
Abstract
Conservation translocations can restore populations and prevent extinction of threatened species. Sourcing adequate genetic diversity is an essential consideration when planning reintroductions, because it influences individual fitness and long-term persistence of populations, yet available populations of threatened species may lack diversity. We estimated population genetic parameters for one of Australia’s most threatened mammals, the northern bettong, Bettongia tropica, to select source populations for reintroduction. Individuals from sites across the species’ extant range in the Wet Tropics of north Queensland were genotyped, using 6,133 informative SNPs. We found that samples clustered into four populations: an isolated northern population at Mt Spurgeon and three connected southern populations in the Lamb Range. Most of the species’ genetic diversity was dispersed across the Lamb Range populations in approximately equal proportions. Populations showed an isolation-by-distance effect, even over short distances within continuous habitat. Admixture of populations was high at distances < 7 km but low at distances > 11 km, and there was asymmetrical gene flow between the two closest neighboring populations. All populations had small effective sizes and experienced drift, but connectivity appears to have mitigated drift and stabilized population sizes within the Lamb Range. The Mt Spurgeon population had a very small effective population size and low genetic diversity. We use our findings to weigh up the risks and benefits of mixing sources for reintroduction, and we recommend a mixed source approach. We do not currently recommend sourcing individuals from Mt Spurgeon and conservation efforts to preserve this population are urgently required.
Similar content being viewed by others

Introduction
Reintroduction, via the translocation of wild animals, is a potentially powerful conservation tool increasingly being used to aid recovery in declining species (IUCN 2013; Taylor et al. 2017). The benefits of conservation translocations include: increasing the range extent, increasing total population size or number of populations, improving metapopulation stability, increasing connectivity and gene flow, restoring ecosystem processes and function, and restoring cultural values to a landscape (Noble et al. 2001; IUCN 2013; Armstrong et al. 2015). We are in the midst of a global biodiversity crisis and such benefits are sorely needed (WWF 2020). However, reintroductions are not always successful at reestablishing self-sustaining, viable populations (Fischer and Lindenmayer 2000; Moseby et al. 2011; Short 2016). Thorough understanding of the species’ biology and knowledge of specific factors that affect establishment success and long-term viability are necessary prior to reintroduction to allow informed decisions that benefit the overall recovery of the species (IUCN 2013).
Genetic factors affect both the fitness of translocated individuals and the long-term survival of reintroduced populations (Di Fonzo et al. 2011). Isolated populations, living in environments that differ, can develop divergent local adaptations and this needs to be considered when choosing population sources to avoid the reintroduction of individuals that are locally maladapted (Houde et al. 2015). Another major consideration for reintroductions is sourcing and maintenance of adequate genetic diversity (Armstrong et al. 2015). Since genetic diversity is the raw material upon which natural selection acts, sourcing and maintaining high diversity will help reintroduced populations adapt to future conditions, increasing their long-term resilience to environmental change (Weeks et al. 2011). Genetic diversity also directly affects population viability in the short-term, as low genetic diversity is associated with inbreeding depression and reduced fitness caused by the increased expression of deleterious recessive alleles (Frankham et al. 2017a; Kyriazis et al. 2021). Furthermore, genetic fitness can interact synergistically with other threats, such as predation (Pacioni et al. 2017). Thus, founder diversity can be an even more important genetic determinant of successful reestablishment than pre-existing local adaptation (Zeisset and Beebee 2013; Maschinski et al. 2013).
One option to increase the diversity of reintroduced populations is to mix sources (Houde et al. 2015). Mixing divergent populations can have a remarkably positive effect on short- to medium-term fitness and population growth (Frankham 2015; Weeks et al. 2017; Fitzpatrick et al. 2020). This ‘hybrid vigor’ effect is thought to be a consequence of increased heterozygosity in F1 and F2 hybrids suppressing the expression of deleterious recessive alleles (Weeks et al. 2011; Hoffmann et al. 2020). However, such mixing also risks outbreeding depression, and populations that are adapted to different local environments may conversely show lower fitness when mixed (Marshall and Spalton 2000; Goldberg et al. 2005; Sagvik et al. 2005; Byrne and Silla 2020). This effect may occur due to watering down of adaptive traits, or genetic incompatibility, or both (Frankham et al. 2011).
Fear of outbreeding depression has caused conservation managers to avoid mixing populations that are genetically distinct. Yet, local adaptation is not the only evolutionary force leading to population divergence, random genetic drift often contributes substantially to population divergence, especially when populations have become small and fragmented. Random drift is important in small populations because the rate of genetic drift is inversely proportional to effective population size, leading to lower genetic diversity, fitness, and adaptive potential, but higher genetic ‘distinctness’ in smaller populations (Weeks et al. 2016). Since admixture following genetic drift of historically connected populations rarely leads to outbreeding depression, the net risk of outbreeding depression is often overestimated in threatened species (Frankham et al. 2011). A recent review showed that the ‘default’ recommendation has been to manage genetically distinct populations separately, yet when both inbreeding and outbreeding depression risks are evaluated, population mixing is more frequently recommended (Liddell et al. 2021). The relative roles of genetic drift and local adaptation in population history should be considered when deciding whether to reintroduce individuals from one or more sources.
