
Abstract
Infectious diseases are a significant burden in global healthcare. Pathogens engage with different host defense mechanisms. However, it is currently unknown if there are disease-specific immune signatures and/or if different pathogens elicit common immune-associated molecular entities to common therapeutic interventions. We studied patients enrolled through the Human Immunology Project Consortium (HIPC), which focuses on immune responses to various infections. Blood samples were collected and analyzed from patients during infection and follow-up time points at the convalescent stage. The study included samples from patients with Lyme disease (LD), tuberculosis (TB), malaria (MLA), dengue virus (DENV), and West Nile virus (WNV), as well as kidney transplant patients with cytomegalovirus (CMV) and polyomavirus (BKV) infections. Using an antibody-based assay, we quantified ~ 350 cell surface markers, cytokines, and chemokines involved in inflammation and immunity. Unique protein signatures were identified specific to the acute phase of infection irrespective of the pathogen type, with significant changes during convalescence. In addition, tumor necrosis factor receptor superfamily member 6 (TNR6), C–C Motif Chemokine Receptor 7 (CCR7), and C–C motif chemokine ligand-1 (CCL1) were increased in the acute and convalescent phases across all viral, bacterial, and protozoan compared to blood from healthy donors. Furthermore, despite the differences between pathogens, proteins were enriched in common biological pathways such as cell surface receptor signaling pathway and response to external stimulus. In conclusion, we demonstrated that irrespective of the pathogen type, there are common immunoregulatory and proinflammatory signals.
Similar content being viewed by others


INTRODUCTION
Infectious diseases caused by communicable pathogens including viruses, bacteria, fungi, and parasites are listed by the World Health Organization as among the leading causes of death worldwide [1]. The host–pathogen dyad is a dynamic system, as the virulence factors and host responses are in constant flux during the course of the infection [2]. Pathogens have evolved numerous strategies to overcome innate and adaptive immunity and escape host immune elimination. This balance of host immune response and pathogen counter-defense contributes to the complexity and diversity of pathological infections [3].
Studying host response toward multiple pathogens has proven useful in determining unique and universal elements of host defense mechanisms [4]. In a recent report, we studied two common arthropod-borne infections: Lyme disease (LD), an extracellular bacterial infection, and West Nile virus infection (WNV), an intracellular viral infection and showed similarities in host responses despite the different vectors and the pathogens [5]. Understanding acute and recovery phase immune signatures of the host when exposed to a variety of broad pathogen families (bacteria, viruses, and protozoa) can provide a greater understanding of host defense and pathogen clearance or persistence mechanisms. Pathogens invade, survive, replicate, and colonize through diverse mechanisms involving both cellular and humoral immunity and with the engagement of both innate and adaptive immunity [3]. These pathogens can exist in circulation or reside in the intracellular niches, and they encounter attacks that include those by host cytotoxic T lymphocytes, macrophage and monocyte systems, natural killer (NK) cells, NK T cells, γδ T cells, cytotoxic cytokines, antibodies, and complement cascade [6,7,8]. Infection with mycobacteria, spirochetes, members of the Herpesviridae family, and fungal infections elicit macrophage hyperactivation and granuloma formation, which localize or clear the infection [9]. Once an infection is established, its clearance or persistence in tissue and cellular niches is interconnected to a balance of host immunity, host genetics, pathogen virulence, pathogen mutations, and other innate survival mechanisms [10, 11]. Outcomes are determined by factors related to both the host and pathogen and the extent of host–pathogen interactions.
We hypothesized that phylogenetically diverse microbes (bacterial, viral, protozoal) might share common pathogen subtype-specific host–pathogen interactions and host defense mechanisms while also eliciting unique host immune signatures specific to an individual pathogen. We also predicted that commonalities of immune and molecular profiles might exist across the acute and chronic/recovery phases of infections, regardless of inherent variations across pathogen species. We have conducted a pilot proteomic study on paired blood samples collected from patients, with Lyme disease (LD), tuberculosis (TB), malaria (MLA), cytomegalovirus (CMV), polyomaviridae (BKV), dengue virus (DENV), and West Nile virus (WNV), during infection and later in the convalescent stage. This study tracks the pathways that are crucial for intracellular pathology (all pathogens are intracellular except for Borrelia burgdorferi that causes LD), microbial defense, and the associated post-infection changes in the host. Our proteomic analyses of diverse infection-induced signatures provide a unique opportunity to identify meta-signatures that characterize host–pathogen interactions, inflammatory responses, and the post-inflammatory milieu.
Our study design incorporates longitudinal, paired samples from these diverse patients within each infection group with different infections and interrogates immune pathways that support clearance of a range of pathogens over time. The diversity of infections allows us to identify both common and unique immune pathways. This study improves our understanding of host–pathogen dynamics that could aid in the development of vital disease diagnostics, interventions, and vaccines that are more needed then ever given the global increase in the incidence of emerging and reemerging infectious diseases.
RESULTS
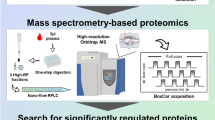
Characteristics of Patients with Bacterial (LD and TB), Protozoan (MLA), and Viral (CMV, BKV, DENV, WNV) Infection. Patients’ age, gender, and the time points of sample collection across seven distinct infections and overall study schematic are presented in Fig. 1 and Table 1. Based on the types of pathogen and diseases, we used either serum (LD, WNV) or plasma (CMV, DENV, TB, BKV, and MLA) isolated from patients’ blood with each infection for the scioCD antibody microarray assay. Blood samples collected 1 month apart from healthy adult individuals (HC) were used as controls. Also, patients with CMV and BKV have undergone kidney transplantation. Samples were collected at two different time points, one at the time of infection or acute phase and the other at the convalescent/recovery phase, the time at which the patients were checked for signs of recovery after they were diagnosed with the disease in the acute phase.

Scheme illustrating specimen collection per infection and protein expression analysis platform. a Barplot demonstrating the dispersion of sample-collection timepoints, gender, and disease state across seven distinct diseases (BKV, cytomegalovirus, dengue, Lyme disease, malaria, tuberculosis, and West Nile virus) and healthy controls. b Overview of the infections studied, specimens collected, and platform used for the data acquisition. scioCD antibody microarray platform profiled the expression of 345 proteins at any given time in plasma or serum isolated from blood.
