
Summary
Tepotinib (MSC2156119J) is an oral, potent, highly selective MET inhibitor. This open-label, phase I study in healthy volunteers (EudraCT 2013-003226-86) investigated its mass balance (part A) and absolute bioavailability (part B). In part A, six participants received tepotinib orally (498 mg spiked with 2.67 MBq [14C]-tepotinib). Blood, plasma, urine, and feces were collected up to day 25 or until excretion of radioactivity was <1% of the administered dose. In part B, six participants received 500 mg tepotinib orally as a film-coated tablet, followed by an intravenous [14C]-tepotinib tracer dose (53–54 kBq) 4 h later. Blood samples were collected until day 14. In part A, a median of 92.5% (range, 87.1–96.9%) of the [14C]-tepotinib dose was recovered in excreta. Radioactivity was mainly excreted via feces (median, 78.7%; range, 69.4–82.5%). Urinary excretion was a minor route of elimination (median, 14.4% [8.8–17.7%]). Parent compound was the main constituent in excreta (45% [feces] and 7% [urine] of the radioactive dose). M506 was the only major metabolite. In part B, absolute bioavailability was 72% (range, 62–81%) after oral administration of 500 mg tablets (the dose and formulation used in phase II trials). In conclusion, tepotinib and its metabolites are mainly excreted via feces; parent drug is the major eliminated constituent. Oral bioavailability of tepotinib is high, supporting the use of the current tablet formulation in clinical trials. Tepotinib was well tolerated in this study with healthy volunteers.
Similar content being viewed by others

Introduction
The mesenchymal–epithelial transition (MET) factor is a receptor tyrosine kinase with a single high-affinity ligand, hepatocyte growth factor (HGF) [1]. HGF binding leads to dimerization and autophosphorylation of MET, activating multiple downstream signaling pathways, including mitogen-activated protein kinase, phosphatidylinositol 3-kinase, signal transducer and activator of transcription protein, and nuclear factor kappa-light-chain-enhancer of activated B cells [1].
Oncogenic MET may not only initiate or promote tumorigenesis, but may also drive resistance to other therapies [2, 3]. Accordingly, exon 14 skipping and high-level amplification of MET are considered primary tumor-driving alterations in non-small cell lung cancer (NSCLC) [2, 4]. In addition, MET amplification is recognized as a secondary oncogenic driver alteration mediating resistance to epidermal growth factor receptor inhibition [2, 5]. Several inhibitors of the MET pathway are currently in development as potential antitumor therapies [2, 3, 6, 7].
Tepotinib (MSC2156119J) is an oral, highly selective, potent adenosine triphosphate-competitive inhibitor of MET, with >1000-fold selectivity for MET over 236 out of 241 other kinases tested, and >200-fold selectivity over the other five [8]. In the first-in-man trial, tepotinib doses of up to 1400 mg/day were administered in patients with solid tumors without reaching the maximum tolerated dose; 500 mg once daily was subsequently established as the recommended phase II dose (RP2D) based on a translational modeling approach [9]. Tepotinib alone or in combination has been shown to be active in various preclinical tumor models [8, 10,11,12,13,14,15,16]. It is currently being studied in clinical trials involving patients with hepatocellular carcinoma (NCT01988493, NCT02115373) [17, 18] and NSCLC (NCT01982955, NCT02864992, NCT03940703) [16, 19,20,21,22].
The purpose of the present study (EudraCT 2013-003226-86) was the characterization of the pharmacokinetics (PK), drug disposition, and metabolism of tepotinib in healthy volunteers. In part A of the study, [14C]-tepotinib was administered orally to evaluate the absorption, distribution, metabolism, and excretion of tepotinib. The availability of [14C]-labeled tepotinib, in conjunction with recent advances in the clinical use of microtracer doses and ultrasensitive bioanalytical methods [23], allowed the evaluation of the PK of tepotinib after intravenous (IV) dosing and its absolute bioavailability in part B of this study. For orally administered drugs such as tepotinib, characterization of the PK profile following IV dosing is of highest value for correct interpretation of distribution and elimination [24]. The combination of both investigations (mass balance and absolute bioavailability) in this study represented an opportunity to obtain a comprehensive understanding of the PK of tepotinib, supporting the ongoing formulation development and its phase II program.
Materials and methods
Study design
This was an open-label, single-center, phase I study in two parts, designed to investigate the mass balance, metabolism, and absolute bioavailability of tepotinib in two groups of six healthy male volunteers (EudraCT 2013-003226-86). Part A of the study was designed to evaluate the mass balance of tepotinib, identify major routes of elimination, determine the unbound fraction of tepotinib in plasma and its blood/plasma distribution, and identify and quantify metabolites of tepotinib in plasma, urine, and feces. Part B was designed to determine the absolute bioavailability of the phase II tablet formulation of tepotinib.
Part A
In part A of the study, six healthy volunteers received a single oral dose of 498 mg tepotinib spiked with 2.67 MBq [14C]-tepotinib (five capsules) on day 1. Participants were hospitalized from day 1 to day 16 of the study, and further assessed on days 19, 22, and 25 (trial end). Participants fasted for at least 10 h overnight prior to oral tepotinib administration, taken with a high-calorie, high-fat-content breakfast. Blood samples for quantitation of tepotinib and total radioactivity in blood and plasma were taken pre-dose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 30, 34, 48, 60, 72, 96, 120, 144, 168, 192, 216, 240, 264, 312, 360, 432, and 576 h after dosing. Separate blood samples were taken pre-dose and at 4, 12, 24, 60, 120, and 240 h post-dose for metabolite identification, and at 4, 8, 12, and 24 h post-dose for determination of the unbound fraction of tepotinib in plasma. Pre-dose feces and urine samples were collected on day 1 and day 2, respectively. Urine was collected at 4- to 12-h intervals on day 1 and every 24 h thereafter up to day 16. Feces were collected every 24 h up to day 16. Provision was made to collect further samples up to day 39 if >1% of the administered dose was detectable in the total (urine and feces) excreta per day on two consecutive measurements.
