planning in the prevention and the early diagnosis is needed in order to improve patient outcomes by early intervention and avoiding disease complications. This can be achieved by increasing the public’s and the health care provider’s awareness in PIDs, their clinical features and presentations. Moreover, newborn screening for SCID is an emerging and cost-effective tool in the early Identification of cases [39, 40]. Other key areas of concern include BCG vaccine schedule, consanguinity and the high risk for autosomal recessive inherited disease, and outreach clinical and educational programs to suburban areas for PID patients’ care and management.
References
Edgar J, Buckland M, Guzman D, Conlon N, Knerr V, Bangs C, et al. The United Kingdom Primary Immune Deficiency (UKPID) Registry: report of the first 4 years’ activity 2008–2012. Clin Exp Immunol. 2013;175:68–78.
The French national registry of primary immunodeficiency diseases. Clin Immunol. 2010; 135:264–72.
Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007;27:517–24.
Errante PR, Franco JL, Espinosa-Rosales FJ, Sorensen R, Condino-Neto A. Advances in primary immunodeficiency diseases in Latin America: epidemiology, research, and perspectives. Ann N Y Acad Sci. 2012;1250:62–72.
Ishimura M, Takada H, Doi T, Imai K, Sasahara Y, Kanegane H, et al. Nationwide survey of patients with primary immunodeficiency diseases in Japan. J Clin Immunol. 2011;31:968–76.
Al Herz W. Primary immunodeficiency disorders in kuwait: first report from Kuwait national primary immunodeficiency registry (2004–2006). J Clin Immunol. 2008;28:186–93.
Al-Tamemi S, Elnour I, Dennison D. Primary immunodeficiency diseases in Oman: five years’ experience at Sultan Qaboos University Hospital. WAO J. 2012;5:52–6.
Reda S, Afifi H, Amine M. Primary immunodeficiency diseases in Egyptian children: a single-center study. J Clin Immunol. 2009;29:343–51.
Barbouche MR, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, et al. Primary immunodeficiencies in highly consanguineous North African populations. Ann N Y Acad Sci. 2011;1238:42–52.
Aghamohammadi A, Mohammadinejad P, Abolhassani H, Mirminachi B, Movahedi M, Gharagozlou M, et al. Primary immunodeficiency disorders in Iran: update and new insights from the third report of the national registry. J Clin Immunol. 2014;34(4):478–90.
Gathmann B, Goldacker S, Klima M, Belohradsky BHG, Notheis G, Ehl S. The German national registry for primary immunodeficiencies (PID). Clin Exp Immunol. 2013;173(2):372–80.
Naidoo R, Ungerer L, Cooper M, Pienaar S, Eley B. Primary immunodeficiencies: a 27-year review at a Tertiary Paediatric Hospital in Cape Town. South Afr J Clin Immunol. 2011;31:99–105.
Gathmann B, Binder N, Ehl S, Kindle G, ESID Registry Working Party. The European internet-based patient and research database for primary immunodeficiencies: update 2011. Clin Exp Immunol. 2012;167(3):479–91.
Javier F, Moore C, Sorensen R. Distribution of primary immunodeficiency diseases diagnosed in a pediatric tertiary hospital. Ann Allergy Asthma Immunol. 2000;84:25–30.
Al Attas R, Rahi A. Primary antibody deficiency in Arabs: first report from Eastern Saudi Arabia. J Clin Immunol. 1998;5(18):368–71.
Suliaman F, Al- Ghonaium A, Harfi H. High incidence of severe combined immune deficiency in the Eastern Province of Saudi Arabia. Paediatr Asthma, Allergy Immunol. 2006;19(1):14–8.
Al-Odaib AN, Abu-Amero KK, Ozand PT, Al-Hellani AM. A new era for preventive genetic programs in the Arabian Peninsula. Saudi Med J. 2003;24(11):1168–75.
Alsmadi O, Al-Ghonaium A, Al-Muhsen S, Arnaout R, Al-Dhekri H, Al-Saud B, et al. Molecular analysis of T-B-NK+ severe combined immunodeficiency and Omenn syndrome cases in Saudi Arabia. BMC Med Genet. 2009;10:116.
Alsum Z, Hawwari A, Alsmadi O, Al-Hissi S, Borrero E, Abu-Staiteh A, et al. Clinical, immunological and molecular characterization of DOCK8 and DOCK8-like deficient patients: single center experience of twenty five patients. J Clin Immunol. 2013;33(1):55–67.
Al-Saud B, Al-Sum Z, Alassiri H, Al-Ghonaium A, Al-Muhsen S, Al-Dhekri H, et al. Clinical, immunological, and molecular characterization of hyper-IgM syndrome due to CD40 deficiency in eleven patients. J Clin Immunol. 2013;33(8):1325–35.
