
Abstract
Taxonomic entities below the species level often pose difficulties for conservation practice, especially when they are ecologically distinct from the nominal species. Genomic tools provide the opportunity to study and potentially resolve such cryptic diversity. The Alcon blue butterfly Phengaris alcon species complex is such a cryptic example, comprising different ecotypes or even subspecies, one of them is the high elevation taxon P. rebeli from the European Alps. We sequenced a first reference genome for Phengaris alcon. Furthermore, we generated whole genome resequence data for individuals of three Swiss ecotypes, i.e., the low elevation P. alcon, the mid elevation and high elevation alpine P. rebeli and integrated genomic data from across Europe to study the relationship among these ecotypes. At a European scale, our results suggest that for the P. alcon complex, biogeography and the evolutionary context of diversification is more multifaceted than previously suggested, falling in the range of more recent ecological speciation. In Switzerland, the three ecotypes were genetically isolated with only limited current gene flow between them. Past gene flow, however, could have given rise to the mid elevation ecotype. Our findings emphasise that high elevation P. rebeli in the Alps should be treated as a distinct species. Our study highlights how the availability of reference genome assemblies allows to address so far open taxonomic questions in conservation research and that broadscale studies are needed to understand the biogeographic history of apparent diversification.
Similar content being viewed by others


Introduction
The ongoing sixth mass extinction is threatening much of the extant biodiversity, negatively impacting species’ abundances and ranges across the tree of life, including insects (Hallmann et al. 2017; Eisenhauer et al. 2019). Conservation policy that aims to counteract their decline is often based on Red Lists that assess the vulnerability of species at a local, national or even global scale (Rodrigues et al. 2006). The problem associated with Red Lists is that they often consider only taxonomically well resolved species (Haig et al. 2006). What constitutes a species is, however, a long-standing debate and many different concepts have been developed and applied (Coyne & Orr 2004). At the core of most species concepts lies the idea that species have to be at least partially reproductively isolated from each other. However, while species have to be completely isolated under classic concepts (Mayr 1942; Coyne & Orr 2004), more recent advances have shown that speciation can also occur in the face of gene flow (Feder et al. 2012). From an evolutionary perspective, speciation is a multifaceted and dynamic process, often seen as a continuum, ranging from populations of the same species with gene flow to reproductively isolated species (Stankowski and Ravinet 2021). Importantly, complete reproductive isolation may be reached when the two species are still genetically compatible to some degree, for example when they become temporally isolated (Taylor and Friesen 2017), occupy ecologically distinct niches (Nosil 2012) and/or evolved increased mate preference limiting gene flow (Merrill et al. 2011). Species may even collapse again if reproductive barriers disappear, e.g., through human alterations of ecosystems (Seehausen et al. 2008).
The species concepts that are applied vary among taxonomic groups. For example, ecological speciation, whereby adaptation to different environments or resources causes the evolution of reproductive barriers but where gene flow is often still abundant, especially at an early stage, is commonly used for freshwater fish (Seehausen and Wagner 2014). On the other hand, taxonomic species delineation in insects is often based on reproductive isolation associated with genetic incompatibilities (Claridge 2017). However, many insect groups include species complexes that comprise different subspecies or lineages, which may even ecologically differ. In such cases, taxonomy may amalgamate species complexes into single taxa, especially when taxonomy is only based on morphology or few genetic markers. This is problematic for conservation because the overall taxon may, for example, be classified as being of low concern for conservation due to the wide geographic occurrence of the entire species complex or because their management is based on ecological assumptions of a single subspecies (Bickford et al. 2007). Although the concept of evolutionary significant units (ESU), representing distinct genetic or ecological lineages within a species, has been suggested for conservation, it is currently not applied to species Red Lists (Stronen et al. 2022). To successfully protect such species complexes, it is therefore necessary to study and, if necessary, resolve them.
Across Europe, almost 500 species of butterflies have been recognized (Wiemers et al. 2018), but to add complexity, numerous taxa include different subspecies, lineages or ecotypes (Settele et al. 2008; Descimon and Mallet 2009). An example is the Phengaris alcon complex whose taxonomic resolution has remained a long-standing debate (Kudrna and Fric 2013; Koubínová et al. 2017). The Phengaris alcon complex encompasses several potential species, subspecies, lineages or ecotypes. Like other species of the genus Phengaris, P. alcon are myrmecophilous, and often differ in their ant host association with Myrmica sp. Ant host specificity may, however, vary and host shifts are recurrent across Europe (Tartally et al. 2019). Taxonomic groups may similarly differ by host-plant use, where females oviposit on different species of the plant family Gentianaceae, mostly on Gentiana. Much of the ongoing taxonomic debate revolves around the distinction between P. alcon (Denis & Schiffermüller, 1775; Lepidoptera, Lycaenidae) and P. rebeli (Hirschke 1905; Lepidoptera, Lycaenidae), which are currently lumped into a single taxon “P. alcon” in the last European checklist (Wiemers et al. 2018). Phengaris alcon populations are widely distributed across Europe (Fig. 1) and are often distinguished by their habitat and/or caterpillar foodplant usage as hygric, xeric or alpine ecotypes (Koubínová et al. 2017). Populations in wet habitats (hygrophilic ecotype) were mainly designated as P. alcon, while populations in dry habitats from outside the Alps (xerophilic ecotype), even from low elevations, were in most cases incorrectly assigned to P. rebeli (Habeler 2008). The latter was, however, originally described from the Styrian Alps occurring only in dry alpine habitats (Habeler 2008). In some cases, the low elevation dry habitat ecotype was designated as a distinct species (P. xerophila; Berger 1946; Lepidoptera, Lycaenidae) or subspecies (P. rebeli xerophila). Based on morphology, the xerophilic ecotype is not consistently distinct from the hygrophilic P. alcon ecotype (Kudrna and Fric 2013). Based on sequence data of four genes, the same study concluded that P. alcon and P. rebeli xerophila are paraphyletic but suggested that the alpine P. rebeli is likely not closely related to the lowland xerophilic ecotypes or any lowland population of P. alcon. The latter is supported by allozyme data, where high elevation P. rebeli from the Eastern Alps were genetically most distinct from other putative P. rebeli from outside of the Alps (Bereczki et al. 2018). The switch of P. alcon from hygric to xeric habitat has likely evolved multiple times across Europe, as has been suggested using genomic markers (Koubínová et al. 2017). However, this study included only a single high elevation P. rebeli individual from the Alps, precluding inferences about the status of alpine P. rebeli.