Part B
On day 1 of part B, six healthy volunteers received a single oral dose of 500 mg tepotinib (one tablet), followed by a delayed IV bolus injection of [14C]-tepotinib (53–54 kBq) 4 h later to cope with the risk of non-linear PK of the IV tracer dose [23]. Participants fasted for at least 10 h overnight prior to oral tepotinib administration, which was taken with a high-calorie, high-fat-content breakfast. Participants were hospitalized from day 1 to day 7 of the study, and further assessed on days 9, 11, and 14 (study end). Blood samples were taken pre-dose and at 0.25, 0.5, 1, 1.5, 2, 3, 4 (just before IV [14C]-tepotinib administration), 6, 8, 10, 12, 16, 24, 30, 34, 48, 60, 72, 96, 120, 144, 192, 240, and 312 h after oral tepotinib administration. Blood samples for the determination of [14C]-tepotinib were taken at 4 h (pre-IV dose) and 4.05, 4.25, 4.5, 5, 5.5, 6, 7, 8, 10, 12, 14, 16, 20, 28, 34, 38, 52, 64, 76, 100, 124, 144, 192, 240, and 312 h, in relation to the oral tepotinib administration.
Participants
Eligible participants were healthy male volunteers aged 18–55 years with a body mass index (BMI) 18–30 kg/m2 and body weight >50 kg. Other inclusion criteria were: no tobacco smoking for ≥6 months before screening and body temperature, blood pressure, and pulse rate within normal ranges. Individuals were excluded from the study if they had any medical condition likely to interfere with study outcomes/assessments, history of clinically relevant disease, any clinically relevant abnormality in screening laboratory parameters/electrocardiogram (ECG), treatment with metabolic enzyme inhibitors/inducers, positivity for hepatitis B surface antigen, hepatitis C virus antibody or human immunodeficiency virus antibody, use of prescription-only medicine/over-the-counter medication within 2 weeks of study initiation, or excessive consumption of xanthine-containing drinks.
Study medication and selection of doses
Oral tepotinib (MSC2156119J, Merck KGaA, Darmstadt, Germany) was formulated as 100 mg film-coated tablets. [14C]-tepotinib, radiolabeled on one carbon per molecule (specific activity, 54.6 mCi/mmol; radiochemical purity, 99.7%), was synthesized by Quotient Bioresearch (Rushden, UK). For part A, tablets were crushed and hard gelatin capsules were prepared to contain approximately 100 mg tepotinib and 0.5 MBq [14C]-tepotinib (~117 μg). For the absolute bioavailability part, a solution for injection was prepared to contain 0.05 MBq [14C]-tepotinib (~11.7 μg) per 1 mL of solution. The definition of the dose of oral [14C]-tepotinib required for determination of mass balance and quantification of tepotinib metabolites was based on preclinical data obtained following oral administration of [14C]-tepotinib to pigmented rats. For dosimetry calculations, the assumption was made that the levels of uptake and retention by respective tissues were the same in man as in experimental animals. Administration of 2.5 MBq (70.1 μCi, 0.585 mg) [14C]-tepotinib chosen for part A of this trial was expected to lead to a radiation burden of 0.5 mSv, which is below 1 mSv, as required by the International Commission on Radiological Protection (ICRP) Category IIa. The dosimetry calculation was carried out by the United Kingdom’s Health Protection Agency Centre for Radiation, Chemical and Environmental Hazards.
The single IV tracer dose of approximately 11.7 μg (in 1 mL) of [14C]-tepotinib (equivalent to 50 kBq [1.4 μCi]), used in part B of the trial to determine the absolute oral bioavailability, was in keeping with regulations regarding tracer doses set forth in ICH M3(R2) [25]. As this dose was expected to lead to a radiation burden below 0.1 mSv, which complies with the ICRP Category I limit of 0.1 mSv, no separate dosimetry was carried out for the IV tracer dose.
The RP2D of tepotinib of 500 mg was chosen to determine PK characteristics at a dose strength that most likely represents the therapeutic dose.
Analytical methods
Bioanalysis of total plasma concentrations of unlabeled tepotinib was performed by Quintiles (Ithaca, New York, USA) using a validated liquid chromatography–tandem mass spectrometry (LC-MS/MS) method. In brief, the isotopically labeled internal standard [2H]-MSC2156119A was added to 20 μL K2-ethylenediaminetetraacetic acid (EDTA) plasma. Tepotinib was extracted by protein precipitation with acetonitrile/water (20 + 80, v/v). Samples were centrifuged and 10 μL aliquots of the supernatant were injected onto an Acquity Charged Surface Hybrid™ (CSH) C18 column (2.1 × 50 mm; 1.7 μM; Waters, Milford, Massachusetts, USA). Separation was carried out using a mobile phase gradient (eluent A: water/formic acid [100 + 1, v/v]; eluent B: acetonitrile/formic acid [100 + 1, v/v]) with a flow rate of 0.6 mL/min. Detection was performed in the positive ionization mode on an AB Sciex API-4000 triple quadrupole mass spectrometer (MS; AB Sciex, Framingham, Massachusetts, USA) with selected reaction monitoring (m/z, 493.3–112.1 for tepotinib and 496.3–115.1 for its deuterated internal standard). The lower limit of quantification (LLOQ) was 5.00 ng/mL, and the calibration standard responses were linear over the range between 5.00 and 2500 ng/mL using a weighted (1/x2) linear regression.
The determination of unbound tepotinib plasma concentrations, following equilibrium dialysis, was performed by Quintiles (Ithaca, New York, USA) using previously qualified LC-MS/MS analytical methods. Plasma (300 μL) was dialyzed against 100 mM sodium phosphate buffer (pH 7.4) in an orbital shaker at 37 °C for 18 h. The deuterated internal standard [2H]-MSC2156119A and 100 mM sodium phosphate buffer (pH 7.4) were added to 5 μL of the dialyzed samples and centrifuged. Supernatant (30 μL) was diluted with acetonitrile/water (50 + 50, v/v) and centrifuged, and 5–20 μL aliquots were injected onto an XBridge C18 column (2.1 × 20 mm; 5 μm; Waters, Milford, Massachusetts, USA). Separation was carried out using a mobile phase gradient (eluent A: 5 mM ammonium bicarbonate in water; eluent B: 5 mM ammonium bicarbonate in methanol) with a flow rate of 1.2 mL/min. Mass spectrometric detection of tepotinib and its internal standard was performed as described above. The LLOQ was 0.015 ng/mL in plasma dialysates and 1.00 ng/mL in plasma. Calibration standards between 0.015 and 30.0 ng/mL (plasma dialysates), and between 1.00 ng/mL and 1000 ng/mL (plasma) were calculated by weighted (1/x2) quadratic regression analysis, respectively.