Distinct and Common Protein Signatures at the Acute Infection Stage Compared to Healthy Controls Were Identified. We identified blood proteins whose expression changed during the acute infection phase when compared to their levels in the blood of healthy normal subjects. Three hundred forty-five blood proteins were analyzed in a multiplexed format with a high intra and intersample reproducibility and < 10% CV. The sensitivity of the assay ranges from 240 aM to 4 fM with a dynamic range of about 5 orders of magnitude. Tumor necrosis factor receptor 6 (TNR6) and C–C motif chemokine ligand 1 (CCL1) were increased in all bacterial, viral, and protozoan infections (Table 2) (full list in Table 2 supplementary). Proteins that were highly elevated in infections compared to controls include intercellular adhesion molecule 1 (ICAM1) [12,13,14,15], TNR6 [16], and vascular cell adhesion molecule 1 (VCAM1) [17] in viral infections; nerve growth factor beta (NGFβ) [18], TNR6 [19], and S100 calcium-binding protein A8/9 (S100A8/9) [20,21,22] in bacteria; CD80, integrin subunit alpha 4 (ITA4) [23], programmed cell death 1 ligand 2 (PD1L2) [24], S100A8/9 [25], transforming growth factor-beta (TGFB1) [26], and TNR6 [27] in protozoan infection. The top 10 proteins specifically increased in each infection type are listed in Fig. 2a. Twenty-one proteins’ expression was significantly changed (6 increased and 15 decreased) in viral infection. Expression of eighteen proteins was significantly changed (8 increased and 10 decreased) in bacterial infection. Twenty-six proteins’ expression was significantly altered (11 increased and 15 decreased) in protozoan infection (Table 2) (full list in Table 2 supplementary). With the cutoff threshold of ≤ 0.05 p value and 1.41-fold change, ICAM1 and CCL27 were the most significantly increased and decreased proteins, respectively, in the viral infection category (Fig. 2b). S100A8/9 and IL5 were the most significantly increased and decreased proteins, respectively, in the bacterial infection category (Fig. 2c). S100A8/9 and TNR16 were the most significantly increased and decreased proteins, respectively, in the protozoan infection category (Fig. 2d).

Analysis to identify significantly increased and decreased proteins and most enriched pathways in the acute infection phase compared to the healthy controls. a The heatmap shows the expression pattern of significant proteins in infection caused by each infectious agents (virus, bacteria, protozoan) with a fold change ≥ 1.4 compared to the healthy controls. b For each category, we identified several uniquely expressed proteins, with which we performed gene set enrichment analysis and identified the significantly affected biological processes at the time of infection.
We performed gene set enrichment analysis on the detected proteins in each sample and identified biological processes that are significantly affected at the time of infection with the different pathogen groups. In bacteria-mediated infections, the most noteworthy biological processes were leukocyte migration (FDR = 1.7E − 12) and immune response (FDR = 6.45E − 12) (Table 3 supplementary). In viral infections, the most noteworthy biological processes were the cell surface signaling pathway (FDR = 12.58E − 13) and the response to external stimuli (FDR = 6.08E − 11) (Table 3 supplementary). In the protozoan category, the most noteworthy biological processes were the cell surface signaling pathway (FDR = 1.7E − 16) and the cytokine-mediated signaling pathway (FDR = 3.35E − 16) (Table 3 supplementary). Apart from these prominent pathways associated with each infection, there are 21 common pathways among all the infections (Table 3 supplementary).
Protein Analysis Identifies Distinct and Common Protein Signatures in Profiled Samples at Convalescent Stage Compared to the Healthy Controls. Next, we compared the expression pattern of significantly increased or decreased proteins at the convalescent stage with a fold change of equal to or higher than 1.41-fold in comparison to healthy controls (Fig. 3a, Table 3) (Table 4 supplementary). C–C motif chemokine ligand 1 (CCL1) and C–C Motif Chemokine Receptor 7 (CCR7) were increased in all bacterial, viral, and protozoan infections. Proteins that were highly elevated in the recovery or convalescent stage compared to healthy controls included CD57, TNR6, and tumor necrosis factor ligand (TNFL6) in viral infection. CCL14 and CD7 were among some of the most decreased proteins in viral infection (Fig. 3b). Angiopoietin 4 (ANGP4), CD9, and immunoglobulin E (IgE) were increased and LFA3, IL5, and CD45RB were among the most decreased proteins in bacterial infection (Fig. 3b). ITA4, CD80, C–C motif chemokine ligand 18 (CCL18), and IL17B were among the increased and PGH1, IL3RB, and IL25 were among the decreased proteins in protozoan infection (Fig. 2 supplementary).

Analysis to identify differentially expressed proteins and most enriched pathways in the convalescent state or recovery phase of infection compared to the healthy controls. a The heatmap shows the expression pattern of top four significantly changed proteins in the convalescent or recovery phase of the infection per infectious agents (virus, bacteria, protozoan) with a fold change ≥ 0.5 compared to the healthy controls. b For each infectious agent, we identified several uniquely expressed proteins, with which we performed gene set enrichment analysis and identified the significantly affected biological processes at the time of recovery.
To assess enriched biological processes at the convalescent state of the infections, we performed gene set enrichment analysis. In bacteria-mediated infections, the most noteworthy biological processes were the immune system process (FDR = 4.49E − 11) and the positive regulation of protein phosphorylation (FDR = 3.52E − 10) (Table 5 supplementary). In viral and protozoan infections, the most noteworthy biological processes were the cell surface signaling pathway (FDR = 3.58E − 8) and the cytokine-mediated signaling pathway (FDR < 1.18E − 8) (Table 5 supplementary). There were 15 common pathways across all the infections (Table 5 supplementary).
Distinct and Common Protein Signatures Comparing Acute and Convalescent Stages of All the 7 Infections. We identified differentially expressed blood proteins in each infection type. There were multiple proteins which were either elevated or depleted at the time of acute infection (Table 6 supplementary). A bubble plot in Fig. 4 shows their names and the degree of their perturbation during acute infection time points. The heatmap in Supplemental Fig. 4a shows the top elevated plasma proteins in the acute phase compared to the convalescent stage for each infection. We have identified the proteins with the highest difference in expression between acute infection and a time point at convalescent phase for each disease and showed the perturbation as longitudinal plots in Fig. 5.

Significantly increased and decreased blood proteins in each infection type were identified by comparing the acute infection phase to the convalescent phase. The bubble plot shows their names and the degree of their perturbation during acute infection time points.

The boxplots display the most differentially changed proteins across the time points of each infection. Non-significant (ns): p < = 1.00e + 00; *1.00e − 02 < p < = 5.00e − 02; **1.00e − 03 < p < = 1.00e − 02; ***1.00e − 04 < p < = 1.00e − 03; ****p < = 1.00e − 04.
We first looked at the proteins identified as being specific to viral infection. IL-9, produced by T cells, has a trend to be upregulated in the acute phase of WNV infection, suggesting that IL-9 may have a role in host defense or viral pathogenicity. In our study, all the CMV-positive patients underwent a kidney transplant and had higher expression of immune checkpoint protein CTLA4 (cytotoxic T lymphocyte-associated protein 4) in the acute infection stage. Expression of ectonucleoside triphosphate diphosphohydrolase 1 (ENTP1) or CD39 protein which catalyzes the hydrolysis of ATP is significantly enhanced in the acute phase of BKV infection. Increased expression of CTLA4 and ENTP1 could be a side-effect of the immunosuppressive drugs that kidney transplant patients are currently prescribed. Plasma BKV positivity is highly associated with co-infection CMV, suggesting possible risk factors for these infections [28]. Chemokine C–C motif chemokine ligand 27 (CCL27) is downregulated in the acute stage of viral infection. Its association with viral infection at the acute or convalescent stage has never been studied in the pathogenesis of viral infections. In DENV, integrin subunit a, integrin subunit alpha 2b (ITA2b or CD41) is crucial for blood coagulation. We observed that ITA2b is significantly downregulated in the acute phase of DENV infection.
Several proteins demonstrated their association with bacterial infection. In LD, immune checkpoint protein B and T lymphocyte attenuator (BTLA) level significantly increased in the acute phase compared to the convalescent period. CD276 level is increased in acute phase of TB compared to convalescent state. Healthy controls have elevated levels of IL-5 compared to individuals with acute bacterial infections. Furthermore, patients with acute bacterial infection continue to have low IL-5 concentrations in the convalescent stage (Fig. 4c supplementary).