Australia has suffered a 10% attrition of its mammal species in the last 250 years, the highest rate of mammal extinctions in the world (Woinarski et al. 2019). Declines are continuing across northern Australia and are affecting most small- to medium-sized mammals (Burbidge and McKenzie 1989; Woinarski et al. 2011, 2015; Woinarski 2015). Reintroduction has had mixed results, and predation has been one of the biggest factors in failed attempts (Short 2009; Moseby et al. 2011). To counter that issue, reintroductions to predator-proof enclosures, or islands, have proved a highly successful strategy (Kanowski et al. 2018; Legge et al. 2018).
Members of the genus Bettongia (Potoroididae) are highly threatened but tend to do well once threats are removed (Moseby et al. 2018). Populations of three of the four extant Bettongia species have been successfully reestablished in Australia in the last 30 years to predator-free havens (Martin et al. 2006; Portas et al. 2016; Legge et al. 2018; Thavornkanlapachai et al. 2019). The fourth species, the northern bettong (B. tropica) is listed as Endangered (Burbidge and Woinarski 2016; DAWE 2022) and is among the top 20 Australian mammals identified as being most at risk of extinction (Geyle et al. 2018). Two B. tropica population localities have disappeared in the last 30 years, leaving the species’ persistence tenuously hanging on the survival of populations in two remaining localities (WWF-Australia 2018). According to Woinarski (2018) there is a 14% risk of the species going extinct within the next 20 years, unless action is taken. Threats causing contemporary decline of B. tropica are likely reduced habitat quality (associated with altered fire regimes, feral cattle, weed invasions, and pigs) and predation by feral cats (Dennis 2001).
The habitat requirements of B. tropica are well understood, making it an ideal candidate for reintroduction. Yet, besides one poorly planned and unsuccessful attempt in 2005 (DEHP 2014), reintroduction has not been implemented for the conservation of this species. Translocation of wild B. tropica individuals to a 950-ha feral and predator-free exclosure managed by the Australian Wildlife Conservancy has been proposed. The proposed site is near Paluma, in the Coane Range, and within the species’ former range, where localised extinction occurred as recently as 2003. Ongoing intensive vegetation management at that site is restoring habitat quality, and all feral animals, predators and competitors will be excluded from within the fenced area prior to reintroduction. Previous genetic work has indicated that there are likely four potential source populations (Pope et al. 2000). However, the necessary detailed information on contemporary population genetics of B. tropica is lacking. This study aims to quantify the genetic diversity and structure of existing populations to make recommendations for sourcing individuals for reintroduction. Our objective is to maximise the chance of reintroduction success, while minimizing the impact on source populations, thereby improving the species’ overall conservation status.
Materials and methods
Populations and sampling
Bettong ear tissue samples (n = 240) were collected during cage trapping at five sites between 2014 and 2021 and refrigerated in 80% ethanol. Four sites were within a continuous band of habitat in the Lamb Range, which is often grouped together as a single locality (e.g., Burbidge and Woinarski 2016). These were: Lower Davies Creek (n = 21), Upper Davies Creek (n = 54), Emu Creek (n = 61), and Tinaroo (n = 84). Upper and Lower Davies Creek are separated by 1.5 km east-west in the north of the Lamb Range; Emu Creek is in the middle of the Lamb Range, 11 km south of Davies Creek and 6.5 km north of Tinaroo, which is at the southern end of the Lamb Range (Fig. 1). Davies Creek, Emu Creek, and Tinaroo were expected to be inhabited by genetically distinct populations based on the findings of Pope et al. (2000). The fifth site, Mount Spurgeon (n = 21), is a geographically isolated population 75 km north of Davies Creek, within Mt Lewis National Park in the Carbine Tablelands. Two additional samples were obtained opportunistically from fresh roadkill within the Lamb Range: one from Tinaroo, and one from Koah, 7 km north of Lower Davies Creek. The Lamb Range and Mt Spurgeon are the only two areas where B. tropica persist (WWF-Australia 2018). Trap locations within these sites are shown in Fig. 1.

Extant and recently extinct Bettongia tropica population localities, and genetic sampling locations for this study. Population names are indicated by the labels. Davies Creek, Emu Creek and Tinaroo occur within a single area of occupied habitat and are therefore also referred to collectively as the ‘Lamb Range populations’. An opportunistic roadkill sample was included from Koah, north of Davies Creek and it is unknown if this individual represents a genetically distinct population. Polygons for extinct populations were modified from Laidlaw et al. (2012)
The degree of genetic differentiation among the Upper and Lower Davies Creek sampled sites was initially unknown. We examined the likely number and composition of genetic populations using methods described in Sect. 2.3 below. This resulted in pooling of Upper and Lower Davies Creek sites into a single population, Davies Creek (n = 75), for subsequent analyses.
Genotyping and quality control filtering
DNA was extracted from samples using a modified salting-out method (Sunnucks and Hales 1996). Library preparation, array-based sequencing, and in silico genotyping of B. tropica individuals for 28,915 Single Nucleotide Polymorphism (SNP) markers were performed by Diversity Arrays Technology (DArT), Canberra, at a depth of 2.5 million reads. Reads were aligned to a Bettongia gaimardi reference genome provided by the Oz Mammals Genomics initiative to improve SNP calling. The following quality control filtering steps were then applied to the data using the R package dartR (Mijangos et al. 2022): loci with < 90% reproducibility (i.e., identical sequences in < 90% of reads) were removed; loci with mean read depth < 10 or > 1000 were removed; all secondary SNPs (more than one SNP on the same read fragment) were removed; individuals were filtered to > 80% call rate to remove any poor-quality samples; SNP loci were filtered to 100% call rate (i.e., loci with any missing data were removed); and finally loci which were significantly out of Hardy-Weinberg equilibrium in one or more population, with a Bonferroni correction applied to the significance threshold (α = 0.05/number of loci), were removed (49 total) to eliminate likely sequencing errors (we defined “populations” for this test based on Pope et al. (2000) and a preliminary assessment of population genetic structure). After filtering, 241 genotypes (individuals) and 6,133 loci remained.