Total radioactivity in blood, plasma, urine, and feces samples, and quick counts in urine and feces samples were determined at the bioanalytical laboratory of PRA (Assen, The Netherlands) using liquid scintillation counting (LSC). Scintillation cocktail Ultima Gold™ (5 mL; Perkin Elmar, Waltham, Massachusetts, USA) was directly spiked into 250 μL plasma and 1000 μL urine samples, respectively, whereas 300 μL aliquots of blood samples were solubilized and decolorized before adding 18 mL of the scintillation cocktail prior to LSC. Feces samples were (quantitatively) homogenized with one or two weight equivalents of water, and an accurately weighted aliquot of approximately 500 mg of the homogenate was dried at 50 °C for at least 3 h. After addition of 100 μL combustion aid, the dried samples were combusted in a sample oxidizer model 307 (Perkin Elmer, Waltham, Massachusetts, USA) using 7 mL CarboSorb® E as an absorber agent for carbon dioxide. At the end of the combustion cycle, the absorber was mixed with 13 mL of the scintillant PermaFluor® E. All scintillation counting using either low-level or normal count modes was carried out in duplicate using a Tri-Carb 3100 TR liquid scintillation analyzer (Perkin Elmer, Waltham, Massachusetts, USA), in compliance with analytical procedures validated by the PRA.
The accelerator mass spectrometry (AMS) analysis of [14C]-tepotinib in plasma samples in part B of the trial was performed by Eckert & Ziegler Vitalea Science (Davis, California, USA) using a validated method that involves isolation of the analyte by ultra-high-performance liquid chromatography (UPLC) fractionation, followed by the quantification of the 14C content in collected fractions using AMS [26]. Aliquots (300 μL) of K3-EDTA plasma samples were fortified with excess (nanograms) of unlabeled tepotinib, prior to extraction in order to create uniform total concentration levels of the analytes in all study samples, and to eliminate the potential for concentration-dependent recovery in the method. Following protein precipitation and further chemical steps, 50 μL of the reconstituted extracts were injected onto a Waters Acquity UPLC instrument equipped with a CSH fluoro-phenyl analytical column (3.0 × 100 mm; 1.7 μm; Waters, Milford, Massachusetts, USA) and an ethylene bridged hybrid C18 (2.1 × 5 mm; 1.7 μm; Waters, Milford, Massachusetts, USA) pre-column. Separation of [14C]-tepotinib from potentially interfering metabolites or degradants that may have been present in the study samples was carried out using a mobile phase gradient (eluent A: 5 mM ammonium acetate in water; eluent B: acetonitrile) with a flow rate of 0.75 mL/min. Liquid chromatography fractions corresponding to tepotinib were collected and prepared for AMS measurement, graphitized with a known amount of carrier carbon using a two-step process that involved oxidation to gaseous CO2 followed by reduction to solid, elemental graphite. Samples were then introduced into the BioMICADAS AMS instrument (PSI/ETH, Zurich, Switzerland). The LLOQ was 0.0883 pg/mL.
Metabolite profiling and identification
Metabolite profiling and structure elucidation in plasma, urine, and feces were performed at the Institute of Drug Metabolism and Pharmacokinetics, Merck KGaA, Grafing, Germany, using a radiometric UPLC method coupled to a high-resolution MS. For urine and feces, metabolites were identified in a single pooled sample per matrix and subject (urine pool of 4–120 h and feces pool of 0–192 h), that covered at least 90% of the total radioactivity of the respective matrix excreted. Intervals with low radioactivity were not used for the generation of the pooled samples to avoid dilution, and to increase the signal-to-noise ratio of the respective chromatograms.
The metabolite structure elucidation was performed using the linear trap quadrupole Orbitrap XL hybrid Fourier transform (FT) MS (Thermo Scientific, Dreieich, Germany) coupled to a Waters Acquity UPLC system. Full-scan FT spectra were acquired for measurement of the masses of molecular ions of parent and metabolites. MS2 fragmentation spectra were acquired after high-energy collisional dissociation of the appropriate molecular ions. Allocation and confirmation of the proposed metabolites to the radioactive peaks were performed using an API4000 QTrap (AB Sciex, Darmstadt, Germany) MS with simultaneous splitting using a fraction collection system (CTC HTS PAL Fraction Collector, Axel Semrau, Sprockhövel, Germany), and offline radioactive measurement using a Topcount NXT® microplate scintillation counter (PerkinElmer, Rodgau, Germany).
Pharmacokinetic analyses
The PK parameters of tepotinib, total radioactivity, and metabolites were evaluated by standard non-compartmental methods using the software Phoenix WinNonlin (v.6.3, Certara INC, Princeton, New Jersey, USA). The maximum plasma concentration (Cmax) and time to reach maximum concentration (tmax) were derived directly from the plasma concentration–time data. Lag time (tlag) was the time prior to the first measurable (non-zero) concentration. The area under the plasma concentration–time curve from 0 to t (last measurable concentration; AUC0–t) was calculated using a mixed log–linear trapezoidal method. Area under the plasma concentration–time curve until the last sampling time (AUCall), which corresponds to AUC0–240 h within the observation period, was used for metabolite calculations. Amounts excreted in urine, feces, and total (urine + feces) were calculated as the sum of amounts (concentration multiplied by volume) for each collection from time 0 to time t. The terminal rate constant (λz) was used to estimate the terminal half-life (t½) (ln 2/λz), and the area under the concentration–time curve from time 0 to infinity (AUC0–∞). Total body clearance (CL) and apparent body clearance (CL/f) were determined by dose/AUC0–∞, and total volume of distribution (Vz) and apparent volume of distribution (Vz/f) were calculated by dose/(AUC0–∞*λz) after IV and oral administration, respectively. Volume of distribution of the central compartment (Vc) was derived by dividing the dose by the initial drug plasma concentration after IV administration. Absolute bioavailability was calculated as F = (AUCoral/doseoral)/(AUCIV/doseIV) × 100. The ratios (%) AUCtepotinib or metabolite/AUCtotal radioactivity were calculated as a percentage of the molar AUCs.