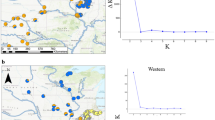
Occurrence and relationship of genotyped individuals of the Phengaris alcon complex from across Europe based on genetic sites that overlapped with the RADseq data of Koubinova et al. (2017). A) Map of Europe depicting the distribution of P. alcon complex (pale blue dots) based on observation data from GBIF (GBIF.org 2023a, b) as well as from the distribution atlas of European butterflies, based on 50 × 50 km grid cells (Kudrna et al. 2015). These sources do not differentiate ecotypes. Map sources: European Comission GISCO: https://ec.europa.eu/eurostat/web/gisco; EU-DEM https://www.eea.europa.eu/data-and-maps/data/. B) Phylogram for all genotyped individuals with grey dots on nodes denoting > 95% bootstrap support based on 1’000 bootstrap replicates in RAxML. C) Principal component analysis for the same individuals. For non-Swiss samples, shapes denote P. alcon ecotypes (squares – xeric, triangles – hygric) and colours the country. For Swiss samples, colours are consistent with Fig. 2. For Italy, Koubinova et al. (2017) indicated one rebeli individual (denoted as “alpine ecotype”) and one individual from the Apennine Mountains, which clusters with individuals from the Balkans
In Switzerland two distinct species P. alcon and P. rebeli were classically recognized until recently (Lepidopterologen Arbeitsgruppe 1987; SwissLepTeam 2010) and evaluated separately in the Swiss Red List (Wermeille et al. 2014). Phengaris alcon occurs at lower elevations, i.e., below 1’000 m.a.s.l. in wetlands and fens. Its primary host plants are Gentiana pneumonanthe and G. asclepiadea and its primary ant host is Myrmica scabrinodis (Bolt et al. 2010). Until recently, two different ecotypes were moreover recognized under the taxon P. rebeli. The first ecotype, to which we refer as “mid elevation P. rebeli” occurs on calcareous dry habitats at mid elevations up to 1’300 m.a.s.l. and regionally higher using G. cruciata as host plants and M. schencki as host ants (Lepidopterologen Arbeitsgruppe 1987). The second ecotype, to which we refer as “high elevation P. rebeli” occurs at high elevations from above the tree line up to 2’200 m.a.s.l. on dry siliceous and calcareous substrate, and likely corresponds to the alpine P. rebeli described by Hirschke (1905). Their known host plants in Switzerland are G. purpurea, G. nivalis, G. campestris (Wipking et al. 2023), but relatively little is known about their host ants, which comprise M. sulcinodis (Jutzeler 1988; Neumeyer et al. 2018).
While both P. alcon and mid elevation P. rebeli lay many eggs on their host plants, the high elevation P. rebeli primarily deposits a single egg per host plant (Wipking et al. 2023). To add complexity, many different local forms or even putative subspecies have been proposed for the P. alcon complex in Switzerland, although most are currently not considered (Table S1; Beuret 1953), but a thorough investigation is lacking. In the face of past taxonomic uncertainties and given that former genetic investigations had only a limited resolution for Switzerland and the Alps in general, we will further refer to these three entities as ecotypes. Importantly, since the last revision of the Swiss Red List, these ecotypes are considered to be a single species P. alcon following the taxonomy of Wiemers et al. (2018), that is based on the findings of Kudrna and Fric (2013) and Koubínová et al. (2017).
To study the genetic differentiation among the three Swiss ecotypes, we first used long read sequencing to generate the first genome assembly of P. alcon. We then generated whole-genome resequencing data for all three ecotypes that exist in Switzerland to study their genetic relationships and to estimate the potential for gene flow among them. To put our Swiss samples in a broader biogeographic context, we further combined our data with former low resolution genomic data for primarily P. alcon from different countries (Koubínová et al. 2017). Finally, we tested for differences in Wolbachia among our sampled individuals. Wolbachia is an endosymbiotic bacterium, which often differs among closely related butterfly species (Lucek et al. 2021) and may also act as a barrier to gene flow (Kaur et al. 2021).
Methods
Phengaris alcon genome sequencing and annotation
We collected caterpillars of Phengaris alcon by screening host plants (G. pneumonanthe) for the presence of eggs on the 31st of August 2021 near Zürich, Switzerland (8.5007°E, 47.2847°N). Plant material was immediately frozen and caterpillars subsequently dissected out. DNA was extracted from a whole caterpillar using a standard phenol/chloroform extraction method (Green and Sambrook 2017). We then sequenced its DNA on the PacBio Sequel II platform (Pacific Biosystems, Menlo Park, CA, USA) using one PacBio SMRT cell. Sequencing and library preparation followed the PacBio Low DNA Input Library protocol and was done by the Functional Genomic Centre Zürich (Zürich, Switzerland).