Our analysis identified TNR16/CD271, receptor expressed by mesenchymal stem cells to be downregulated in the acute and convalescent stages of malaria. However, to date, TNR16/CD271 has not been reported as being associated with malaria.
We report here higher levels of S100A8/A9 at the time of acute microbial infection and its decline at the convalescent stage supporting its antimicrobial and chemotactic property. Since S100A8/A9 is involved in inflammatory processes induced either by pathogens or in chronic inflammation such as tendinopathy [29], it is an indicator of inflammation rather than one diagnostic for a specific pathogen. A change in its concentration in serum from acute to convalescent phase, however, could serve as a potential diagnostic marker for microbial infection. These calcium-binding proteins are highly expressed in neutrophils and cause neutrophil chemotaxis, which are involved in the immune defense [30]. S100A8 is a marker of neutrophil-mediated inflammation, and there are studies suggesting it as a useful biomarker to distinguish bacterial from viral respiratory infections [31].
Pathway analysis demonstrated that regulating the immune system process was the most significantly enriched biological process (p = 3.1E − 22). Biological processes enriched in individual infections and in bacterial and viral infections are listed in Table 7 supplementary. There are 21 biological processes commonly enriched in between bacterial and viral infections (Table 7 supplementary). Three selected proteins and their relative abundance in viral, bacterial, and protozoan infections at the time of acute infection when compared to the convalescent state are presented in Fig. 4c supplementary.
DISCUSSION
We have performed a comprehensive analysis of circulating proteins in human cohorts infected with phylogenetically distinct pathogens, with the goal of identifying common and unique proteins, mediators, and pathways that contribute to the pathophysiology of infection. The high-throughput proteomic profiling used in this study allowed for sample analytic multiplexity without sacrificing sensitivity and data reproducibility. In this unique pilot study design, we used paired samples from seven different human infections caused by multiple viruses, bacteria, and protozoa to assess proteins and infer pathways activated during active infection and recovery and identified common and unique proteomic profiles of responses to a range of different pathogens.
Human viruses are known to develop suitable strategies for modifying apoptosis in virus-infected cells and in virus-primed T cells. Apoptosis-mediated physiological depletion of T lymphocytes due to viral infection silences the immune response [32]. IL-2, IFN-g, or TNF-a is known to induce the co-expression of TNR6 and ICAM1 on activated B cells. This might suggest a role of ICAM1 and ICAM1-mediated signals in the induction of B cell apoptosis [33, 34]. VCAM1 is another cell-adhesion molecule, known to play a role in regulating T cell-mediated inflammation and pathology, found to be up in the acute phase of viral infection [17]. Thus, our findings underscore the potential role of VCAM-1 in regulating the immune response and inflammatory reactions against viral infections.
Both IgE and ANGP4 are up in the acute and recovery phases of bacterial infection. Most LD patients produce specific IgE antibodies [35], and its concentration is usually higher in patients with TB [36]. ANGP4 promotes angiogenesis, which is crucial in tick-borne encephalitis [37, 38]. Increased expression of CSF3 is uniquely associated with acute bacterial infection, which can be explained as CSF3 is known to be a biomarker for the pathogenesis of the cytokine storm in active TB [39]. Remission of tuberculosis infection could be ascertained by the expression of CD9, an exosome marker, one of the highly expressed proteins in the recovery phase of bacterial infection cohort [40].
Previous studies have reported infection-specific proteins that distinguish bacterial infections from viral [41]. TNR6, a TNF-receptor superfamily expressed on the cell surface of activated human T and B lymphocytes [42], was a common protein upregulated during all acute infections and continues to be up during the recovery phase in viral infection (Figs. 2 and 3). TNR6-mediated apoptosis may have a role in the induction of peripheral tolerance, in the antigen-stimulated suicide of mature T cells, or both [43]. More recently, TNR6 has been reported to be involved in inflammation or cell migration [44]. It was interesting to note that TNR6 continued to be upregulated in the recovery phase of viral infection along with its death ligand TNFL6 and ICAM1.
In malarial infection, levels of CD80 and CCL18 are elevated in both the acute and recovery phases. CD80 is expressed on memory B cells essential for maintaining long-term humoral immunity to infectious organisms, including Plasmodium [45, 46]. Host responses against Plasmodium involve humoral and cell-mediated immunity. Higher expression of S100A8/9 protein in the acute phase of the Plasmodium-infected patients was also accompanied by increased abundance of CCL18 in the convalescent stage, a cytokine produced by myeloid cells that helps in T cell-mediated immunity (Figs. 2 and 3) [47] and tissue repair processes at epithelial surfaces. These myeloid cells also manufacture IL17B that facilitates the communication between leukocytes and epithelial cells, enhancing innate defense mechanisms and tissue repair processes at epithelial surfaces [48, 49]. Knowledge of the biological functions of this cytokine is limited. In our study, IL17B is upregulated during the recovery phase of malaria and, therefore, might possess a defensive role. In malaria infection, MSCs counteract parasite-mediated negative regulation of T cell functions [50].
The Ca + 2 and Zn + 2 binding heterodimeric protein S100A8/9 was a common protein family upregulated during active infections in bacteria and malaria, with the greatest decline during convalescence. Under physiological conditions, these proteins are stored in neutrophils, monocytes, and macrophages, secreted upon tissue/cell damage or cellular stress or activation of phagocytes, serving as a danger signal [51, 52]. Here in inflammatory conditions, we observe that irrespective of the pathogen type, a surge in these proteins is a hallmark of acute infection. S100A8/A9 effectively inhibits the growth of bacteria Borrelia at infectious sites during the initial phase of LD [20], allowing time for the recruitment of phagocytes. Subsequently, S100A9 enhances the phagocytic activity of infiltrating leukocytes, accelerating the clearance of pathogens [21]. Also, targeting S100A8/A9 reduces organ injury by decreasing tissue damage in the lung during TB [22]. S100A8/A9 is found to be elevated in the serum of malaria patients, which may contribute to pathogen immune escape or tolerance [25]. Therefore, we conclude that S100A8/A9 is induced in two infectious diseases studied here and may play a modulatory role in acute infection caused by bacteria and protozoa. Further studies would be needed to evaluate this marker in the context of infection recovery vs resistance, which is beyond the scope of this study.