Safety assessment
Treatment-emergent adverse events (TEAEs), changes in laboratory tests, ECG, vital signs, and physical examinations were assessed and graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.0. All safety endpoints across both parts of the study were analyzed descriptively.
Results
Participants
A total of 12 male healthy volunteers were enrolled into the study, starting on December 11, 2013; the last participant’s final visit took place on May 2, 2014. In part A (n = 6), median age was 45.0 years (range, 25–54 years), and median BMI was 27.0 kg/m2 (range, 20.4–28.9 kg/m2). Five participants were Caucasian and one was of mixed parentage Caucasian/black or African/American. In part B (n = 6), median age was 22.5 years (range, 21–24 years), median BMI was 22.85 kg/m2 (range, 21.1–25.7 kg/m2), and all participants were Caucasian. All volunteers in both parts of the trial received treatment as scheduled.
Part A
Mass balance and routes of excretion
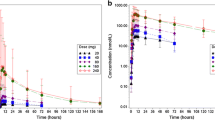
Median recovery of total radioactivity in excreta (feces plus urine) was 92.5% (range, 87.1–96.9%) of the orally administered [14C]-tepotinib dose (Fig. 1). Radioactivity was predominantly excreted in feces (median, 78.7%; range, 69.4–82.5%), and only to a minor extent in urine (median, 14.4%; range, 8.8–17.7%; Table 1). In feces, first radioactivity was detected after 24–48 h, and excretion was noted in all participants up to 360 h post-dose. In urine, radioactivity was observed within the first collection period of 4 h, and up to 360 h after dosing (with values below the LLOQ at 312 and 336 h after dosing in two participants). For all subjects, collection was stopped already at day 16 because <1% of the [14C]-tepotinib dose was recovered in excreta on two consecutive measurements, i.e. days 15 and 16.

Median cumulative recovery of [14C]-radioactivity in urine and feces and combined (total). Pie chart shows respective geometric mean values
Oral PK of tepotinib and [14C]-tepotinib-related total radioactivity
Concentrations of tepotinib and total radioactivity in plasma increased slowly, with a median tmax of 9 h, and a median tlag of 2 and 2.5 h, respectively (Table 2). Comparison between the plasma concentration–time profile of total radioactivity, and tepotinib (as shown in Fig. 2) suggests the formation of circulating metabolites with elimination rate-limited kinetics – a conclusion that is supported by the longer apparent t½ of 38 h for total radioactivity versus 33 h for tepotinib. Accordingly, higher exposure values for AUC0–t, AUC0–∞, and Cmax, and smaller values for CL/f and Vz/f, were observed for total radioactivity compared with tepotinib (Table 2). A median of only 14.4% of the radioactive dose was excreted via the kidneys. This is consistent with the small geometric mean value of 1.2 L/h estimated for the renal clearance of total radioactivity, which indicates that renal elimination contributes to the overall clearance of tepotinib, and its metabolites only to a minor extent. The blood:plasma ratio of total radioactivity was approximately 0.8 over time, and the respective concentration–time profile observed for total radioactivity in blood paralleled with that observed in plasma measured at the same time points. This blood:plasma ratio is also noted for Cmax and AUC (Table 2), indicating that some radioactivity is distributed into blood cells when taking into account the median hematocrit of 0.445 observed in the six healthy volunteers on day 1 pre-dose. The unbound fraction of tepotinib in plasma was evaluated at four time points around tmax, (i.e. 4, 8, 12, and 24 h after oral administration), with median values ranging between 1.8% and 2.4%. The lowest and highest free fractions observed in individual participants were 1.65% and 3.25%, respectively. The ex vivo protein binding data are in agreement with results derived from in vitro studies, in that the unbound fraction of tepotinib in human plasma ranged from 1.6–3.4% over the concentration range between 0.3 and 10 μM [27]. Tepotinib has been shown to bind primarily to plasma proteins [28]; human serum albumin and α-acid glycoprotein were previously identified as the main binding partners [29].

Mean plasma concentration–time curve for tepotinib and total radioactivity (linear scale [top panel] and log-linear scale [bottom panel]) (n = 6)
Metabolite identification and profiling in plasma and excreta
A total of ten phase I and phase II metabolites were detected. This included the chiral metabolite M506, which was elucidated to be formed by keto oxidation of the piperidine ring of tepotinib in the alpha position of the nitrogen. Of note, the formation of the respective enantiomers of this chiral metabolite was quantified in an additional analysis, applying an enantioselective LC-MS/MS method. M668 was likely formed by direct N-glucuronidation. Demethylation on the methyl-piperidine moiety formed metabolite M478. M524–1, −2, and − 3; M522–1 and − 2; and the two diastereomeric N-oxides M508–1 and − 2 were formed by further oxidation, or oxidation and demethylation. Figure 3 shows the proposed structures of tepotinib metabolites, including quantitative information in plasma, feces, and urine. In plasma, proposed structures could be allocated to all three radioactive peaks observed. These consisted of tepotinib and two metabolites, M506 and M668, which accounted for 55%, 41%, and 4% of the AUCall of total 14C-radioactivity, respectively. M506 had a median tmax of 24 h (range, 24–60 h). Its apparent median t½ was 92 h (range, 66–145 h), based on data from four healthy volunteers. The minor plasma metabolite M668 had a median tmax of 12 h (range, 12–24 h) and an apparent t½ of 55.8 h, based on data from a single volunteer.