All obtained HiFi reads were used for assembly with hifiasm 0.16.0 using standard parameters (Cheng et al. 2021). To identify contigs of non-lepidopteran origin, we compared each contig against the NCBI nucleotide collection using Blast + 2.9.0 (Camacho et al. 2009). A total of 28 contigs, accounting for 0.6% of the initial assembly length, were subsequently filtered out. Out of these filtered contigs, one contig represented the complete Wolbachia genome, which we kept as a separate assembly. We estimated the completeness of our filtered P. alcon genome with the Busco pipeline 5.1.2 (Manni et al. 2021), which was performed against the lepidoptera_odb10 database containing 5286 BUSCO groups of highly conserved single copy orthologs. We next identified repeats using RepeatMasker 4.1.3 (www.repeatmasker.org) and performed gene annotation with Braker2 (Brůna et al. 2021) on the soft masked assembly. For the latter, we took the BUSCO odb10_arthropoda protein database as a reference (Kriventseva et al. 2019). We only kept gene predictions showing support by hints, using the script selectSupportedSubsets.py of the Braker2 pipeline.
Genomics of the Swiss Phengaris alcon complex
We collected 35 specimens of the Phengaris alcon complex between 2016 and 2021, covering the three ecotypes, i.e., the low elevation P. alcon, the mid elevation and the high elevation alpine P. rebeli, respectively (Table S2). In some cases, whole bodies were collected, while in other cases, only one leg or a caterpillar was obtained. Tissue was stored in absolute ethanol prior to DNA extraction, for which we used the Qiagen Blood & Tissue Kit (Qiagen AG, Hombrechtikon, Switzerland) following the manufacturer’s protocol. Because DNA yield was low for legs and caterpillars, we performed whole genome amplifications with the Qiagen REPLI-g kit. Paired-end sequencing libraries were prepared at the Department of Biosystems Science and Engineering (DBSSE) of ETH Zürich in Basel, followed by whole-genome sequencing (WGS) on an Illumina NovaSeq 6000. Samples were sequenced on two lanes of S4 flow cells.
We processed demultiplexed raw sequence reads as follows: First, we trimmed poly-G tails with fastp (Chen et al. 2018) and mapped all retained reads against our Phengaris alcon reference assembly, using bwa v0.7.17 (Li 2013). We then employed SAMtools v.1.13 (Li et al. 2009) to remove unmapped, unpaired, or duplicated reads. We called variants for all individuals combined with BCFtools v.1.12 (Danecek and McCarthy 2017). Finally we used VCFtools 0.1.16 (Danecek et al. 2011) to remove (i) non-biallelic SNPs, (ii) indels, (iii) SNPs with genotype quality score < 30, (iv) SNPs with more than 20% missing data, (v) SNPs with minor allele frequencies (MAF) < 0.03, and vii.) SNPs with depths < 6 or > 40. This resulted in a dataset containing 494’213 SNPs. Six individuals were subsequently filtered out due to high levels of missingness (Table S2), resulting in 29 individuals for analyses.
We inferred population structure in a first step with Admixture 1.3.0, which implements a likelihood approach to estimate ancestry (Patterson et al. 2012). We ran Admixture first across all individuals, varying the values for K, i.e., the number of assumed populations, from 1 to 10 and performed a cross-validation test to determine the optimal value of K. To remove effects driven by increased linkage disequilibrium, we only considered SNP sites that were at least 1’000 bps away from each other for Admixture (NSNPs= 111’414). In a second step we used a principal component (PC) analysis as implemented in PLINK 1.90b (Chang et al. 2015) to visualize the genetic relationship among individuals. Subsequent statistical analyses on the resulting PC scores were done using linear models in R 4.1.1 (R Core Team 2021).
To infer the phylogenomic structure across all retained specimens, we first used RAxML 8.2.11 (Stamatakis 2014), implementing a generalised time-reversible (GTR) model with optimised substitution rates and a gamma model of rate heterogeneity. We further applied an ascertainment bias correction to account for the fact that we only used polymorphic SNP positions with the ASC_GTRGAMMA function implemented in RAxML. Statistical support was assessed using 1’000 bootstrap replicates followed by a thorough maximum likelihood search.
Next, we estimated the level of pairwise genetic differentiation (FST) among our three Phengaris ecotypes using VCFtools, pooling individuals from across the distribution range for P. alcon, P. rebeli mid elevation and P. rebeli high elevation, respectively. For each comparison, we defined regions of accentuated genetic divergence as 10’000 bps windows for which the average FST > 0.95 and contained more than two SNPs. We subsequently extracted the gene sequence for these regions and used NCBI blastx (Boratyn et al. 2013) to identify their correspondence to other Lepidoptera.
Lastly, we assessed the potential for gene flow among our sampled individuals. First by reconstructing a network using SplitsTree 5 with a GTR model (Huson and Bryant 2006) using all available SNPs. Secondly, we estimated recent migration rates among our three ecotypes with BA3-SNPs 3.0.4 (Mussmann et al. 2019), a modification of BayesAss (Wilson and Rannala 2003) that allows the handling of large SNP datasets. For this, we assessed the optimal mixing parameters for migration rates (deltaM = 0.2688), allele frequencies (delta = 0.5500), and inbreeding coefficients (deltaF = 0.0188) by running ten repetitions in BA3-SNP-autotune 3.0.4 as recommended by Mussmann et al. (2019). Subsequently, BA3-SNPs was run with the predefined mixing parameters for 50 million generations, sampling every 100th generation. Because BA3-SNPs is computationally intensive for very large datasets (Mussmann et al. 2019), we had to run this analysis on a thinned dataset, using only SNPs that were at least 10’000 bps apart (NSNPs=30’320).