Divergence & population structure
Principal Component Analysis (PCA) was used to visualize the distribution of the species’ genetic variance along ordination axes (Pearson 1901; Jolliffe 2003). Population groupings were inferred from Discriminant Analysis of Principal Components (DAPC) using the R packages SambaR v.1.08 (Jong et al. 2020) and adegenet v2.1.5 (Jombart 2008). First, to ascertain the number of genetic populations, de novo K-means clustering was performed. For this step, 185 principal components explaining 95% of the total variance were retained, as recommended by Jombart & Collins (2015), and possible numbers of clusters up to K = 10 were explored. The optimal number of clusters was determined by examining the resulting BIC vs. K plot for a minimum value or ‘elbow’ in the curve. Geographic population boundaries were determined from inferred clusters, and redefined populations were used in all subsequent analysis.
DAPC was then rerun to identify possible migrants by assigning individuals to inferred genetic clusters. For this second step, the number of principal components used for clustering was based on both a-score and cross-validation methods: the minimum number to retain was demined by plotting the increase in successful cross-validation predictions with additional PCs, while the maximum number to retain was determined by the maximum value in the a-score spline plot. This resulted in eight principal components being retained, which together explained ~ 25% of the total variance. The number of discriminant functions was set to three (K-1). This approach was designed to optimize the trade-off between including sufficient information for discrimination, and overfitting or instability of group assignments. Genetic clusters were assumed to equate to natal populations, and disagreement between capture and assigned population was interpreted as signifying migration (i.e., migration by the individual in question or by a recent ancestor).
Population divergence was estimated from Nei and Chesser’s sample size bias corrected genome-wide FST ‘GST’ (Nei and Chesser 1983) calculated using R package FinePop2 v0.4 (Kitada et al. 2021) and by comparing private, fixed, and mean frequency differences in alleles between each pair of populations. For these comparisons, each of the Lamb Range samples were rarefied to the size of the smaller Mt Spurgeon sample (n = 21) 1000 times (bootstrapped) and averaged. Unrarefied private and fixed alleles were also calculated between each population and a pooled sample containing the reset of the populations to identify the number of alleles unique to each. Weir and Goudet’s (2017) measure of population-specific FST was calculated in the R package FinePop2 (Kitada et al. 2021). This statistic measures deviation of each population from the hypothetical ancestral population (Kitada et al. 2021). To evaluate the role of isolation-by-distance in population divergence, a Mantel test was performed on pairwise individual Euclidean genetic and geographic distances.
To examine the relative roles of past diversity loss due to genetic drift versus accumulation of potentially adaptive variation in shaping population differences, genetic diversity metrics were regressed against population-specific FST. Weeks et al. (2016) demonstrated that a close relationship between population-specific FST and genetic diversity (linear for heterozygosity and quadratic for allelic richness) occurs when population differences are primarily a consequence of drift. Conversely, if population divergence is due to new mutations and local adaptation, then population-specific FST will have a weaker relationship with genetic diversity (Weeks et al. 2016). Expected heterozygosity and allelic richness calculations are described in Sect. 2.4 below.
Structure and contemporary gene flow between populations was assessed using individual admixture coefficients estimated using the R package LEA-3.3.2 (Frichot and François 2015). Least-squares estimates of ancestry proportions were iteratively estimated using nonnegative matrix factorization (sNMF), for values of K ancestral populations from two up to the optimal number of clusters determined by minimum cross-entropy. This method was chosen for admixture analysis over the functionally similar program STRUCTURE (Pritchard et al. 2000) because it is faster and can be better at detecting structure in the presence of inbreeding (Frichot et al. 2014), which may occur in small B. tropica populations. Admixture proportions were used to corroborate migrants identified by DAPC.
Genetic diversity
Population level genetic diversity was assessed using the following diversity metrics: allelic richness (AR), Shannon index (SI), observed heterozygosity (HO), and expected heterozygosity (HE), calculated in DartR (Mijangos et al. 2022). Multilocus observed heterozygosity (MLH) of each individual was estimated, and a Kruskal-Wallis rank sum test followed by a Wilcoxon test with continuity correction was used to compare MLH between populations.
Since estimates of allelic richness are sensitive to sample size (Shafer et al. 2015), allelic richness of each Lamb Range population was also calculated from a rarefied subsample equal in size to the smaller Mt Spurgeon sample (21). Rarefication of individuals was bootstrapped using 1000 permutations to create probability distributions for allelic richness that were approximately normal, against which population differences could be tested. ANOVA was used to compare rarefied allelic richness among the three Lamb Range populations. In pairwise comparisons between Mt Spurgeon and each of the Lamb Range populations, the rarefied Lamb Range distribution was treated as a ‘null’ distribution against which the single, unrarefied allelic richness estimate for Mt Spurgeon was tested. If the Mt Spurgeon estimate fell outside the 95% prediction interval, then we considered it significantly different (see Dyer 2017).