Proposed metabolic pathway for tepotinib. Structural formulae are postulates and have yet to be confirmed by nuclear magnetic resonance analysis and/or chemical synthesis, except for those proven by synthesis: racemic M506 (MSC2569775), M508–1 (MSC2489309), M508–2 (MSC2489308), and M478 (MSC2157042)
In feces, 93% of the recovered radioactivity could be structurally identified. The parent compound excreted in feces accounted for 45% of the total administered radioactive dose. The des-methyl metabolite M478 accounted for 9%, the direct N-glucuronide M668 for 6%, and the racemic metabolite M506 for about 3%. Altogether, the oxidized metabolites M524–1, −2, and − 3, and M522–1 and − 2 each accounted for ≤2% of the administered dose.
In urine, 91% of the recovered radioactivity could be allocated to chemical structures. Most prominent was the parent drug tepotinib, comprising 7% of the total radioactive dose. The N-oxide metabolites M508–1 and − 2 accounted for about 2% and 3% of the total administered dose, respectively.
Overall, 52% of the total radioactive administered dose excreted in feces or urine was associated with the parent compound and approximately 23% and 7% with phase I and phase II metabolites, respectively (Fig. 3).
Part B
Oral and IV PK of tepotinib
Six healthy volunteers received a single oral dose of 500 mg tepotinib followed by an IV bolus injection of [14C]-tepotinib (actual individual doses ranged from 53 to 54 kBq) 4 h later. Figure 4 suggests a triphasic decline of plasma concentrations, including a sharp drop during the first 15 min, followed by a second decline phase of up to 6–12 h before a final mono-exponential decrease in concentrations. The initial sharp decline in concentrations is mirrored by the small geometric mean value of 34.6 L estimated for Vc, which indicates that tepotinib rapidly distributes out of the central compartment (Table 3). In comparison to Vc, a much higher geometric mean value for Vz (573.6 L) was observed, which points to pronounced distribution of tepotinib into peripheral compartments. The geometric mean CL was 12.8 L/h, and a geometric mean elimination t½ of 31.2 h could be estimated from the terminal slope of the plasma concentration–time profile after bolus injection of the IV [14C]-tracer dose. After parallel single oral administration of 500 mg tepotinib in the current phase II tablet formulation, tlag was 0.5 h and tmax was 10 h. Tepotinib concentrations declined in a mono-exponential fashion with an apparent t½ of 28.3 h, similar in range to the t½ values estimated after IV dosing. Geometric mean values for Vz/f and CL/f of 726.0 L and 17.8 L/h, respectively (Table 3), were slightly larger after oral than after IV dosing.

Mean plasma concentration–time curves for tepotinib after oral and IV administration (log-linear scale)
Bioavailability
At the current recommended phase I dose of tepotinib of 500 mg/day [9, 30], and using the tablet formulation being administered in ongoing clinical phase II trials, tepotinib had a geometric mean absolute bioavailability of 72% (range, 62–81%), with low inter-individual variability (geometric CV, 10.8%).
Safety
The single oral dose of 500 mg tepotinib was well tolerated. Nine of 12 healthy volunteers experienced 19 TEAEs, 17 of which were grade one in severity (according to NCI-CTCAE). The grade two TEAEs were abdominal pain and presyncope. All TEAEs had resolved by the end of the study, except for one incidence of grade one arthralgia reported by a subject in part B; however, this was considered to be unrelated to the study medication. Eight TEAEs were judged to be treatment related; diarrhea and abdominal pain were reported in two patients each. There were no withdrawals due to TEAEs and neither deaths, serious adverse events, nor other significant adverse events were reported.
Discussion
This phase I, open-label study was conducted to investigate the mass balance, to identify and profile metabolites, and to assess the absolute bioavailability of tepotinib in two groups of healthy male volunteers. During early stages of clinical development, knowledge of absolute bioavailability is often missing, although this information can be extremely helpful to guide development of oral drugs [30]. The availability of [14C]-labeled material, as part of a standard mass balance study, is an opportunity to investigate absolute bioavailability in parallel with minimal additional effort [23]. The use of a tracer IV dose reduces formulation work and, in line with ICH M3(R2) [31], may also not require generation of additional preclinical IV data to cover administration in humans. To optimize the approach, both investigations can even be performed in the same group of participants [32]. However, this approach was not applied in this study, due to practical considerations related to uncertainties regarding the duration of the excreta collection periods in part A of the study, which turned out to be shorter than initially expected.
Following single oral administration of 498 mg tepotinib spiked with 2.67 MBq [14C]-tepotinib to male healthy volunteers, the overall median recovery of the radioactive dose in excreta was 92.5%. For a study to provide appropriate information about the excretion of a compound, the European Medicines Agency recommends that recovery of radioactivity in urine and feces should be at least 90% of the radioactive dose [33]. Therefore, we conclude that the study provides a good and reliable assessment of the mass balance of tepotinib and, consequently, of its elimination pathways.
Radioactivity was predominantly recovered in feces, accounting for 78% of the radioactive dose, while only approximately 14% of radioactivity was excreted via urine. Unchanged tepotinib was identified as the main source of radioactivity, accounting in total for 52%, and in feces for 45%, of the radioactive dose. Recovery of a high fraction of unchanged drug in feces, after oral administration, in a mass balance study often suggests incomplete absorption from the gastrointestinal tract. However, the advantage of the design of this study is that the results of part B provided insight into gastrointestinal drug absorption. In this case, the high absolute bioavailability of tepotinib of 72%, determined in part B, pointed to a high absorbed fraction of orally dosed tepotinib, which indicates that incomplete absorption was not the main root cause for the high level of radioactivity associated with unchanged tepotinib recovered in feces. In support of this, preclinical results from an excretion study with [14C]-tepotinib in bile-canaliculated rats indeed showed that a predominant fraction of radioactivity is excreted unchanged via the bile. Since tepotinib is known to be a substrate of the drug efflux transporter P-glycoprotein [29], active transport of the parent drug (via bile) may also represent a relevant elimination pathway of tepotinib in humans. Interestingly, none of the individual concentration–time profiles derived from tepotinib or total radioactivity showed any secondary peaks. This may suggest that neither tepotinib nor its metabolites (potentially released during its intestinal passage, e.g. formed by hydrolysis of the [direct N-]glucuronide M668 or reduction of the N-oxide M508), are re-absorbed at relevant quantities. However, we cannot exclude the absence of distinct secondary peaks being due to a slow absorption rate.