The biogeographic context of the Swiss Phengaris alcon complex
To set our Swiss samples into a broader biogeographic context, we aligned the dataset of Koubínová et al. (2017) comprising single-end RAD sequences for 25 P. alcon and one P. rebeli specimen (designated as “alpine” ecotype in Koubínová et al. (2017)) from outside Switzerland against our reference assembly. Alignment, SNP calling and filtering were done as for the WGS dataset, and one P. alcon specimen from the Italian Alps had to be removed due to missingness. We subsequently extracted the overlapping SNPs between both datasets (NSNPs = 883) for our subsequent analyses. As for the Swiss specimens, we inferred the phylogenomic structure across all individuals with RAxML and visualized the population structure with a PCA in PLINK.
Wolbachia
We annotated the obtained Wolbachia assembly with the Rapid Annotation using Subsystem Technology (Rast) web service version 2.0 (Aziz et al. 2008). To test if P. alcon and P. rebeli also have different Wolbachia strains, we aligned all our genomic data against our Wolbachia assembly. Alignment, variant calling and filtering were performed as outlined above. We then established the relationship of Wolbachia among our Phengaris host individuals using RAxML with 1’000 bootstrap replicates.
Results
The Phengaris alcon genome
Our assembled P. alcon genome was 590.9 Mbps long (N50 = 1.52 Mbps) and contained 987 contigs with an average GC content of 36.6%. The BUSCO analysis found a 97.2% completeness score with 4’922 complete and single copy BUSCO groups, 215 complete and duplicated BUSCOs, 27 fragmented BUSCOs, and 122 missing BUSCOs. The gene prediction yielded 22’625 transcripts for 20’454 genes.
Structure, relationship and divergence of the Swiss Phengaris alcon complex
Three genetic clusters were the best K for Admixture (Fig. 2A), with the second being K = 2. Admixture separated the three Swiss Phengaris ecotypes, where past gene flow seems to have occurred, especially between the mid elevation P. rebeli and the high elevation P. rebeli and, to a lesser extent P. alcon. When assuming only two genetic clusters (K = 2) mid elevation P. rebeli are intermediates between P. alcon and high elevation P. rebeli with more genomic contribution from P. alcon (Fig. S1). Similarly, the leading PC axis accounting for 21.9% of the total variance separated P. alcon and P. rebeli high elevation, where individuals of the mid elevation P. rebeli fell in between (Fig. 2B). While gene flow between the two P. rebeli ecotypes occurred along a west to east transect (Fig. 2C), the RAxML analysis resolved the three Swiss Phengaris ecotypes each as distinct clusters (Fig. 2D).

Genetic structure of the Phengaris alcon and P. rebeli across Switzerland. A) Admixture analysis assuming three genetic clusters (K). B) Principal component (PC) analysis for the same individuals with respective Admixture proportions for K = 3 indicated. C) Map of Switzerland with the genotyped individuals and their respective admixture proportions (K = 3) indicated. D) Phylogram for all genotyped individuals with grey dots on nodes denoting > 95% bootstrap support based on 1’000 bootstrap replicates in RAxML. Colours are as follows: Yellow – P. alcon, blue – P. rebeli mid elevation, green – P. rebeli high elevation
Average genomic divergence (FST) varied among our three comparisons (P. alcon vs. P. rebeli mid elevation: FST=0.168; P. rebeli high elevation vs. P. rebeli mid elevation: FST=0.178; P.alcon vs. P. rebeli high elevation: FST=0.223; Fig. 3). Among these comparisons, a total of 9, 39 and 59 10 kb windows showed accentuated genomic divergence, corresponding to 7, 16 and 31 outlier genes (Tables S3-S5). Few genes could be annotated with blastx, suggesting that outliers could be linked to immune response, life history or pheromones (Tables S3-S5).

Pairwise genetic differentiation (FST) across the P. alcon genome assembly comparing the three studied groups. Contigs are highlighted in alternating shading. Each dot represents the average FST within 10 kb windows. Red lines indicate the average genome-wide FST. Blue dots highlight regions of accentuated genomic differentiation (see Methods)
The SplitsTree network confirmed the relationship among the three Swiss Phengaris ecotypes outlined by the aforementioned analyses and highlighted that gene flow occurs primarily within each ecotype (Fig. 4A). Similarly, estimates of contemporary gene flow from BayesAss are very limited, consistent with a current isolation of the three Phengaris ecotypes (Fig. 4B, Table S6-S5).

In a broader biogeographic context, the Swiss P. alcon formed a distinct clade, where mid-elevation P. rebeli clustered more closely than high-elevation P. rebeli (Fig. 1). However, bootstrap support for the reduced genomic dataset was generally lower, especially among samples from Eastern Europe (Fig. 1B). The putative high-elevation P. rebeli of Koubínová et al. (2017) from the Italian Alps similarly clustered with the Swiss high-elevation P. rebeli. Also, the putative xeric P. alcon from the Italian Alps were in between the two Swiss P. rebeli ecotypes. The distinction between the Swiss Phengaris lineages was similarly driving the leading PC axis, accounting for 18.0% of the total variance, when the second axis, accounting for 13.3% of the variance, separated individuals from Eastern Europe (Fig. 1C). Consistent with Koubínová et al. (2017), xeric and hygric P. alcon occur independently from each other in distant European clades.