Observed and expected heterozygosity were also compared within each population. Fisher’s exact tests of Hardy-Weinberg Equilibrium (HWE) were performed for loci within each population, according to the method of Wigginton et al. (2005) and using DartR (Mijangos et al. 2022). However, the loci most strongly deviating from HWE (i.e., that were significant after applying a Bonferroni correction), were previously removed from the dataset during data filtering; see Sect. 2.1 above. Global (across loci, within-population) HWE was determined by comparing the number of significant (P < 0.05) loci remaining within each population against the number expected to be significant due to chance (i.e., a null binomial distribution with 5% probability of ‘success’ for HWE tests at alpha = 0.05 and number of ‘trials’ = number variable loci), as suggested by Waples et al. (2015). Global disequilibrium was determined at a one-tailed significance level of P < 0.05. Population differences were investigated by identifying whether loci deviations from HW proportions were characterized by a heterozygosity excess or deficit, using Nei’s (1986) inbreeding coefficient, FIS.
Effective population size
Effective population size (Ne) was estimated using the Linkage Disequilibrium (LD) method implemented in Ne Estimator v2.1 (Do et al. 2014). This method estimates Ne from LD measured as the squared correlation coefficient (r2) between alleles at pairs of loci. The processes of genetic drift and inbreeding cause the level of LD to be higher in smaller effective populations, enabling effective population size to be estimated accurately from genetic data (Waples 2006; Waples and Do 2010; Gilbert and Whitlock 2015). Since the 6,133 loci used to produce Ne estimates are distributed across only 22 B. tropica chromosomes, some loci will be physically linked, confounding LD, and biasing Ne estimates downward. To account for this bias, the chromosome correction factor of Waples et al. (2016) was applied to the output of Ne Estimator. Sample size bias was automatically adjusted for within Ne Estimator (Waples 2006; Do et al. 2014), and minor allele frequency was filtered to > 0.02 within the software settings (Waples and Do 2010).
To investigate the possible effect of gene flow on Ne estimates, Ne was calculated both including and excluding migrants for populations where migration was detected. Probable first and second generation (F0 & F1) migrants were identified as individuals with < 60% local ancestry according to the LEA admixture analysis. Discrepancies between effective size estimates with and without migrants were subsequently used to estimate migration rate by rearranging Equation A1 in Waples and England (2011) to:
Where \(m\) = immigration rate, \({\widehat{N}}_{EPP}\)= estimated effective pool of parents (including parents of immigrants), and \({\widehat{N}}_{e}\) = estimated local effective size (without migrants). For simplicity only two populations were considered (i.e., n = 2 in Waples and England’s original formulation), equilibrium migration was assumed, and the contribution of individuals with immigrant ancestry ≤ 40% to the parent pool were ignored.
Results
Delineation and structure of potential source populations
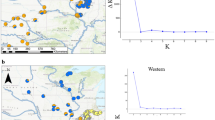
The data were best explained by four population clusters, indicated by the lowest point in the BIC curve produced by step one of the DAPC analysis, and by the lowest cross-entropy score from the LEA analysis. However, both the BIC and cross-entropy values for K = 4 were only slightly lower than for K = 3 (Fig. 2). No genetic division between Upper and Lower Davies Creek was observed by DAPC (Fig. 2) or PCA (Fig. 3), therefore these sites were pooled into a single ‘Davies Creek’ population. Genetic population boundaries otherwise matched sample sites.

Discriminant Analysis of Principal Components (DAPC) of Bettongia tropica genetic diversity. De novo K-means clustering was first used to determine the number of genetic populations (a), with optimal number of clusters determined at the lowest BIC value plotted (i.e., at K = 4). For this step 186 Principal Components were retained which explained 95% of the total variance. Individuals were then assigned to the four clusters, and (b) shows a confusion matrix of inferred vs. expected clusters from the capture locations. Misassigned (potential migrant) individuals are circled. For this second DAPC step eight Principal Components were retained which explained ~ 25% of the total variance

Principal component analysis of Bettongia tropica genetic distance showing individuals (plotted on the two most explanatory ordination axes) and population labels
Mt Spurgeon was genetically distant from the three Lamb Range populations (Fig. 3). There were five fixed allele differences between the pooled Lamb Range populations and Mt Spurgeon, and the pooled Lamb Range populations had 3,311 private alleles (i.e., they were not observed in Mt Spurgeon). A substantially smaller number of alleles were private to Mt Spurgeon (268), which was approximately the same as the number of alleles unique to the Davies Creek population (231). Tinaroo and Emu Creek populations had 19 and 41 private alleles, respectively, but together they had 289. In pairwise comparisons between each of the populations, there were approximately four times more alleles private to the Lamb Range populations than to Mt Spurgeon even after rarefying to account for small sample size at Mt Spurgeon (Table 1). GST values were also higher in all comparisons between Mt Spurgeon and the Lamb Range populations than in comparisons between populations within the Lamb Range (Table 1). There was little evidence of contemporary gene flow between Mt Spurgeon and the Lamb Range; individuals from Mt Spurgeon had < 1% Lamb Range ancestry, and Lamb Range individuals had < 7% Mt Spurgeon ancestry (Fig. 4).