Ten metabolites of tepotinib were identified in this clinical trial. The identification of metabolites in plasma and excreta was almost complete, and far above the recommended threshold of 80% [25]. Overall, the influence of metabolism on the elimination of tepotinib appears to be modest. Only approximately 30% of the radioactive dose can be related to the structurally identified metabolites in excreta; a value much smaller than the 52% related to unchanged drug. Although the drug-metabolizing enzymes involved in the formation of all of the identified metabolites have not yet been completely determined, it can nevertheless be concluded from the data that the contribution of a single enzyme, e.g. cytochrome P450 isoforms or uridine diphosphate-glucuronosyltransferases, to the sum of either phase I or II metabolic pathways does not account for more than 25% of tepotinib’s overall elimination. In addition, the low total clearance observed after IV administration in relation to human liver blood flow further suggests that tepotinib is subject to only minor pre-systemic metabolism (hepatic and/or intestinal) following oral administration. Based on the results of this study, we consider that there is a low risk for clinically significant alterations to the PK of tepotinib, either by inhibition or induction of drug-metabolizing enzymes through the use of concomitant medications (drug–drug interactions) or by functional polymorphisms of these enzymes.
M506 was the only major metabolite detected in plasma (i.e. >10% of total drug-related exposure, as specified in current guidelines) [25, 31, 34]. While M506 accounted for 41% of the total radioactivity exposure in plasma, it only accounted for 3% of radiolabeled drug in excreta, which, also in the absence of any related secondary metabolite, indicates that this is not an important route of elimination (Fig. 3). Interestingly, M506 was not identified in previous in vitro metabolism studies with [14C]-labeled tepotinib using hepatocytes from multiple species, including humans. The trace amount of M506 and its slow formation rate in hepatocytes likely impeded its detection by LSC. The apparent discrepancy between the prominent appearance of this metabolite in the systemic circulation, and the small fraction excreted may be related to a number of factors. M506 was found to be highly protein-bound, with a low affinity for blood cells. Thus, a low volume of distribution, combined with a low clearance, might explain the relative over-representation of M506 in plasma. Further results from ongoing investigations will provide more information about the PK of this chiral metabolite, and will also contribute to a better understanding of the relevance of enantiomers of M506.
In summary, tepotinib has high absolute bioavailability (72%) with low inter-individual variability, providing consistent and predictable dose delivery, and relative insensitivity to factors that can increase variability, such as drug–drug interactions. This contrasts with many small-molecule kinase inhibitors that have poor oral bioavailability and, therefore, also have variable and less predictable PK [30]. Moreover, the late tmax, in conjunction with the long t½ of tepotinib of 30 h, results in small peak–trough fluctuations at steady-state, and should enable continuous and constant inhibition of the MET receptor with once-daily dosing. Tepotinib has a large volume of distribution, indicating preferential partitioning into tissues, and indeed has been consistently found at higher concentrations in xenograft tumors than in the plasma of mice [8], which further supports its target inhibition. Overall, the PK of tepotinib as assessed in this study showed very favorable characteristics for further development in clinical trials.
In this study, a single 500 mg dose of tepotinib was well tolerated by healthy volunteers; although 75% experienced TEAEs, most were mild and resolved rapidly. Tepotinib has also been found to be well tolerated in phase I/Ib and phase II clinical trials in patients with solid tumors, which have further characterized its side-effect profile [9, 17, 18, 21, 35, 36]. Ongoing phase II trials in NSCLC and hepatocellular carcinoma will add further insight into the side-effect profile and efficacy of tepotinib [16,17,18, 20,21,22, 35].
Conclusions
This study has demonstrated that the tablet formulation of tepotinib at the RP2D of 500 mg once daily has high oral bioavailability, supporting its use in phase II studies. Tepotinib exhibits low-to-moderate clearance and a high volume of distribution, resulting in a long plasma t½. Thus, its PK profile is considered favorable for once-daily dosing to ensure continuous target inhibition. Hepatic elimination is the major pathway in humans and tepotinib is primarily excreted unchanged via feces, and only to a minor extent via urine. Ten phase I and II metabolites were identified in total (plasma and excreta), with chiral M506 being the only major circulating metabolite that warrants further characterization. The results also indicate that the potential for drug–drug interactions affecting the PK of tepotinib is low, irrespective of whether concomitant medications induce or inhibit drug-metabolizing enzymes.