Wolbachia
Our assembled Wolbachia genome was 1’436’415 bps long and has a GC content of 34.0%. The RAST annotated features include 1’435 putative protein-coding genes with 37 RNAs elements. A total of 327 genes (23%) could be assigned to a subsystem, i.e., proteins grouped by a relationship in function.
All of our genotyped Phengaris individuals from Switzerland were infected by Wolbachia. Generally, each Phengaris ecotype contained distinct Wolbachia lineages, where P. alcon showed evidence for two Wolbachia lineages (Fig. 5). One individual from south western Switzerland for each of the P. rebeli ecotypes further harboured an additional Wolbachia lineage.

Relationship of Wolbachia occurring in Swiss Phengaris alcon and P. rebeli. Phylogram for all genotyped individuals with grey dots on nodes denoting > 95% bootstrap support based on 1’000 bootstrap replicates in RAxML. Colours denote hosts: Yellow – P. alcon, blue – P. rebeli mid elevation, green – P. rebeli high elevation
Discussion
Taxonomic entities below the species level, such as sub-species or evolutionary significant units (ESUs), represent difficulties for conservation practice, especially if they are ecologically distinct or represent cryptic species, as they are generally not recognized in national or international Red Lists and therefore by public stakeholders (Mace 2004). The genetic tools to study such cryptic diversity have been a limiting factor for a long time. This is because existing markers often provide a very limited resolution, especially in the case of evolutionary young species as well as due to the lack of reference genomes that allow for deeper genomic investigations. With the advent of third generation sequencing approaches, reference genomes or assemblies are now becoming available even for non-model organisms, providing unprecedented opportunities for conservation research (Theissinger et al. 2023). Adding population-level genomic data, such reference genomes have the power to resolve some of the longstanding taxonomic issues. Using genomic data, we aimed to shed some light on the current debate about the taxonomic status of P. alcon and P. rebeli, focusing especially on Switzerland. Overall, we show that in Switzerland mid elevation P. alcon and high elevation P. rebeli can be seen as genetically and ecologically distinct entities and that combined with the European level, the biogeography as well as evolutionary context of diversification within the P. alcon complex seem to be more complicated than previously suggested (Koubínová et al. 2017).
The evolution of barriers to gene flow that lead to reproductive isolation is a key requirement for speciation (Coyne & Orr 2004). Especially under ecological speciation, interspecific gene flow is often still predominant at an early stage of the speciation process and may even fuel the formation of novel species (Feder et al. 2012; Seehausen et al. 2014). The latter has for example been suggested for Coenonympha butterflies, where hybridization between low-elevation C. arcania and alpine C. gardetta triggered the evolution of two hybrid species in the Alps, i.e. C. darwiniana and C. macromma (Capblancq et al. 2019). Our genomic inferences suggest that in Switzerland, low-elevation P. alcon and high elevation P. rebeli form distinct genetic clusters (Fig. 2) with little to no current gene flow between them (Fig. 4). The high elevation P. rebeli in Switzerland are likely a representative of the P. rebeli described by Hirschke (1905) from the Styrian Alps, given that it clusters with other P. rebeli from the eastern Alps (Fig. 1). Conversely, the third ecotype that we studied, i.e., the mid elevation P. rebeli from the Alps, seems to have contact zones between the high elevation P. rebeli along a west-east transect across the Alps (Fig. 2). On the one hand, geographically extensive gradients of genetic intergradation have been found in other alpine butterflies such as Erebia manto, which was associated with colonization of the Alps from distinct glacial refugia (Schmitt et al. 2014; Jospin et al. 2023). On the other hand, mid elevation P. rebeli seem to be genetically intermediate between P. alcon and the high elevation P. rebeli, being more closely related to the former (Figs. 2 and 3). Mid elevation P. rebeli form nevertheless a distinct genetic cluster (Fig. 2) and evidence for current gene flow is limited (Fig. 4). Conversely, past historical gene flow might have been possible as P. alcon used to be more widely distributed, including in the western part of Switzerland, but these populations have become extinct over the last century (Lepidopterologen Arbeitsgruppe 1987; Wermeille et al. 2014). The mid elevation P. rebeli that we studied might therefore represent a distinct glacial lineage of P. rebeli or potentially a hybrid lineage similar to Coenonympha. Given that mid elevation P. rebeli occur on calcareous dry habitats, it could also represent a xeric ecotype that is often associated with P. alcon. Indeed, xeric ecotype individuals from Italy cluster close with our mid elevation P. rebeli (Fig. 1). Although larger sample sizes and a broader sampling are required to disentangle the different scenarios, a hybrid origin of the mid elevation P. rebeli seems to be likely, given the intermediate placing of this ecotype in all our analyses (Figs. 1, 2, 3 and 4).
The current genetic isolation between Swiss P. alcon and the two P. rebeli ecotypes is further evidenced by their association with different Wolbachia strains, where both P. rebeli ecotypes have different strains (Fig. 5). In two cases, we observed potential recent host shifts, i.e. individuals with distinct Wolbachia strains, which has been observed in other systems of alpine butterflies (Lucek et al. 2021). Interestingly, we also detected two Wolbachia strains within P. alcon, which could reflect additional population structure. Different Wolbachia strains may cause reproductive isolation upon secondary contact through different processes (Kaur et al. 2021), consequently our findings implicate that potential conservation measures should also incorporate an assessment of the endosymbiotic community. Albeit our outlier analysis was limited by the number of genotyped individuals, the genomic differentiation was heterogenous across the genome with some regions of accentuated differentiation (Fig. 3), which could reflect putative islands of selection (Feder et al. 2012). Adaptation to different ant hosts has especially been associated with chemical mimicry (Akino et al. 1999). Our analysis indeed suggests that such differences could have a genetic basis, however, further investigations with larger sample sizes are required to pinpoint the genomic architecture of differentiation among P. alcon and P. rebeli.