Admixture of Bettongia tropica individuals. LEA least-squares estimates of ancestry proportions were iteratively estimated for K ancestral populations from K = 2 up to K = 4, which was the optimal number of clusters determined by DAPC. Columns represent individual probability of ancestry (0–1) belonging to each of the colored clusters. At K = 4, clusters represent sampled populations
There was also genetic structure observed within the Lamb Range. This is illustrated by PCA axis two in Fig. 3, which explains > 10% of the total variation. There was a significant isolation-by-distance effect revealed by the Mantel test (R2 = 0.623, P < 0.001), even after Mt Spurgeon individuals were removed from the analysis (R2 = 0.427, P < 0.001; Fig. 5). Davies Creek was the most genetically distinct among Lamb Range populations according to GST, private alleles, and the PCA (Table 1; Fig. 3). There was 100% concordance between individuals captured at Davies Creek and DAPC assignment to the Davies Creek genetic cluster (i.e., no individuals were flagged as potential immigrants; Fig. 2). According to the LEA analysis only eight out of 75 (10%) of Davies Creek individuals showed admixture > 20%, and the greatest admixture of a Davies Creek individual was 38% non-Davies ancestry (23% Emu Creek, 13% Tinaroo, 1.7% Mt Spurgeon; Fig. 4).

Isolation-by-distance of B. tropica individuals based on Mantel tests of Euclidean genetic distance and natural log geographic distance. Grey (dark) circles = pairwise comparisons within the Lamb Range, pink (light) triangles = comparisons containing individuals from Mt Spurgeon. Two tests were performed: one with all the samples (solid line), and another with Lamb Range individuals only (dashed line). Mt Spurgeon individuals were excluded from this second test because the Mt Spurgeon population was both genetically distant and far away, and therefore may have disproportionately influenced the results
There was some discordance between capture population and DAPC assignment of individuals to Emu Creek and Tinaroo. One individual captured at Emu Creek was ‘misassigned’ to the Tinaroo cluster, whilst 14 individuals captured at Tinaroo were ‘misassigned’ to the Emu Creek cluster, totalling 15 migrants (Fig. 2). For 11 of these 15 individuals, the LEA admixture analysis also showed that they had higher non-local than local ancestry. The four individuals for which the two analysis methods disagreed were all caught at Tinaroo. However most (12 out of 15) DAPC identified migrants had LEA ancestry proportions in the range 40–60%, thus these individuals are likely admixed second-generation migrants and classification as ‘migrants’ may be arbitrary. Overall, mean Emu Creek ancestry at Tinaroo was 24.9% and mean Tinaroo ancestry at Emu Creek was 16.8%.
Genetic diversity and effective size of potential source populations
Unadjusted allelic richness was similar among all Lamb Range populations (Davies Creek = 1.88; Emu Creek = 1.86; Tinaroo = 1.85), but lower in Mt Spurgeon (1.46). On average, rarefaction reduced allelic richness estimates for the Lamb Range populations by 4–5% (Fig. 6a). Mt Spurgeon allelic richness fell outside the 95% prediction intervals of all Lamb Range population rarified null distributions, and no permutation produced an allelic richness estimate lower than Mt Spurgeon. The three Lamb Range populations were also significantly different from each other according to the ANOVA of rarified allelic richness (P < 0.001), with a Tukey’s HSD test indicating that Davies Creek > Emu Creek > Tinaroo (Fig. 6a; Table 2). Mt Spurgeon was also lower in heterozygosity and Shannon diversity than the Lamb Range populations, while the three Lamb Range populations had similar levels of heterozygosity and Shannon diversity (Table 2).

Genetic diversity of the four Bettongia tropica populations. (a) Rarefied allelic richness (box and whiskers) and unadjusted sample allelic richness (triangles). Rarefication of the three larger samples to the smallest sample was bootstrapped, giving the whisker range. (b) Individual multilocus heterozygosity (MLH). Black points = ‘mild’ outliers (> 1.5 * IQR above or below the hinge); red asterisks = ‘extreme’ outliers (> 3 * IQR above or below the hinge). Notches show approximate 95% confidence interval around medians
There was a significant difference in individual multilocus heterozygosity (MLH) between populations revealed by the Kruskal-Wallis rank sum test (χ2 = 59.417, DF = 3, P < 0.001), again due to individuals from Mt Spurgeon having lower heterozygosity than individuals from Lamb Range populations (Wilcoxon signed-rank P < 0.001). There were no significant differences in pairwise comparisons of MLH between any of the Lamb Range populations (p > 0.57). Within each population, individuals varied in MLH, and there were three extreme outliers (defined as < Q1–3 * IQR or > Q3 + 3 * IQR; Mcgill et al. 1978). There was one individual with unusually high heterozygosity in each of the Davies Creek and Tinaroo samples, and one individual with unusually low heterozygosity in the Tinaroo sample (Fig. 6b).
Globally, all populations were in Hardy-Weinberg Equilibrium (P = 0.847–0.999). There were a small percentage of loci remaining out of equilibrium within each population (2.7–4.7%; Table 2), however these were not more frequent than expected by chance. Amongst significant loci there was a mean heterozygosity deficit, whereas across all loci there was a mean heterozygosity excess. This was true in all populations (Table 2).