In the acute phase of all the infections compared to the healthy control, we have identified 21 common upregulated signaling pathways (Fig. 2 supplementary, Table 3 supplementary)—cell surface receptor signaling, response to external stimulus, and cytokine-mediated signaling pathway are few to mention. Understanding of such interactions between the pathogen and host is critical for guiding novel therapies and understanding the factors that lead to the development of active disease. Cell surface receptor signaling is a crucial step where once the bacteria adhere to the surface they exert greater resistance toward clearance by immune factors and establish pathogenesis [53]. In TB, M. tuberculosis binds to cell surface receptors and shifts the balance from the host‐protective apoptotic cell death program toward a lytic form of host cell death to orchestrate the infection process to facilitate its growth, dissemination, and entry into latency [54]. In case of LD, B. burgdorferi activates several intracellular pathogen recognition receptors including cell surface TLRs, which collectively contribute to inflammatory signaling [55, 56]. In malaria, Plasmodium’s entry into the host erythrocytes to establish its pathogenesis requires multiple molecular interactions between the surface proteins of merozoites and cell surface receptors on the host including TLRs and glycosylphosphatidylinositol-mediated signaling [57, 58]. Viruses overcome intrinsic cellular barriers by inducing distinct classes of receptor-mediated endocytosis coupled with receptor-mediated signaling [59]. Virus–receptor interactions play a key regulatory role in viral host range and viral pathogenesis [60]. Integrins are known to function as the cellular receptor for WNV [60], gB and gH/gL are essential for CMV entry [61], sulfated glycosaminoglycans (GAGs), lectins, glycosphingolipid (GSL), proteins with chaperone activity, laminin-binding proteins, are known to be crucial for DENV [62].
Downregulation of IL-5, CCL27, and TNR-16 proteins is unique to bacterial, viral, and malarial infections, respectively, and could provide pathogen-specific monitoring biomarkers for early and recovering infections. TNR-16/CD271 is a surface marker of MSCs and is downregulated in the acute and convalescent phases of malaria (Fig. 3 supplementary). This suggests reduced capacity to combat immune modulation by the pathogen.
In the acute phase of malaria, P-glycoprotein homolog 1 (PGH1) is downregulated, a protein of Plasmodium falciparum, which has been linked to chloroquine, mefloquine, and halofantrine resistance [63]. This may indicate that our cohort is sensitive to antimalarials. In the convalescent phase of malaria, BMP5 a ligand of the TGF-beta superfamily that can lead to the recruitment and activation of SMAD family transcription factors [64] is downregulated. More research is needed to determine whether TGF-beta levels could be a candidate marker for malarial infection or disease severity.
In the acute phase of bacterial infection, TNR11, a TNF-receptor superfamily, is downregulated. They are essential regulators of the interaction between T cells and dendritic cells. Downregulation of TNR11 may impair the differentiation of naive CD4 + T cells toward Th2 cells contributing to the chronic tissue-destructive T cell activity [65]. In the convalescent phase of bacterial infection, CD45RB, CD276, and CCL27 are downregulated. CD45 is a receptor tyrosine phosphatase essential for TCR signaling [66], CD276 belongs to the immunoglobulin superfamily that causes T cell activation and IFN-γ production [67], and CCL27 is a cytokine pivotal role in T cell-mediated inflammation [68]. This indicates that during bacterial infection and recovery, T cell response and activation are low, which may indicate that it has a minor role in defending against bacteria. It was interesting to note that CCL27 and BMP7 are down in the acute phase of viral infection as this may result in amplified virus infection. CCL27-knockout mice have overreactive skin inflammatory responses in a model of psoriasis [69]. BMP7 is a potent antagonist of TGF-β1 and an antifibrotic [70]. In the convalescent phase, IL17C and CCL1 got our attention as both were significantly downregulated. IL17C is a proinflammatory cytokine that reinforces innate immune barriers and stimulates highly inflammatory TH17 cells involved in the pathogenesis of several diseases, including infectious [71]. CCL1 is involved in inflammatory processes through leukocyte recruitment and could play a crucial role in angiogenesis and other viral and tumoral processes [72]. BTLA is the most significantly elevated protein in acute Lyme disease infections. Its expression is high on terminally differentiated B and T cells, where it is known to repress the activation of signal transduction. Moreover, BTLA expression may indicate terminally exhausted lymphocytes. BTLA shares similarities with other immune checkpoints such as PD-1 and CTLA-4 which are the targets of the currently used immunotherapies [73].
Several similar studies have reported protein and gene transcripts associated with the pathogens included in our study [5, 74,75,76]. These studies include basic transcriptomics studies to LC–MS-based platforms to modified aptamer-binding technology (“SOMAmers”). For example, Penn-Nicholson et al. identified signatures for TB including 5-protein signature that included C9, IGFBP-2, CD79A, MXRA-7, and NrCAM. These proteins were elevated in TB progressors compared to non-progressors [74]. The SomaScan Assay included ~ 3000 proteins and the platform in this study included ~ 350 proteins. Of them, only 40 proteins were common in the two platforms. Perhaps because of the study design and the assay platforms the TB-specific proteins identified in Penn-Nicholson et al. and ours do not overlap. The markers proteins. Liu et al. performed [75] reported an 8-gene marker panel to improve clinical prediction of severe dengue progression that included LTF, UQCRQ, CKAP4 as increased and ARNTL, PDGFRB, TGFBR3, RASSF5, GDPD5 as decreased [75]. These transcripts also were not significant in our study, partly, it was a gene expression study, and we profiled proteins, and we did not seek progression markers in our study. Recently, we have published protein signatures on LD and WNV cohorts for their specific markers by using two LC–MS-based proteomic protocols which also identified a different unique set of proteins [5]. The non-overlap of markers is due to the unique antibody-based platform specifically designed to capture cell surface marker and cytokine profiling. The unique discoveries made in these studies could complement each other and provide an overall picture of the pathobiology for further validation.
We acknowledge a few limitations in this study. We used serum and plasma to profile host proteins in different diseases. Even though previous proteomic studies have suggested essentially the same proteomic profiles for serum and plasma, there are differences in clotting factors and fibrinogens, which may affect our ability to compare different cohorts and diseases [77]. Another limitation of our study is the small sample size. The study was part of an ancillary project where the objective was to assess the usefulness of “omic” interrogation of circulating proteins in the blood of different infectious agents. The samples were collected from studies associated with the NIH/NIAID HIPC consortium. As we have listed the studies and centers in the “Materials and Methods” section describing the study subjects and samples, this presents a unique opportunity to assess proteomic profiling of the biobanked blood samples. Since the data presented here was generated from a set of pathogens by design and due to the limited sample numbers, the specificity of the markers identified may not represent all viral, all bacterial, and all microbial infections.
In conclusion, we evaluated the common and unique proteomic profiles of responses to infection by a range of different pathogens. These observations and markers will help generate hypotheses that can be tested by designing studies with larger cohorts to validate the clinical significance of these markers.