Lipstein EA, Vorono S, Browning MF, Green NS, Kemper AR, Knapp AA, et al. Systematic evidence review of newborn screening and treatment of severe combined immunodeficiency. Pediatrics. 2010;25:1226–35.
Al-Herz W, Al-Mousa H. Combined immunodeficiency: the Middle East experience. J Allergy Clin Immunol. 2013;131(3):658–60.
The Central Department of Statistics and Information (CDSI) website: www.cdsi.gov.sa. Accessed 15 Mar 2015.
Ouederni M, Vincent QB, Frange P, Touzot F, Scerra S, Bejaoui M, et al. Major histocompatibility complex class II expression deficiency caused by a RFXANK founder mutation: a survey of 35 patients. Blood. 2011;118(19):5108–18.
Al-Mousa H, Al-Shammari Z, Al-Ghonaium A, Al-Dhekri H, Al-Muhsen S, Al-Saud B, et al. Allogeneic stem cell transplantation using myeloablative and reduced-intensity conditioning in patients with major histocompatibility complex class II deficiency. Biol Blood Marrow Transpl. 2010;16(6):818–23.
Buckley RH. Variable phenotypic expression of mutations in genes of the immune system. J Clin Invest. 2005;115:2974.
Woroniecka M, Ballow M. Office evaluation of children with recurrent infection. Paediatr Clin N Am. 2000;47(6):1211–24.
Buckley R. J Allergy Clin Immunol. 2004;113(4):793–800.
Seymour B, Miles J, Haeney M. Primary antibody deficiency and diagnostic delay. J Clin Pathol. 2005;58:546–7.
Gathmann B, Grimbacher B, Beauté J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157 Suppl 1:3–11.
Ehlayel MS, Bener A, Laban MA. Primary immunodeficiency diseases in children: 15 year experience in a tertiary care medical center in Qatar. J Clin Immunol. 2013;33(2):317–24.
Bousfiha AA, Jeddane L, El Hafidi N, Benajiba N, Rada N, El Bakkouri J, et al. First report on the Moroccan registry of primary immunodeficiencies: 15 years of experience (1998–2012). J Clin Immunol. 2014;34(4):459–68.
Plotkin SA, Orenstein WA, Offit PA. Vaccines. 6th ed. Edinburgh: Elsevier/Saunders; 2013.
Alrabiaah AA, Alsubaie SS, Bukhari EI, Gad A, Alzamel FA. Outbreak of Bacille Calmette-Guérin-related lymphadenitis in Saudi children at a university hospital after a change in the strain of vaccine. Ann Saudi Med. 2012;32(1):4–8.
Bernatowska E, Wolska-Kusnierz B, Pac M, Kurenko-Deptuch M, Zwolska Z, Casanova J, et al. Disseminated bacillus calmette-guérin infection and immunodeficiency. Emerg Infect Dis. 2007;13(5):799–801.
I˙kinciogˇulları A, Dogˇu F, iftci E, nal E, Ertem M, Reisli I, et at. An intensive approach to the treatment of disseminated BCG infection in a SCID patient. Bone Marrow Transplantation. 2002; 30, 45–47.
Paiman S, Siadati A, Mamishi S, Tabatabaie P, Khotaee G. Disseminated mycobacterium bovis infection after BCG vaccination. Iran J Allergy Asthma Immunol. 2006;5(3):133–7.
Marciano B, Huang C, Joshi G, Rezaei N, Carvalho B, Allwood Z, et al. BCG vaccination in patients with severe combined immunodeficiency: complications, risks, and vaccination policies. J Allergy Clin Immunol. 2014;133:1134–41.
Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115(2):391–8.
Chan K, Davis J, Pai SY, Bonilla FA, Puck JM, Apkon M. A Markov model to analyze cost-effectiveness of screening for severe combined immunodeficiency (SCID). Mol Genet Metab. 2011;104:383–9.
Acknowledgments
We would like to thank the following individuals: Dr. Sultan Al- Sedairy Charmaine of the Research Center for the ongoing support. Dr. Edward Devol chairman of the Biostatistics, Epidemiology, and Scientific Computing Department (BESCD) for the support and continuing collaboration, Mansoor Baig for the software development, Shazia sobhani for the database development and management, Samia Alhashim for the statistical analysis, bader Alhablani and Saleh Alageel for the administrative support, we also like to thank our registrars Manal Mohammed and Ahsan Yassen.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Al-Saud, B., Al-Mousa, H., Al Gazlan, S. et al. Primary Immunodeficiency Diseases in Saudi Arabia: a Tertiary Care Hospital Experience over a Period of Three Years (2010–2013). J Clin Immunol 35, 651–660 (2015). https://doi.org/10.1007/s10875-015-0197-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-015-0197-6