Effective population size (Ne) estimates were small for all B. tropica populations. Full sample estimates ranged from 53 to 71 in the Lamb Range and 31 in Mt Spurgeon (Table 2). Mt Spurgeon’s estimate of Ne was significantly lower (determined by non-overlapping jackknife 95% CIs) than estimates in Davies Creek and Tinaroo, but not Emu Creek. There were no significant differences in NE estimates among Lamb Range populations. For subsequent analyses, 26 admixed individuals with > 40% migrant ancestry were identified and excluded from Tinaroo, and nine from Emu Creek. All Davies Creek and Mt Spurgeon individuals had > 60% local ancestry, however the roadkill sample from Koah was removed as it may belong to a genetically differentiated but otherwise unsampled population. Excluding migrants reduced Ne estimates for both Emu Creek (53 down to 38) and Tinaroo (70 down to 51), which meant that migration rate could be estimated between these populations using Eq. 1. Immigration rate was high in both populations, but slightly higher at Emu Creek (0.176) than Tinaroo (0.164).
Population-specific FST was highly correlated with allelic richness and expected heterozygosity (Fig. 7). A linear model best fit the relationship between He and FST, with the correlation being almost perfect (R2 > 0.999; P < 0.001; DF = 2). A quadratic model best fit the relationship between rarefied AR and population specific FST, which was also extremely tight (R2 > 0.999; P = 0.001; DF = 1).

Relationship between genetic diversity and genetic uniqueness of four Bettongia tropica populations. Two measures of genetic diversity were used: (a) expected heterozygosity and (b) rarefied allelic richness. Population-specific FST was used to measure genetic uniqueness. A high correlation indicates genetic drift (loss of diversity) is driving the genetic divergence of populations
Discussion
Reestablishing B. tropica in the southern part of its former range could greatly improve the species’ conservation status. Understanding source population genetics improves the chance of successful reintroduction and establishment of a genetically diverse population. We found four, existing, genetically differentiated populations, from which individuals could potentially be sourced for reintroduction. However, these populations had differing levels of genetic diversity, and there were differences in the degree to which populations were genetically isolated. One population, occurring at Mt Spurgeon, had a particularly small effective size and low genetic diversity. We use our findings to develop recommendations for reintroduction.
Genetic characteristics of extant B. tropica populations
Genetic diversity was similar across the three Lamb Range populations, but lower at Mt Spurgeon. Genetic diversity is important for fitness and adaption, and low genetic diversity is associated with inbreeding depression (Alho et al. 2009; Armstrong et al. 2015; énos et al. 2016; Frankham et al. 2017a; Weeks et al. 2017). The heterozygosity estimate for Mt Spurgeon is very low compared to most estimates for wild mammal populations found in the literature, with a few exceptions from endangered species such as arctic ringed seals, polar bears, black bears and western barred bandicoots (Kjeldsen et al. 2016; White et al. 2018). The Lamb Range populations have similar heterozygosity to other endangered marsupial species that have suffered major population declines such as natural (non-admixed) burrowing bettong (Bettongia lesueur) populations (White et al. 2018; Rick et al. 2019) and reintroduced or captive greater bilby (Macrotis lagotis) populations (Lott et al. 2020). The Lamb Range average heterozygosity is slightly lower than estimates for species with large or outbred populations (Kjeldsen et al. 2016). Nevertheless, cross-study comparisons of SNP heterozygosity should be viewed cautiously, and inference is limited by ascertainment and sample size biases (Schmidt et al. 2020).
All B. tropica populations had small effective sizes, suggesting that they may be vulnerable to further diversity loss through genetic drift. This has implications for the long-term survival of these populations and for management, including reintroduction. The Mt Spurgeon estimate fell below the ‘50’ minimum viable size threshold for preventing immediate inbreeding effects (Harmon and Braude 2010; Jamieson and Allendorf 2012). Populations will theoretically lose heterozygosity at a rate of 1/(2*Ne) due to drift, thus an Ne estimate of 31 predicts a loss of 1.61% heterozygosity per generation, potentially explaining the low diversity observed at Mt Spurgeon. Although larger than Mt Spurgeon, effective population size estimates in the Lamb Range were well below the ‘500’ effective individuals theoretically required for long-term population viability. Moreover, Frankham et al. (2014) argue that the 50/500 rule is too low, and in fact more than 100 effective individuals are required to keep fitness decline due to inbreeding depression below 10%. Yet, despite the implications of having small effective sizes, populations in the Lamb Range appear to have been stable over the last 20 years (Whitehead 2018).
Removing migrants from the Emu Creek and Tinaroo samples reduced effective size estimates. Fewer immigrants may have had proportionally higher effect on Emu Creek effective size estimates compared to Tinaroo because of the smaller relative size of that population. Over-estimation of local effective size from linkage disequilibrium is expected when migration levels are high and migrant individuals are included in the sample, because the parent pool being sampled is actually the metapopulation, not just the local population (Waples and England 2011). Whilst using ‘local’ estimates (i.e., excluding migrants) is necessary to avoid double counting when inferring census size from effective size, we argue that for our objective of assessing drift risk, ‘metapopulation’ estimates (i.e., including migrants) are more suitable because they implicitly capture the mitigating effect of gene flow on drift. Indeed, we found that even though Emu Creek and Mt Spurgeon have similar local effective sizes, Emu has retained significantly higher genetic diversity and thus is less susceptible to inbreeding depression. Gene flow greatly increases population persistence (Mims et al. 2019), therefore connectivity likely plays a crucial role in maintaining genetic diversity and preventing decline of the Lamb Range populations, and explains the population stability.