Former attempts to resolve the P. alcon species complex on a broader geographic scale concluded that P. alcon and P. rebeli are paraphyletic and show only a limited level of geographic structure, but excluding the high elevation P. rebeli from the Alps (Kudrna and Fric 2013). A more recent genomic study similarly showed that dry (xeric) and wetland (hygric) ecotypes of P. alcon evolved repeatedly across Europe (Koubínová et al. 2017). Combining the dataset of the latter study with our own, we confirmed this notion (Fig. 1). However, our inferences suggest that biogeographic history of P. alcon/P. rebeli across Europe is more complex than previously suggested with at least three major genetic clusters being present: the first represents P. alcon from the Balkans as well as the individual from the Italian Apennine mountains and comprises both the dry and wetland ecotypes. The two other clusters are represented by P. alcon from Switzerland and the high elevation P. rebeli from the Alps, where the mid elevation P. rebeli and individuals from the Italian Alps are intermediate between the two. Surprisingly, Spanish P. alcon cluster close to Swiss P. alcon, which could suggest that they derive from a similar glacial refugia and that the high elevation P. rebeli could have persisted in a different refugium east of the Alps. Similar biogeographic patterns have been found for other butterflies and have often been attributed to genetic divergence during phases of allopatry through the glacial cycles (Schmitt et al. 2006; Kühne et al. 2017; Lucek et al. 2020; Cupedo and Doorenweerd 2022). Given the broad distribution of the P. alcon complex across Europe (Fig. 1), further genomic investigations seem thus necessary to understand the biogeographic context of this species complex at the continental scale.
Taken together, our results highlight that high elevation P. rebeli are genetically distinct from P. alcon in Switzerland, consistent with a former genetic study from the Eastern Alps (Bereczki et al. 2018), forming a distinct genetic cluster at the European level. While evolutionary biologists use the speciation continuum to describe the dynamic and often ongoing process of speciation, taxonomy requires the delineation of the terminal branches of this process into species. In the shadow of taxonomic uncertainties, the P. alcon species complex has been suggested as a showcase to apply the ESU concept for conservation (Casacci et al. 2014). In the light of our findings and given that ESUs are currently not recognized by public stakeholders in most cases (IUCN Standards and Petitions Committee 2022), we recommend treating the high elevation P. rebeli from the Alps as a distinct species. We uncovered a large cryptic diversity of P. alcon across Europe, even though its biogeographic context requires a broader geographic sampling. This is especially true for the mid elevation P. rebeli ecotype, which could be a more recently evolved ecotype or glacial lineage. Repeated ecotype formation within P. alcon across Europe (Koubínová et al. 2017) as well as speciation of the high elevation P. rebeli occurred likely over a much more recent evolutionary time frame than for many other butterfly species (Ebdon et al. 2021). Because intrinsic barriers to gene flow are often absent or not strong enough to prevent admixture, they may be especially vulnerable to future range or habitat changes (Jardim de Queiroz et al. 2022). Lastly, our study highlights how the availability of reference genome assemblies allows to address so far open taxonomic problems in conservation research by providing a much higher resolution (Theissinger et al. 2023).
Data availability
Genomic data is available from NCBI: Genome assembly and associated data (BioProject: PRJNA1063918), short-read data (BioProject: PRJNA1064043). Genome annotations and Wolbachia assembly are available from Zenodo: https://doi.org/10.5281/zenodo.10555743.
References
Akino T, Knapp JJ, Thomas JA, Elmes GW (1999) Chemical mimicry and host specificity in the butterfly Maculinea rebeli, a social parasite of Myrmica ant colonies. Proc R Soc B 266, 1419–1426
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA et al (2008) The RAST server: Rapid annotations using Subsystems Technology. BMC Genomics 9:75
Bereczki J, Pecsenye K, Varga Z, Tartally A, Tóth JP (2018) Maculinea rebeli (Hirschke) - a phantom or reality? Novel contribution to a long-standing debate over the taxonomic status of an enigmatic Lycaenidae butterfly: Maculinea rebeli - a phantom or reality? Syst Entomol 43:166–182
Beuret H (1953) Die Lycaeniden Der Schweiz. F. Straub, Basel, Switzerland
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K et al (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155
Bolt D, Neumeyer R, Rey A, Sohni V (2010) Kleiner Moorbläuling (Lepidoptera: Lycaenidae: Phengaris alcon) und Echte Knotenameisen (Hymenoptera: Formicidae: Myrmica) am Pfannenstiel (Kanton Zürich, Schweiz). Entomologica Helv 3:27–48
Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N et al (2013) BLAST: a more efficient report with usability improvements. Nucleic Acids Res 41:W29–W33
Brůna T, Hoff KJ, Lomsadze A, Stanke M, Borodovsky M (2021) BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP + and AUGUSTUS supported by a protein database. NAR Genomics and Bioinformatics 3:lqaa108