MATERIALS AND METHODS
Study Subjects and Samples. The study includes 72 blood (serum or plasma) samples collected from 32 subjects infected with seven different pathogens, obtained through collaboration with six leading academic centers in the US, that participated in the Human Immunology Project Consortium (HIPC) (Fig. 1a). The research has been carried out in accordance with the institutional review board approval, and all the participants provided written informed consent. We received BK virus nephropathy (BKV) samples (n = 4, male = 2, female = 2, mean age = 53.75 years) from kidney transplant recipients consented at UCLA through IRB#11–001387. Cytomegalovirus blood samples (n = 5, male = 2, female = 3, mean age = 38 years) were collected from kidney transplant recipients enrolled at UCSF through IRB approved study #14–13,573 and NIH 3U19AI1281913 study entitled “Mapping Immune Responses to CMV in Renal Transplant Recipients.” Study samples from patients with malaria (n = 4, male = 2, female = 2, mean age = 32.5 years) were obtained from Seattle Children’s Research Institute through IRB#20,162,556. The samples were collected as a part of the Immunization by Mosquito with Radiation Attenuated Sporozoites (IMRAS) trial (NCT01994525). The samples in the study were from the four infection control subjects in the IMRAS trial, i.e., received mosquito bites but no attenuated parasites (i.e., vaccine) during the vaccination phase, and then were challenged with wild-type parasites and became infected. The time points are acute infection (day 5/6 post-challenge infection) and convalescent (day 112 post-challenge infection and following drug treatment ~ day 11 to resolve the infection). Samples from the tuberculosis cohort (n = 5, male = 4, female = 1, mean age = 38.25 years) were obtained from La Jolla Institute for Immunology through IRB#VD-143. Dengue study participants (n = 5, male = 2, female = 3, mean age = 9.4 years) in Managua, Nicaragua, were consented through UC Berkeley IRB #2010–06-1649 and the Nicaraguan Ministry of Health IRB# NIC-MINSA/CNDR CIRE-01/10/06–13. The blood samples were collected as part of the Pediatric Hospital-based Dengue Study in Nicaragua, with multiple funding sources since 2005 (ongoing). Two recent funding sources are NIH grants P01 AI106695 (EH) and U19 AI118610 (EH/AS—HIPC). Samples from the West Nile virus cohort (n = 4, male = 2, female = 2, mean age = 55.75 years) studies were obtained through H-305333 and 0510000728 at Baylor College of Medicine and Yale School of Medicine. The Lyme disease cohort (n = 5, male = 2, female = 3, mean age = 61.6 years) was obtained through IRB# 1,112,009,475 at Yale School of Medicine. Both WNV and LD samples were collected as a part of HIPC grant entitled “Systems Immune Profiling of Divergent Responses to Infection” (NIH award AI 089992). Healthy control (n = 4, male = 2, female = 2, mean age = 43.75 years) samples were also included for comparison. All samples were stored at −20° C until use.
ScioCD Assay. In brief, 72 blood samples (Fig. 1a) were labeled at an adjusted protein concentration for 2 h with the fluorescent dye scioDye 2 (Sciomics, Neckargemünd, Germany). Sample labeling and incubation were performed as previously described in detail [78, 79]. In brief, we prepared a reference sample by pooling all samples included and labeled with a second dye (scioDye 1). After 2 h, the labeling reaction was stopped, and the buffer exchanged to phosphate-buffered saline (PBS). For improved assay robustness and differentiation power, each sample was competitively incubated together with a common reference sample on one microarray slide in a reference-based dual-color approach as described in detail before [80]. The samples were then assayed on scioCD antibody microarrays (Sciomics) targeting 351 different proteins by 517 antibodies, each in four replicates. Array surfaces were blocked with scioBlock (Sciomics) on a Hybridization Station 4800 PRO (Tecan, Grödig, Austria), and the samples were subsequently incubated competitively with the reference sample using a dual-color approach. After incubation for 3 h, the slides were thoroughly washed with 1 × PBSTT (phosphate-buffered saline containing Tween and Triton), rinsed with 0.1 × PBS as well as with water and subsequently dried with nitrogen.
Data Acquisition and Analysis. Slide scanning was conducted using a Powerscanner (Tecan, GmbH, Grödig, Austria) with constant instrument laser power and photomultiplier settings. Spot segmentation was performed with GenePix Pro 6.0 (Molecular Devices, Union City, USA). The median signal intensities of the spots were used. The background was not subtracted as an earlier one, and continuous assessments proved for antibody microarray analyses that background subtraction is not reducing but inducing artifacts. The acquired raw data were analyzed using the linear models for microarray data (limma) package of R-Bioconductor after uploading the median signal intensities. Isoforms in the dataset were merged and replaced with mean value resulting in 345 unique proteins (Table 1 supplementary). For normalization, a specialized invariant Lowess method was applied [80]. A linear model was fitted using limma, and differences between sample groups were calculated based on the fitted group means generated by the linear model and are presented as log-fold changes (logFC) calculated for the basis 2.
Data Analysis. Data was analyzed with the objective of identifying differences between (1) active infection state and healthy controls, (2) convalescent state and healthy controls, and (3) active infection state and convalescent state. Preprocessing and analysis were conducted by using Python (3.8.8) and R (4.0.4). Acquired raw data were analyzed using the linear models for the microarray data (LIMMA 3.46) package of R-Bioconductor after uploading the median signal intensities. For normalization, a specialized invariant Lowess method was applied. For analysis of the samples, a multi-factorial linear model was fitted with LIMMA resulting in a two-sided t-test or F-test based on moderated statistics. Multiple contrasts were defined between disease timepoints and disease classes to discern protein signatures. Differential expression results were represented by using the R packages ggplot2 (3.3.5) and pheatmap (1.0.12). Next to infection type, the sample type was accounted for using the information as an additional factor in the linear model. All presented p values were adjusted for multiple testing by controlling the false discovery rate according to Benjamini and Hochberg. Proteins were defined as differential for logFC > 0.5 (1.4 fold change) and an adjusted p value < 0.05. Moreover, descriptive statistics were captured by using the Python packages matplotlib (3.3.4), numpy (1.19.5), pandas (1.2.0), scipy (1.7.2), and seaborn (0.11.1). Comparisons for disease signature were defined by observing differences between the initial acute disease timepoint and the final recovery timepoint. The healthy control cohort (n = 4) was normalized using the R package sva (3.38.0) to standardize observed variation with Combat. Differences in protein abundance between samples or sample groups are presented as log-fold changes (logFC) calculated for the base 2. Gene set enrichment was conducted on the statistically significant proteins (p value <= 0.05) through the Python package gseapy (0.10.4) observed from the comparison of acute infection timepoint with the healthy control as well as the disease in comparison to the healthy control. Combined score is described as c = log(p) * z, where c is the combined score, p = is the Fisher exact test p value, and z is the z-score for deviation from expected rank (81). Gseapy utilized the human catalog from KEGG 2021, GO Biological Process 2021, as well as GO Molecular Function 2021. The study schematic is presented in Fig. 1b.
Data Availability
Proteomics data generated from this study has been deposited in IMMPORT database with accession number SDY2008, under workspace ID 6886 “HIPC Multicenter Acute Infection IOF.”
References
Mortality, G.B.D., and C. Causes of Death. 2015. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 385 (9963): 117–171.
Casadevall, A., and L.A. Pirofski. 2000. Host-pathogen interactions: Basic concepts of microbial commensalism, colonization, infection, and disease. Infection and Immunity. 68 (12): 6511–6518.
Thakur, A., H. Mikkelsen, and G. Jungersen. 2019. Intracellular pathogens: Host immunity and microbial persistence strategies. Journal of Immunology Research 2019: 1356540.
McGuire, M.K., A.Z. Randall, A.E. Seppo, K.M. Jarvinen, C.L. Meehan, D. Gindola, et al. 2020. Multipathogen analysis of IgA and IgG antigen specificity for selected pathogens in milk produced by women from diverse geographical regions: The INSPIRE study. Frontiers in Immunology 11: 614372.
Boada, P., B. Fatou, A.A. Belperron, T.K. Sigdel, K.K. Smolen, Z. Wurie, et al. 2022. Longitudinal serum proteomics analyses identify unique and overlapping host response pathways in Lyme disease and West Nile virus infection. Frontiers in Immunology 13: 1012824.