Genetic distinctness was correlated with population genetic diversity, suggesting drift is the primary driver of genetic structure in this species. In fact, almost the entire variance in population divergence, measured by population-specific FST, was explained by genetic drift. Mt Spurgeon is the most geographically isolated population, and as expected, was also the most genetically distinct. However, the genetic uniqueness of Mt Spurgeon was predominantly characterized by an absence of genetic diversity found in the Lamb Range. In other words, many loci heterozygous in other populations were homozygous in all individuals sampled at Mt Spurgeon. This suggests strong drift has led to a loss of ancestral diversity and fixation at Mt Spurgeon since the time it diverged from its last common ancestor with the Lamb Range populations. Given the small effective size, it is unsurprising that little novel, potentially adaptive, variation has accrued at Mt Spurgeon. This adds to the growing body of evidence that maladaptive drift is a dominant force in the genetic differentiation of threatened species (Coleman et al. 2013; Weeks et al. 2016; Casey et al. 2018).
Genetic and geographic distances were much shorter between the Lamb Range populations, and we found genetic evidence of recent migration among populations. The three Lamb Range populations occur in continuous habitat and individuals are known to be capable of dispersing at least several kilometers. However, we also found an isolation-by-distance effect occurring over short distances, confirming the findings of Pope et al. (2000). Populations were panmictic at 1.5 km (Lower – Upper Davies Creek), but structure appeared at 7 km (Emu Creek – Tinaroo), and then jumped to a much greater level of isolation at 11 km (Davies Creek – Emu Creek). Gene flow is insufficient for panmixia across the Lamb Range.
The observed patterns of B. tropica genetic structure may, in part, reflect their historic biogeography. Pope et al. (2000) showed there was a deep split in B. tropica mitochondrial haplotypes within the Lamb Range, dividing northern B. tropica populations (i.e., Davies Creek, Mt Spurgeon and the now extinct Mt Windsor population) from southern populations (Tinaroo, and the now extinct Coane Range population), with admixture at Emu Creek (see Fig. 1). Pope et al. hypothesized this divergence was associated with periods of contraction of Wet Tropics rainforest during the Pleistocene, with secondary contact occurring relatively recently around 5000–7000 years ago when rainforest expanded (Moritz et al. 2009; Graham et al. 2010). It’s possible that some of this relic structure remains in the nuclear genome, if migration has been extremely limited over millennia. Conversely, if migration has occurred consistently at the rate we observed, it would have presumably dissolved historic structure within this timeframe. We found that gene flow favored dispersal from Emu Creek south to Tinaroo, however this is opposite to the northward migration bias that was recorded in the late 1990s (Pope et al. 2000), suggesting that dispersal patterns are in flux and migration may have occurred in pulses rather than at a steady equilibrium rate. Alternatively, structure could be evolving dynamically, if very strong drift (due to the small effective size) is consistently causing diversity loss at a rate faster than is replenished by gene flow. Population history of B. tropica is likely complex and is yet unresolved.
Recommendations for reintroduction
Based on our genetic findings and existing information, we argue that the best strategy would be a ‘mixed source’ approach to reintroduction (Houde et al. 2015), whereby individuals are sourced from all three Lamb Range populations, rather than sourcing all individuals from a single population. This recommendation is supported by four arguments.
First, there are no closely related, or environmentally matched, populations available that are likely to confer pre-existing adaptation to the proposed reintroduction site. Until 2003, a natural B. tropica population occurred at the site, however, since the species’ regional extinction from the Coane Range, the nearest extant population is 250 km north, at Tinaroo in the Lamb Range. Pope et al. (2000), who sampled 10 individuals from the Coane Range population prior to its extinction, did find mitochondrial sequences more closely related to Tinaroo than the other populations, and Houde et al. (2015) recommended that if population ancestry could be identified, then the closest ancestral match should be used as the source to reestablish an extinct population. However, this is based on the assumption of genetic similarity, and genomic patterns are often discordant with mitochondrial patterns due to complex population histories of isolation and admixture (Singhal and Moritz 2013), as appears to be the case for B. tropica (Pope et al. 2000). Moreover, the environmental differences, and therefore divergent selection pressures, are far greater between the Coane Range and Lamb Range than they are within the Lamb Range. For example, the Coane Range is drier and more seasonally variable than the Lamb Range (Bateman et al. 2012). Thus, it is unlikely that the shared ancestry between Coane Range and Tinaroo (Lamb Range) populations would endow Tinaroo animals with fitness advantages over the other Lamb Range populations. In the absence of a source population with conferrable local adaptations, maximising diversity is the recommended approach for selecting sources (Houde et al. 2015).
Second, while we found the Lamb Range populations each have higher genetic diversity and larger effective size than Mt Spurgeon, they are still small and have each lost a (different) portion of the species’ diversity through genetic drift. The reintroduced population would most likely benefit from sourcing individuals from each of the populations so that more of the species’ diversity is captured, making it better equipped to adapt (Weeks et al. 2011). Offspring of mixed parents may also have increased fitness due to genetic rescue, helping the new population establish and increase quickly to a self-sustaining size (Frankham 2015; Weeks et al. 2017; MacFarlane 2019; Fitzpatrick et al. 2020). Rapid increase in size after founding from a few individuals is important to avert major genetic drift and allow purging of deleterious recessive alleles (Hoffmann et al. 2020). Other examples of bettong genetic rescue attest to the benefit of mixing, across subspecies (Rick et al. 2019).