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K et al (2009) BLAST+: architecture and applications. BMC Bioinformatics 10:421
Capblancq T, Mavárez J, Rioux D, Després L (2019) Speciation with gene flow: evidence from a complex of alpine butterflies (Coenonympha, Satyridae). Ecol Evol 9:6444–6457
Casacci LP, Barbero F, Balletto E (2014) The evolutionarily significant unit concept and its applicability in biological conservation. Italian J Zool 81:182–193
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4:7
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890
Cheng H, Concepcion GT, Feng X, Zhang H, Li H (2021) Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18:170–175
Claridge MF (2017) Insect species - concepts and practice. In: Foottit RG, Adler PH (eds) Insect biodiversity. John Wiley & Sons, Ltd, Chichester, UK, pp 527–546
R Core Team (2021) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
Coyne JA, Orr HA (2004) Speciation. Oxford University Press, Oxford, UK
Cupedo F, Doorenweerd C (2022) Mitochondrial DNA-based phylogeography of the large ringlet Erebia Euryale (Esper, 1805) suggests recurrent Alpine-Carpathian disjunctions during Pleistocene (Nymphalidae, Satyrinae). Nota Lepid 45:65–86
Danecek P, McCarthy SA (2017) BCFtools/csq: haplotype-aware variant consequences. Bioinformatics 33:2037–2039
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Descimon H, Mallet J (2009) Bad species. In: Settele J, Shreeve TG, Konvicka M, Dyck VH (eds) Ecology of butterflies in Europe. Cambridge University Press, Cambridge, UK, pp 219–249
Ebdon S, Laetsch DR, Dapporto L, Hayward A, Ritchie MG, Dincӑ V et al (2021) The pleistocene species pump past its prime: evidence from European butterfly sister species. Mol Ecol 30:3575–3589
Eisenhauer N, Bonn A, Guerra A, C (2019) Recognizing the quiet extinction of invertebrates. Nat Commun 10:50
Feder JL, Egan SP, Nosil P (2012) The genomics of speciation-with-gene-flow. Trends Genet 28:342–350
GBIF.org (2023a) GBIF Occurrence Download https://doi.org/10.15468/dl.dehz84
GBIF.org (2023b) GBIF Occurrence Download https://doi.org/10.15468/dl.fsgk5u
Green MR, Sambrook J (2017) Isolation of High-Molecular-Weight DNA using organic solvents. Cold Spring Harb Protoc 2017: pdb.prot093450
Habeler H (2008) Die subalpin-alpinen Lebensräume Des Bläulings Maculinea rebeli (Hirschke 1904) in den Ostalpen (Lepidoptera, Lycaenidae). Joannea Zoolgica 10:143–164
Haig SM, Beever EA, Chambers SM, Draheim HM, Dugger BD, Dunham S et al (2006) Taxonomic considerations in listing subspecies under the U.S. Endangered species Act. Conserv Biol 20:1584–1594
Hallmann CA, Sorg M, Jongejans E, Siepel H, Hofland N, Schwan H et al (2017) More than 75% decline over 27 years in total flying insect biomass in protected areas. PLoS ONE 12:e0185809
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267
IUCN Standards and Petitions Committee (2022) Guidelines for Using the IUCN Red List Categories and Criteria. Version 15.1. Prepared by the Standards and Petitions Committee. Downloadable from https://www.iucnredlist.org/documents/RedListGuidelines.pdf
Jardim de Queiroz L, Doenz CJ, Altermatt F, Alther R, Borko Š, Brodersen J et al (2022) Climate, immigration and speciation shape terrestrial and aquatic biodiversity in the European Alps. Proceedings of the Royal Society of London B 289: 20221020
Jospin A, Chittaro Y, Bolt D, Demergès D, Gurcel K, Hensle J et al (2023) Genomic evidence for three distinct species in the Erebia manto complex in Central Europe (Lepidoptera, Nymphalidae). Conserv Genet 24:293–304
Jutzeler D (1988) Fund Von I (Hirschke 1904) Im Glarnerland (Lepidoptera, Lycaenidae). Mitt Entomol Ges. Basel 38:124–125
Kaur R, Shropshire JD, Cross KL, Leigh B, Mansueto AJ, Stewart V et al (2021) Living in the endosymbiotic world of Wolbachia: a centennial review. Cell Host Microbe 29:879–893
Koubínová D, Dincă V, Dapporto L, Vodă R, Suchan T, Vila R et al (2017) Genomics of extreme ecological specialists: multiple convergent evolution but no genetic divergence between ecotypes of Maculinea alcon butterflies. Sci Rep 7:13752
Kriventseva EV, Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simão FA et al (2019) OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res 47:D807–D811
Kudrna O, Fric ZF (2013) On the identity and taxonomic status of Lycaena alcon rebeli Hirschke 1905 — a long story of confusion and ignorance resulting in the fabrication of a ghost species (Lepidoptera: Lycaenidae). Nachr Des Entomologischen Vereins Apollo 34:117–124
Kudrna O, Pennerstorfer J, Lux K (2015) Distribution atlas of European butterflies and skippers. Wissenschaftlicher Verlag Peks e.K., Schwanfeld, Germany
Kühne G, Kosuch J, Hochkirch A, Schmitt T (2017) Extra-mediterranean glacial refugia in a Mediterranean faunal element: the phylogeography of the chalk-hill blue Polyommatus coridon (Lepidoptera, Lycaenidae). Sci Rep 7:43533
Lepidopterologen Arbeitsgruppe (1987) Tagfalter Und Ihre Lebensräume. Schweiz Und Angrenzende Gebiete. Arten, Gefährdung, Schutz. Schweizerischer Bund für Naturschutz, Basel, Switzerland
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Lucek K, Butlin RK, Patsiou T (2020) Secondary contact zones of closely-related Erebia butterflies overlap with narrow phenotypic and parasitic clines. J Evol Biol 33:1152–1163