References
Drilon A, Cappuzzo F, Ou SI, Camidge DR (2017) Targeting MET in lung Cancer: will expectations finally be MET? J Thorac Oncol 12:15–26. https://doi.org/10.1016/j.jtho.2016.10.014
Wu YL, Soo RA, Locatelli G, Stammberger U, Scagliotti G, Park K (2017) Does c-met remain a rational target for therapy in patients with EGFR TKI-resistant non-small cell lung cancer? Cancer Treat Rev 61:70–81. https://doi.org/10.1016/j.ctrv.2017.10.003
Bouattour M, Raymond E, Qin S, Cheng AL, Stammberger U, Locatelli G, Faivre S (2018) Recent developments of c-met as a therapeutic target in hepatocellular carcinoma. Hepatology 67:1132–1149. https://doi.org/10.1002/hep.29496
Van Der Steen N, Giovannetti E, Pauwels P, Peters GJ, Hong DS, Cappuzzo F, Hirsch FR, Rolfo C (2016) cMET exon 14 skipping: from the structure to the clinic. J Thorac Oncol 11:1423–1432. https://doi.org/10.1016/j.jtho.2016.05.005
Ahsan A (2016) Mechanisms of resistance to EGFR tyrosine kinase inhibitors and therapeutic approaches: an update. Adv Exp Med Biol 893:137–153. https://doi.org/10.1007/978-3-319-24223-1_7
Garajova I, Giovannetti E, Biasco G, Peters GJ (2015) C-met as a target for personalized therapy. Transl Oncogenomics 7:13–31. https://doi.org/10.4137/TOG.S30534
Miranda O, Farooqui M, Siegfried JM (2018) Status of agents targeting the HGF/c-met Axis in lung Cancer. Cancers 10:280. https://doi.org/10.3390/cancers10090280
Bladt F, Faden B, Friese-Hamim M, Knuehl C, Wilm C, Fittschen C, Gradler U, Meyring M, Dorsch D, Jaehrling F, Pehl U, Stieber F, Schadt O, Blaukat A (2013) EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-met inhibitors. Clin Cancer Res 19:2941–2951. https://doi.org/10.1158/1078-0432.ccr-12-3247
Falchook GS, Kurzrock R, Amin HM, Xiong W, Fu S, Piha-Paul S, Janku F, Eskandari G, Catenacci DV, Klevesath M, Bruns R, Stammberger U, Johne A, Bladt F, Friese-Hamim M, Girard P, El Bawab S, Hong DS (2020) First-in-man phase I trial of the selective MET inhibitor tepotinib in patients with advanced solid tumors. Clin Cancer Res 26:1237–1246. https://doi.org/10.1158/1078-0432.CCR-19-2860
Bladt F, Friese-Hamim M, Ihling C, Wilm C, Blaukat A (2014) The c-met inhibitor MSC2156119J effectively inhibits tumor growth in liver cancer models. Cancers (Basel) 6:1736–1752. https://doi.org/10.3390/cancers6031736
Sohn J, Liu S, Parinyanitikul N, Lee J, Hortobagyi GN, Mills GB, Ueno NT, Gonzalez-Angulo AM (2014) cMET activation and EGFR-directed therapy resistance in triple-negative breast cancer. J Cancer 5:745–753. https://doi.org/10.7150/jca.9696
Bill KL, Garnett J, Ma X, May CD, Bolshakov S, Lazar AJ, Lev DC, Pollock RE (2015) The hepatocyte growth factor receptor as a potential therapeutic target for dedifferentiated liposarcoma. Lab Investig 95:951–961. https://doi.org/10.1038/labinvest.2015.62
Scorsone K, Zhang L, Woodfield SE, Hicks J, Zage PE (2014) The novel kinase inhibitor EMD1214063 is effective against neuroblastoma. Investig New Drugs 32:815–824. https://doi.org/10.1007/s10637-014-0107-4
Mikami K, Medova M, Nisa L, Francica P, Gluck AA, Tschan MP, Blaukat A, Bladt F, Aebersold DM, Zimmer Y (2015) Impact of p53 status on radiosensitization of tumor cells by MET inhibition-associated checkpoint abrogation. Mol Cancer Res 13:1544–1553. https://doi.org/10.1158/1541-7786.mcr-15-0022
Friese-Hamim M, Bladt F, Locatelli G, Stammberger U, Blaukat A (2017) The selective c-met inhibitor tepotinib can overcome epidermal growth factor receptor inhibitor resistance mediated by aberrant c-met activation in NSCLC models. Am J Cancer Res 7:962–972
Yang JC-H, Ellers-Lenz B, Straub J, Johne A, Wu Y-L (2019) INSIGHT 2: tepotinib plus osimertinib in patients with EGFR-mutant NSCLC having acquired resistance to EGFR TKIs due to MET amplification: a phase II trial in progress study. Ann Oncol 30(suppl 9):ix181. https://doi.org/10.1093/annonc/mdz437.061
Decaens T, Barone C, Assenat E, Wermke M, Fasolo A, Merle P, Blanc J-F, Grando V, Bruns R, Straub J, Zhao C, Faivre S (2018) Efficacy and safety of the met inhibitor tepotinib in patients (pts) with advanced MET+ hepatocellular carcinoma (HCC) previously treated with sorafenib. Ann Oncol 29(suppl 8):ix48. https://doi.org/10.1093/annonc/mdy432.004
Ryoo B-Y, Ren Z, Kim T-Y, Pan H, Rau K-M, Choi HJ, Park J-W, Kim JH, Yen C-J, Kim B-H, Zhou D, Straub J, Zhao C, Qin S (2018) Phase 2 trial of tepotinib vs sorafenib in Asian patients (pts) with advanced hepatocellular carcinoma (HCC). Ann Oncol 29(suppl 8):viii207. https://doi.org/10.1093/annonc/mdy282.005
Yang JC-H, Zhou J, Kim D-W, Ahmad AR, Soo RA, Bruns R, Straub J, Johne A, Scheele J, Park K, Wu Y-L (2019) Tepotinib + gefitinib vs chemotherapy in MET-amplified EGFR-mutant non-small cell lung cancer (NSCLC): predefined subgroup analysis of a phase 1b/2 study. Cancer Res 79(13 suppl):CT193. https://doi.org/10.1158/1538-7445.AM2019-CT193
Wu Y-L, Cheng Y, Zhou J et al. (2019) Long-term outcomes to tepotinib plus gefitinib in patients with EGFR-mutant NSCLC and MET dysregulation: 18 month follow-up. J Thorac Oncol 10(suppl):S284. https://doi.org/10.1016/j.jtho.2019.08.571
Sakai H, Felip E, Cortot AB, Veillon R, Griesinger F, Patel J, Horn L, Mazieres J, De Castro Carpeno J, Morise M, Sakamoto T, Bruns R, Scheele J, Straub J, Paik P (2018) Tepotinib in patients with advanced non-small lung cancer (NSCLC) and MET exon 14-skipping mutations: phase II data. Ann Oncol 29(suppl 9):ix152. https://doi.org/10.1093/annonc/mdy425.004
Paik P, Cortot A, Felip E, Sakai H, Mazieres J, Horn L, Griesinger F, Bruns R, Scheele J, Straub J, Veillon R (2019) A phase 2 trial of tepotinib in patients with non-small cell lung cancer (NSCLC) harboring MET alterations: the VISION study. Ann Oncol 30(suppl 2):ii66. https://doi.org/10.1093/annonc/mdz063.080
Lappin G (2016) Approaches to intravenous clinical pharmacokinetics: recent developments with isotopic microtracers. J Clin Pharmacol 56:11–23. https://doi.org/10.1002/jcph.569