Zhao, Y., L. Lin, Z. Xiao, M. Li, X. Wu, W. Li, et al. 2018. Protective role of gammadelta T cells in different pathogen infections and its potential clinical application. Journal of Immunology Research. 2018: 5081634.
Ismail, N., J.P. Olano, H.M. Feng, and D.H. Walker. 2002. Current status of immune mechanisms of killing of intracellular microorganisms. FEMS Microbiology Letters. 207 (2): 111–120.
Shepherd, F.R., and J.E. McLaren. 2020. T cell immunity to bacterial pathogens: mechanisms of immune control and bacterial evasion. International Journal of Molecular Sciences 21 (17).
Hilhorst, M., T. Shirai, G. Berry, J.J. Goronzy, and C.M. Weyand. 2014. T cell-macrophage interactions and granuloma formation in vasculitis. Frontiers in Immunology 5: 432.
Nikolich-Zugich, J. 2008. Ageing and life-long maintenance of T-cell subsets in the face of latent persistent infections. Nature Reviews Immunology 8 (7): 512–522.
Kaufmann, S.H., and B.D. Walker. 2006. Host-pathogen interactions. Current Opinion in Immunology 18 (4): 371–373.
Sedmak, D.D., D.A. Knight, N.C. Vook, and J.W. Waldman. 1994. Divergent patterns of ELAM-1, ICAM-1, and VCAM-1 expression on cytomegalovirus-infected endothelial cells. Transplantation 58 (12): 1379–1385.
Girmanova, E., I. Brabcova, J. Klema, P. Hribova, M. Wohlfartova, J. Skibova, et al. 2012. Molecular networks involved in the immune control of BK polyomavirus. Clinical and Developmental Immunology 2012: 972102.
Yeh, T.M., S.H. Liu, K.C. Lin, C. Kuo, S.Y. Kuo, T.Y. Huang, et al. 2013. Dengue virus enhances thrombomodulin and ICAM-1 expression through the macrophage migration inhibitory factor induction of the MAPK and PI3K signaling pathways. PLoS ONE 8 (1): e55018.
Dai, J., P. Wang, F. Bai, T. Town, and E. Fikrig. 2008. Icam-1 participates in the entry of West Nile virus into the central nervous system. Journal of Virology 82 (8): 4164–4168.
Varanasi, V., A.A. Khan, and A.V. Chervonsky. 2014. Loss of the death receptor CD95 (Fas) expression by dendritic cells protects from a chronic viral infection. Proceedings of the National Academy of Sciences USA 111 (23): 8559–8564.
Ou, R., M. Zhang, L. Huang, R.A. Flavell, P.A. Koni, and D. Moskophidis. 2008. Regulation of immune response and inflammatory reactions against viral infection by VCAM-1. Journal of Virology 82 (6): 2952–2965.
Minnone, G., F. De Benedetti, L. Bracci-Laudiero. 2017. NGF and its receptors in the regulation of inflammatory response. International Journal of Molecular Sciences 18 (5).
O’Donnell, J.A., C.L. Kennedy, M. Pellegrini, C.J. Nowell, J.G. Zhang, L.A. O’Reilly, et al. 2015. Fas regulates neutrophil lifespan during viral and bacterial infection. Journal of Leukocyte Biology 97 (2): 321–326.
Lusitani, D., S.E. Malawista, and R.R. Montgomery. 2003. Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of Borrelia burgdorferi. Infection and immunity. 71 (8): 4711–4716.
Wang, S., R. Song, Z. Wang, Z. Jing, S. Wang, and J. Ma. 2018. S100A8/A9 in inflammation. Frontiers in Immunology 9: 1298.
Gopal, R., L. Monin, D. Torres, S. Slight, S. Mehra, K.C. McKenna, et al. 2013. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. American Journal of Respiratory and Critical Care Medicine 188 (9): 1137–1146.
Jian, J.Y., S.I. Inoue, G. Bayarsaikhan, M. Miyakoda, D. Kimura, K. Kimura, et al. 2021. CD49d marks Th1 and Tfh-like antigen-specific CD4+ T cells during Plasmodium chabaudi infection. International Immunology 33 (8): 409–422.
Karunarathne, D.S., J.M. Horne-Debets, J.X. Huang, R. Faleiro, C.Y. Leow, F. Amante, et al. 2016. Programmed death-1 ligand 2-mediated regulation of the PD-L1 to PD-1 axis is essential for establishing CD4(+) T cell immunity. Immunity 45 (2): 333–345.
Kim, T.S., Y.J. Kang, J.Y. Kim, S. Lee, W.J. Lee, Y. Sohn, et al. 2015. Up-regulated S100 calcium binding protein A8 in Plasmodium-infected patients correlates with CD4(+)CD25(+)Foxp3 regulatory T cell generation. Malaria Journal 14: 385.
Drewry, L.L., and J.T. Harty. 2020. Balancing in a black box: Potential immunomodulatory roles for TGF-beta signaling during blood-stage malaria. Virulence. 11 (1): 159–169.
Punsawad, C., P. Viriyavejakul, C. Setthapramote, and S. Palipoch. 2015. Enhanced expression of Fas and FasL modulates apoptosis in the lungs of severe P. falciparum malaria patients with pulmonary edema. International Journal of Clinical and Experimental Pathology 8 (9): 10002–10013.
Toyoda, M., D.P. Puliyanda, N. Amet, L. Baden, V. Cam, R. Radha, et al. 2005. Co-infection of polyomavirus-BK and cytomegalovirus in renal transplant recipients. Transplantation 80 (2): 198–205.
Crowe, L.A.N., M. McLean, S.M. Kitson, E.G. Melchor, K. Patommel, H.M. Cao, et al. 2019. S100A8 & S100A9: Alarmin mediated inflammation in tendinopathy. Science and Reports 9 (1): 1463.
Ryckman, C., K. Vandal, P. Rouleau, M. Talbot, and P.A. Tessier. 2003. Proinflammatory activities of S100: Proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. The Journal of Immunology 170 (6): 3233–3242.
Havelka, A., K. Sejersen, P. Venge, K. Pauksens, and A. Larsson. 2020. Calprotectin, a new biomarker for diagnosis of acute respiratory infections. Science and Reports 10 (1): 4208.
Wattre, P., V. Bert, and D. Hober. 1996. Apoptosis and human viral infections. Annales de Biologie Clinique (Paris) 54 (5): 189–197.
Moller, P., C. Henne, A. Schmidt, A. Eichelmann, F. Leithauser, S. Bruderlein, et al. 1992. Expression of APO-1, a cell surface molecule mediating apoptosis, during normal B cell ontogeny and in B cell tumors. Co-expression and coregulation of APO-1 and ICAM-1 (CD54) in germinal central cells. Verhandlungen der Deutschen Gesellschaft fur Pathologie 76: 237–242.
Moller, P., C. Henne, F. Leithauser, A. Eichelmann, A. Schmidt, S. Bruderlein, et al. 1993. Coregulation of the APO-1 antigen with intercellular adhesion molecule-1 (CD54) in tonsillar B cells and coordinate expression in follicular center B cells and in follicle center and mediastinal B-cell lymphomas. Blood 81 (8): 2067–2075.