Lucek K, Bouaouina S, Jospin A, Grill A, de Vos JM (2021) Prevalence and relationship of endosymbiotic Wolbachia in the butterfly genus Erebia. BMC Ecol Evol 21:95
Mace GM (2004) The role of taxonomy in species conservation. Phil Trans Royal Soc Lond B 359:711–719
Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM (2021) BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 38:4647–4654
Mayr E (1942) Systematics and the origin of species. Columbia UniversityPress, New York, USA
Merrill RM, Gompert Z, Dembeck LM, Kronforst MR, McMillan WO, Jiggins CD (2011) Mate preference across the speciation continuum in a clade of mimetic butterflies. Evolution 65:1489–1500
Mussmann SM, Douglas MR, Chafin TK, Douglas ME (2019) BA3-SNPs: contemporary migration reconfigured in BayesAss for next‐generation sequence data. Methods Ecol Evol 10:1808–1813
Neumeyer R, Rey A, Sommerhalder J (2018) Neues Vom Taxon Phengaris alcon rebeli (Hirschke 1904) auf Der Hochebene Obersand (GL). Entomo Helv 11:79–88
Nosil P (2012) Ecological speciation. Oxford University Press, Oxford, UK
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y et al (2012) Ancient admixture in human history. Genetics 192:1065–1093
Rodrigues A, Pilgrim J, Lamoreux J, Hoffmann M, Brooks T (2006) The value of the IUCN Red List for conservation. Trends Ecol Evol 21:71–76
Schmitt T, Hewitt GM, Muller P (2006) Disjunct distributions during glacial and interglacial periods in mountain butterflies: Erebia Epiphron as an example. J Evol Biol 19:108–113
Schmitt T, Habel JC, Rödder D, Louy D (2014) Effects of recent and past climatic shifts on the genetic structure of the high mountain yellow-spotted ringlet butterfly Erebia manto (Lepidoptera, Satyrinae): a conservation problem. Glob Change Biol 20:2045–2061
Seehausen O, Wagner CE (2014) Speciation in freshwater fishes. Annual Rev Ecologym Evol Syst 45:621–651
Seehausen O, Takimoto G, Roy D, Jokela J (2008) Speciation reversal and biodiversity dynamics with hybridization in changing environments. Mol Ecol 17:30–44
Seehausen O, Butlin RK, Keller I, Wagner CE, Boughman JW, Hohenlohe PA et al (2014) Genomics and the origin of species. Nat Rev Genet 15:176–192
Settele J, Kudrna O, Harpke A, Kühn I, van Swaay C, Verovnik R et al (2008) Climatic Risk Atlas of European Butterflies. BioRisk 1: 1–712
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Stankowski S, Ravinet M (2021) Defining the speciation continuum. Evolution 75:1256–1273
Stronen AV, Norman AJ, Vander Wal E, Paquet PC (2022) The relevance of genetic structure in ecotype designation and conservation management. Evol Appl 15:185–202
SwissLepTeam (2010) Die Schmetterlinge (Lepidoptera) Der Schweiz: eine kommentierte, systematisch-faunistische Liste. Fauna Helv 25
Tartally A, Thomas JA, Anton C, Balletto E, Barbero F, Bonelli S et al (2019) Patterns of host use by brood parasitic maculinea butterflies across Europe. Phil Trans Royal Soc B 374:20180202
Taylor RS, Friesen VL (2017) The role of allochrony in speciation. Mol Ecol 26:3330–3342
Theissinger K, Fernandes C, Formenti G, Bista I, Berg PR, Bleidorn C et al (2023) How genomics can help biodiversity conservation. Trends Genet S0168952523000203
Wermeille E, Chittaro Y, Gonseth Y (2014) Rote Liste Der Tagfalter Und Widderchen: Papilionidea, Hesperioidea Und Zygaenidae: Gefährdete Arten Der Schweiz, stand 2012. Bundesamt für Umwelt BAFU, Schweiz
Wiemers M, Balletto E, Dincă V, Fric ZF, Lamas G, Lukhtanov V et al (2018) An updated checklist of the European butterflies (Lepidoptera, Papilionoidea). ZooKeys 811:9–45
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wipking W, Jaun A, Wymann H-P (2023) Larval food-plant of alpine populations of the high mountain ant blue Phengaris rebeli Hirschke, 1904 in the Bernese Oberland (Switzerland) (Lepidoptera: Lycaenidae). Entomol Zeitschr 133: 1-4
Acknowledgements
We are grateful for the cantons Bern, Graubünden, Valais, Vaud, Zug and Zürich for providing sampling permits. We are further indebted to Rolf Kugler for providing specimens and Emmanuel Rey for making the European distribution map.
Funding
Sequencing of the reference genome assembly supported by the Canton of Zürich through project funding to GD. The resequencing was funded by the Stiftung Emilia Guggenheim-Schnurr of the Naturforschenden Gesellschaft in Basel. KL was funded by a Swiss National Science Foundation Eccellenza Project The evolution of strong reproductive barriers towards the completion of speciation (PCEFP3_202869).
Open access funding provided by University of Neuchâtel
Author information
Authors and Affiliations
Contributions
Conceptualization: KL, NW; Funding acquisition: KL, GD; Formal analyses: KL, LB, CC; Resources: YC, AE, AJ, BJ, HPW GD; Writing – Original Draft preparation: KL; Writing – Review & Editing: KL, LB, CC, YC, AE, AJ, BJ, NW, HPW, GD.
Corresponding author
Ethics declarations
Conflict of interest
No conflicts of interests occurred.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lucek, K., Blattner, L., Cornet, C. et al. Towards a genomic resolution of the Phengaris alcon species complex. Conserv Genet (2024). https://doi.org/10.1007/s10592-024-01605-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10592-024-01605-x