Hinderling PH, Papoian T (2020) Why collecting pharmacokinetic information after intravenous drug administration is important. Clin Pharmacol Drug Dev 9:146–150. https://doi.org/10.1002/cpdd.763
European Medicines Agency (2009) ICH guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. https://www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals. Accessed 18 Dec 2019
Vogel JS, Giacomo JA, Schulze-Konig T, Keck BD, Lohstroh P, Dueker S (2010) Accelerator mass spectrometry best practices for accuracy and precision in bioanalytical (14) C measurements. Bioanalysis 2:455–468. https://doi.org/10.4155/bio.10.13
Dorsch D, Schadt O, Stieber F, Meyring M, Gradler U, Bladt F, Friese-Hamim M, Knuhl C, Pehl U, Blaukat A (2015) Identification and optimization of pyridazinones as potent and selective c-met kinase inhibitors. Bioorg Med Chem Lett 25:1597–1602. https://doi.org/10.1016/j.bmcl.2015.02.002
Schadt O, Blaukat A (2017) Tepotinib. In: Chackalamannil S, Rotella D, Ward SE (eds) Comprehensive medicinal chemistry III, 3rd edn. Elsevier, Oxford, pp 178–203
Merck KGaA. Data on File, Darmstadt, Germany
Herbrink M, Nuijen B, Schellens JH, Beijnen JH (2015) Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat Rev 41:412–422. https://doi.org/10.1016/j.ctrv.2015.03.005
US Food and Drug Administration (2012) Guidance for industry. M3(R2) nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. Questions and answers. http://www.gmp-compliance.org/guidemgr/files/UCM292340.PDF. Accessed 18 Dec 2019
von Richter O, Massimini G, Scheible H, Udvaros I, Johne A (2016) Pimasertib, a selective oral MEK1/2 inhibitor: absolute bioavailability, mass balance, elimination route, and metabolite profile in cancer patients. Br J Clin Pharmacol 82:1498–1508. https://doi.org/10.1111/bcp.13078
European Medicines Agency (2012) Guideline on the investigation of drug interactions. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf. Accessed 18 Dec 2019
US Food and Drug Administration (2016) Safety testing of drug metabolites: guidance for industry. Center for Drug Evaluation and Research (CDER), US Food and Drug Administration. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079266.pdf. Accessed 18 Dec 2019
Cheng Y, Zhou J, Lu S, Zhang Y, Zhao J, Pan H, Chen Y-M, Chian C-F, Bruns R, Johne A, Scheele J, Wu Y-L (2018) Phase II study of tepotinib + gefitinib (TEP+GEF) in MET-positive (MET+)/epidermal growth factor receptor (EGFR)-mutant (MT) non-small cell lung cancer (NSCLC). Ann Oncol 29(suppl 8):viii493. https://doi.org/10.1093/annonc/mdy292
Shitara K, Yamazaki K, Tsushima T, Naito T, Matsubara N, Watanabe M, Sarholz B, Johne A, Doi T (2019) Phase I trial of the MET inhibitor tepotinib in Japanese patients with solid tumors. Anticancer Drugs (submitted)
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) (2016) International conference on harmonization (ICH) E6 guideline for good clinical practice (GCP). https://www.ich.org/page/efficacy-guidelines. Accessed 18 Dec 2019
European Commission (2001) European union clinical trial directive (EU CTD). https://ec.europa.eu/health/human-use/clinical-trials/directive_en. Accessed 18 Dec 2019
Acknowledgments
The trial was sponsored by Merck KGaA, Darmstadt, Germany. The authors would like to thank the volunteers and their families, investigators, co-investigators, and the study teams at each of the participating centers and at Merck KGaA, Darmstadt, Germany. We are also grateful to Mr. Roland Neugebauer and Ms. Eleanor Harrison for their excellent assistance in the non-compartmental PK analysis of this study and to Mr. Thomas Scheller for the metabolite profiling and identification work. Michael Meyring is currently an employee of Nuvisan GmbH, Grafing, Germany. Medical writing assistance was provided by Andrew Noble and Sandra Cuscó PhD, Bioscript Science, Macclesfield, UK, and funded by Merck KGaA, Darmstadt, Germany.
Data accessibility statement
Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck KGaA’s Data Sharing Policy. All requests should be submitted in writing to Merck KGaA’s data sharing portal, which can be found at https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. When Merck KGaA has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck KGaA will endeavor to gain agreement to share data in response to requests.
Funding
This study was funded by Merck KGaA, Darmstadt, Germany.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AJ, HS, and AB are employed by Merck KGaA, Darmstadt, Germany, which is developing tepotinib. MM and PW were employed by Merck KGaA, Darmstadt, Germany, when the study was conducted. JJvL is employed by PRA, which was contracted by Merck KGaA, Darmstadt, Germany, to conduct parts of the reported research. Tepotinib is currently under clinical investigation and has not been approved by any regulatory authority.
Ethical approval
This study was conducted in accordance with the principles of the Declaration of Helsinki, in compliance with the International Conference on Harmonisation (ICH) E6 Guideline for Good Clinical Practice (GCP) [37], and was compliant with the European Union Clinical Trial Directive (EU CTD) [38]. The study protocol and the informed consent form (ICF) were approved by the Independent Ethics Committee for Clinical Pharmacology of the Foundation Beoordeling Ethiek Biomedisch Onderzoek (‘Evaluation of the Ethics of Biomedical Research’; Assen, The Netherlands).
Informed consent
All participants signed the ICF before any trial-related procedures were started. The study was conducted by Pharmaceutical Research Association (PRA) Health Sciences (Zuidlaren, The Netherlands).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Johne, A., Scheible, H., Becker, A. et al. Open-label, single-center, phase I trial to investigate the mass balance and absolute bioavailability of the highly selective oral MET inhibitor tepotinib in healthy volunteers. Invest New Drugs 38, 1507–1519 (2020). https://doi.org/10.1007/s10637-020-00926-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-020-00926-1