Benach, J.L., B.L. Gruber, J.L. Coleman, G.S. Habicht, and M.G. Golightly. 1986. An IgE response to spirochete antigen in patients with Lyme disease. Zentralblatt für Bakteriologie, Mikrobiologie und Hygiene. Series A: Medical Microbiology, Infectious Diseases, Virology, Parasitology 263 (1–2): 127–132.
Ohrui, T., K. Zayasu, E. Sato, T. Matsui, K. Sekizawa, and H. Sasaki. 2000. Pulmonary tuberculosis and serum IgE. Clinical and Experimental Immunology 122 (1): 13–15.
Jones, N., and D.J. Dumont. 2000. Tek/Tie2 signaling: New and old partners. Cancer and Metastasis Reviews 19 (1–2): 13–17.
Bogovic, P., A. Kastrin, S. Lotric-Furlan, K. Ogrinc, T. Avsic Zupanc, M. Korva, et al. 2022. Comparison of laboratory and immune characteristics of the initial and second phase of tick-borne encephalitis. Emerging Microbes & Infections 11 (1): 1647–1656.
Boni, F.G., I. Hamdi, L.M. Koundi, K. Shrestha, and J. Xie. 2022. Cytokine storm in tuberculosis and IL-6 involvement. Infection, Genetics and Evolution 97: 105166.
Khonina, N.A., S.D. Nikonov, S.V. Shpilevskii, O. Leplina, E. Shevela, L.V. Sakhno, et al. 2000. Specific features of immunity in patients with different forms of pulmonary tuberculosis. Problemy Tuberkuleza 1: 30–32.
Oved, K., A. Cohen, O. Boico, R. Navon, T. Friedman, L. Etshtein, et al. 2015. A novel host-proteome signature for distinguishing between acute bacterial and viral infections. PLoS ONE 10 (3): e0120012.
Oehm, A., I. Behrmann, W. Falk, M. Pawlita, G. Maier, C. Klas, et al. 1992. Purification and molecular cloning of the APO-1 cell surface antigen, a member of the tumor necrosis factor/nerve growth factor receptor superfamily. Sequence identity with the Fas antigen. Journal of Biological Chemistry 267 (15): 10709–10715.
Ji, G., A. Gu, F. Hu, S. Wang, J. Liang, Y. Xia, et al. 2009. Polymorphisms in cell death pathway genes are associated with altered sperm apoptosis and poor semen quality. Human Reproduction 24 (10): 2439–2446.
Devel, L., N. Guedeney, S. Bregant, A. Chowdhury, M. Jean, and P. Legembre. 2022. Role of metalloproteases in the CD95 signaling pathways. Frontiers in Immunology 13: 1074099.
Brown, S.L., J.J. Bauer, J. Lee, E. Ntirandekura, and J.S. Stumhofer. 2022. IgM(+) and IgM(-) memory B cells represent heterogeneous populations capable of producing class-switched antibodies and germinal center B cells upon rechallenge with P. yoelii. Journal of Leukocyte Biology 112 (5): 1115–1135.
De, S.L., S. May, K. Shah, M. Slawinski, S. Changrob, S. Xu, et al. 2021. Variable immunogenicity of a vivax malaria blood-stage vaccine candidate. Vaccine. 39 (19): 2668–2675.
Schutyser, E., A. Richmond, and J. Van Damme. 2005. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. Journal of Leukocyte Biology 78 (1): 14–26.
Rutz, S., X. Wang, and W. Ouyang. 2014. The IL-20 subfamily of cytokines–from host defence to tissue homeostasis. Nature Reviews Immunology 14 (12): 783–795.
Blumberg, H., D. Conklin, W.F. Xu, A. Grossmann, T. Brender, S. Carollo, et al. 2001. Interleukin 20: Discovery, receptor identification, and role in epidermal function. Cell 104 (1): 9–19.
Kalkal, M., M. Tiwari, R.S. Thakur, V. Awasthi, V. Pande, D. Chattopadhyay, et al. 2021. Mesenchymal stem cells: A novel therapeutic approach to enhance protective immunomodulation and erythropoietic recovery in malaria. Stem Cell Reviews and Reports 17 (6): 1993–2002.
Sedaghat, F., and A. Notopoulos. 2008. S100 protein family and its application in clinical practice. Hippokratia 12 (4): 198–204.
Xia, C., Z. Braunstein, A.C. Toomey, J. Zhong, and X. Rao. 2017. S100 proteins as an important regulator of macrophage inflammation. Frontiers in Immunology 8: 1908.
Vinod, V., S. Vijayrajratnam, A.K. Vasudevan, and R. Biswas. 2020. The cell surface adhesins of Mycobacterium tuberculosis. Microbiological Research 232: 126392.
Stutz, M.D., M.P. Clark, M. Doerflinger, and M. Pellegrini. 2018. Mycobacterium tuberculosis: Rewiring host cell signaling to promote infection. Journal of Leukocyte Biology 103 (2): 259–268.
Petnicki-Ocwieja, T., and A. Kern. 2014. Mechanisms of Borrelia burgdorferi internalization and intracellular innate immune signaling. Frontiers in Cellular and Infection Microbiology 4: 175.
Tang, X., Y. Cao, G. Arora, J. Hwang, A. Sajid, C.L. Brown, et al. 2021. The Lyme disease agent co-opts adiponectin receptor-mediated signaling in its arthropod vector. Elife 10.
Soni, R., D. Sharma, P. Rai, B. Sharma, and T.K. Bhatt. 2017. Signaling strategies of malaria parasite for its survival, proliferation, and infection during erythrocytic stage. Frontiers in Immunology 8: 349.
Salinas, N.D., and N.H. Tolia. 2016. Red cell receptors as access points for malaria infection. Current Opinion in Hematology 23 (3): 215–223.
Grove, J., and M. Marsh. 2011. The cell biology of receptor-mediated virus entry. Journal of Cell Biology 195 (7): 1071–1082.
Maginnis, M.S. 2018. Virus-receptor interactions: The key to cellular invasion. Journal of Molecular Biology 430 (17): 2590–2611.
Vanarsdall, A.L., and D.C. Johnson. 2012. Human cytomegalovirus entry into cells. Current Opinion in Virology 2 (1): 37–42.
Hidari, K.I., and T. Suzuki. 2011. Dengue virus receptor. Tropical Medicine and Health 39 (4 Suppl): 37–43.
Reed, M.B., K.J. Saliba, S.R. Caruana, K. Kirk, and A.F. Cowman. 2000. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 403 (6772): 906–909.
Kotepui, K.U., P. Kwankaew, F.R. Masangkay, A. Mahittikorn, and M. Kotepui. 2022. Transforming growth factor-beta concerning malarial infection and severity: a systematic review and meta-analysis. Infectious Diseases and Tropical Medicine 7 (10).
van Roon, J.A., C.A. Glaudemans, J.W. Bijlsma, and F.P. Lafeber. 2003. Differentiation of naive CD4+ T cells towards T helper 2 cells is not impaired in rheumatoid arthritis patients. Arthritis Research & Therapy 5 (5): R269–R276.
Lim, B., R.M. Sutherland, Y. Zhan, G. Deliyannis, L.E. Brown, and A.M. Lew. 2006. Targeting CD45RB alters T cell migration and delays viral clearance. International Immunology 18 (2): 291–300.
Chapoval, A.I., J. Ni, J.S. Lau, R.A. Wilcox, D.B. Flies, D. Liu, et al. 2001. B7–H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nature Immunology 2 (3): 269–274.
Morales, J., B. Homey, A.P. Vicari, S. Hudak, E. Oldham, J. Hedrick, et al. 1999. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proceedings of the National Academy of Sciences USA 96 (25): 14470–14475.
Davila, M.L., M. Xu, C. Huang, E.R. Gaddes, L. Winter, M.T. Cantorna, et al. 2022. CCL27 is a crucial regulator of immune homeostasis of the skin and mucosal tissues. iScience 25 (6): 104426.
Hao, Z.M., M. Cai, Y.F. Lv, Y.H. Huang, and H.H. Li. 2012. Oral administration of recombinant adeno-associated virus-mediated bone morphogenetic protein-7 suppresses CCl(4)-induced hepatic fibrosis in mice. Molecular Therapy 20 (11): 2043–2051.
Nies, J.F., and U. Panzer. 2020. IL-17C/IL-17RE: Emergence of a unique axis in T(H)17 biology. Frontiers in Immunology 11: 341.
Tamguney, G., J. Van Snick, and H. Fickenscher. 2004. Autocrine stimulation of rhadinovirus-transformed T cells by the chemokine CCL1/I-309. Oncogene 23 (52): 8475–8485.
Demerle, C., L. Gorvel, and D. Olive. 2021. BTLA-HVEM couple in health and diseases: Insights for immunotherapy in lung cancer. Frontiers in Oncology 11.
Penn-Nicholson, A., T. Hraha, E.G. Thompson, D. Sterling, S.K. Mbandi, K.M. Wall, et al. 2019. Discovery and validation of a prognostic proteomic signature for tuberculosis progression: A prospective cohort study. PLoS Medicine 16 (4): e1002781.
Liu, Y.E., S. Saul, A.M. Rao, M.L. Robinson, O.L. Agudelo Rojas, A.M. Sanz, et al. 2022. An 8-gene machine learning model improves clinical prediction of severe dengue progression. Genome Medicine 14 (1): 33.
Sigdel, T.K., P. Boada, M. Kerwin, P. Rashmi, D. Gjertson, M. Rossetti, et al. 2023. Plasma proteome perturbation for CMV DNAemia in kidney transplantation. PLoS ONE 18 (5): e0285870.
Nambu, M., S. Nishiumi, T. Kobayashi, T. Masuda, S. Ito, M. Yoshida, et al. 2020. Effects of differences in pre-analytical processing on blood protein profiles determined with SWATH-MS. Journal of Proteomics 223: 103824.
Jansa, V., T. Klancic, M. Pusic, M. Klein, E. Vrtacnik Bokal, H. Ban Frangez, et al. 2021. Proteomic analysis of peritoneal fluid identified COMP and TGFBI as new candidate biomarkers for endometriosis. Science and Reports 11 (1): 20870.
Luan, F.L., F. Trillsch, A. Henger, F. Eichinger, S. Norman, H. Appelman, et al. 2009. A pilot study of gene expression-based categorization of pancreas transplant biopsies. Transplantation 87 (2): 222–226.
Schroder, C., A. Jacob, S. Tonack, T.P. Radon, M. Sill, M. Zucknick, et al. 2010. Dual-color proteomic profiling of complex samples with a microarray of 810 cancer-related antibodies. Molecular and Cellular Proteomics 9 (6): 1271–1280.
Chen, E.Y., C.M. Tan, Y. Kou, Q. Duan, Z. Wang, G.V. Meirelles, et al. 2013. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14: 128.
Acknowledgements
We thank Dr. Angel Balmaseda for facilitating samples collected as part of the Pediatric Hospital-based Dengue Study in Nicaragua. We thank Aruna Dharshan De Silva, Faculty of Medicine, General Sir John Kotelawala Defense University, Sri Lanka; Dhammika Vidanagama, National Tuberculosis Reference Laboratory, Sri Lanka; Bandu Gunasena National Hospital for Respiratory Diseases, Sri Lanka, for recruitment of active TB patients. We thank Xiaomei Wang for helping with WNV cohort.
Funding
The authors would like to acknowledge the funding support from NIH/NIAID (HIPC IOF) Grant U19-AI118610 to MS, NIH/NIAID Grant U19-AI118610-05 to AF-S and EH, NIH NIAID Grant U19AI118626 to BP, NIH/NIAID Grant U19-AI-128913 to MS and ER, NIH/NIAID Grant AI089992 to RRM and LKB, NIH NIAID Grant U19AI118626 to Bjoern Peters, NIH/NIAID Grant P01 AI106695 to EH, and NIH NIAID Grant R01 AI099631 to Angel Balmaseda.
Author information
Authors and Affiliations
Contributions
Tara Sigdel (Conceptualization, Data curation, Writing), Swastika Sur (Data curation, Writing), Patrick Boada (Data curation, Formal analysis, Writing) Suzanne M. McDermott (Resources, Data curation, Writing) Cecilia Arlehamn (Resources, Data curation, Writing), Kristy O. Murray (Resources, Data curation, Writing), Linda K. Bockenstedt (Resources, Data curation, Writing), Maggie Kerwin (Methodology, Data curation, Writing), Elaine Reed (Resources, Data curation, Writing), Eva Harris (Resources, Data curation, Writing), Ken Stuart (Resources, Data curation, Writing), Bjoern Peters (Resources, Data curation, Writing), Ana Sesma (Resources, Data curation, Writing), Ruth R. Montgomery (Resources, Data curation, Writing), Minnie M. Sarwal (Conceptualization, Resources, Data curation, Supervision, Writing).
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
This study was performed in line with the principles of the Declaration of Helsinki. The research was carried out in accordance with the institutional review board approval, and all the participants provided written informed consent. The detailed information is as follows. We received BK virus nephropathy (BKV) samples from kidney transplant recipients consented at UCLA through IRB#11–001387. Cytomegalovirus blood samples and healthy control samples were collected from kidney transplant recipients enrolled at UCSF through IRB approved study #14–13573 and NIH 3U19AI1281913 study entitled “Mapping Immune Responses to CMV in Renal Transplant Recipients.” Study samples from patients with malaria were obtained from Seattle Children’s Research Institute through IRB#20162556. The samples were collected as a part of the Immunization by Mosquito with Radiation Attenuated Sporozoites (IMRAS) trial (NCT01994525). Study samples from the tuberculosis cohort were obtained from La Jolla Institute for Immunology through IRB#VD-143. Dengue study participants in Managua, Nicaragua, were consented through UC Berkeley IRB #2010–06-1649 and the Nicaraguan Ministry of Health IRB# NIC-MINSA/CNDR CIRE-01/10/06–13. Samples from the West Nile virus cohort studies were obtained through H-305333 and 0510000728 at Baylor College of Medicine and Yale School of Medicine. The Lyme disease cohort was obtained through IRB# 1112009475 at Yale School of Medicine.
Consent for Publication
Informed consent was obtained from all recruited subjects for publishing the results of the study.
Competing Interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Tara K. Sigdel, Swastika Sur, and Patrick Boada are joint first authors
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sigdel, T.K., Sur, S., Boada, P. et al. Proteome Analysis for Inflammation Related to Acute and Convalescent Infection. Inflammation 47, 346–362 (2024). https://doi.org/10.1007/s10753-023-01913-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-023-01913-3