Third, the risk of outbreeding depression from mixing sources is low. We have demonstrated that the population differences within B. tropica are primarily attributable to genetic drift, and drift rarely generates outbreeding depression unless it has led to fixed chromosome differences ((Frankham et al. 2017b). While it is unknown if there is any karyotype variation within B. tropica populations, it seems improbable, given chromosomes across species within this genus are conserved (Sharman et al. 1980). Outbreeding depression is most likely to occur when mixing allopatric populations adapted to different environments (e.g., Byrne and Silla 2020), whereas the Lamb Range populations inhabit similar environments, and all occur within 17 km of each other. Since outbreeding depression can occasionally evolve between isolated populations in similar environments, and this risk increases with increasing time since isolation, Frankham et al. (2011) recommended a 500-year gene flow threshold for mixing populations. Despite evidence of historic isolation of northern and southern Lamb Range B. tropica populations for tens or potentially hundreds of thousands of years in the mitochondrial genome (Pope et al. 2000; Haouchar et al. 2016), we found evidence of recent gene-flow. The range of admixture coefficients present in the three Lamb Range populations indicates not only migration, but survival and presumed fitness, of population hybrids over multiple generations. Furthermore, a 500-year threshold may be overly conservative for bettongs as mixing divergent Bettongia lesueur populations which are estimated to have been separated for more than 8000 years did not result in reduced fitness (Rick et al. 2019). Perhaps this is because most bettongs, including B. tropica and B. lesueur, have small, fragmented populations and therefore much larger genetic rescue effects obscure outbreeding depression upon mixing. Alternatively, mitochondrial divergence may simply not be a good predictor of reproductive isolation (Singhal and Moritz 2013). Even if outbreeding depression were to occur, fitness can be restored by selection within a few generations if the population size is sufficiently large (Frankham et al. 2011, 2017b).
Fourth, reintroductions should not be done at the risk of causing localised extinction by removing too many individuals from natural populations (IUCN 2013). Yet, reintroductions require a minimum number of founding individuals to be successful (Tracy et al. 2011). A total of 50 founding individuals have been proposed for this translocation (Australian Wildlife Conservancy, personal communication, 2020), based on successful reintroductions in the Potoroidae family (Short 2009; Finlayson et al. 2010; Pacioni et al. 2013) and theorical recommendations (Weeks et al. 2011; Jamieson and Allendorf 2012). We found that effective population sizes of all B. tropica populations were small, all less than 100, and therefore removal of 50 individuals from any one population may increase its extinction risk. A mixed source approach allows the impact of removals to be spread across the three populations, thereby reducing extinction risk to any particular population. Given that the Lamb Range populations all have similar levels of diversity, demographic or effective population size and genetic distance should be used as the key determining factors. Selecting more divergent individuals will capture greater diversity, and more individuals should come from the larger population, to ensure that existing populations are not harmed.
We do not, however, recommend sourcing any animals from Mt Spurgeon at this time. This population has low genetic diversity, and therefore would contribute little additional variation, and may even reduce fitness of the founding population by introducing deleterious genetic load (Wilder et al. 2020). Moreover, its very small effective population size means that removing any individuals would be overly risky. Persistence of the Mt Spurgeon population is of utmost conservation importance, and further management interventions should be undertaken to prevent its extinction, including supplementing the population for genetic rescue.
Data Availability
The dataset analysed in this study will be made available on Research Data Australia repository, via publication in Research Data JCU, upon completion of the first author’s thesis. Raw sequence reads will also be published on NCBI Sequence Read Archive (SRA), DNA sequences of SNP fragments on GenBank, and SNP genotypes on European Variation Archive. Prior to thesis completion data are available from the corresponding author on reasonable request.
Code Availability
R code and data are available from https://github.com/StephanieTodd/diversity_struct_Btropica.
References
Alho JS, Lillandt B-G, Jaari S, Merilä J (2009) Multilocus heterozygosity and inbreeding in the siberian jay. Conserv Genet 10:605–609. https://doi.org/10.1007/s10592-008-9588-z
Armstrong D, Hayward M, Moro D, Seddon P (2015) Advances in Reintroduction Biology of Australian and New Zealand Fauna. Csiro Publishing
Bateman BL, Abell-Davis SE, Johnson CN (2012) Climate-driven variation in food availability between the core and range edge of the endangered northern bettong (Bettongia tropica). Australian J Zool 59:177–185
Bérénos C, Ellis PA, Pilkington JG, Pemberton JM (2016) Genomic analysis reveals depression due to both individual and maternal inbreeding in a free-living mammal population. Mol Ecol 25:3152–3168. https://doi.org/10.1111/mec.13681
Burbidge A, McKenzie N (1989) Patterns in the Modern decline of western Australia’s Vertebrate Fauna: causes and conservation implications. Biol Conserv 50:143–189
Burbidge A, Woinarski J (2016) Bettongia tropica. In: Red List of Threatened Species
Byrne PG, Silla AJ (2020) An experimental test of the genetic consequences of population augmentation in an amphibian. Conserv Sci Pract 2:e194. https://doi.org/10.1111/csp2.194
Casey CS, Orozco-terWengel P, Yaya K et al (2018) Comparing genetic diversity and demographic history in co-distributed wild south american camelids. Heredity 121:387–400. https://doi.org/10.1038/s41437-018-0120-z
Coleman RA, Weeks AR, Hoffmann AA (2013) Balancing genetic uniqueness and genetic variation in determining conservation and translocation strategies: a comprehensive case study of threatened dwarf galaxias, Galaxiella pusilla (Mack) (Pisces: Galaxiidae). Mol Ecol 22:1820–1835. https://doi.org/10.1111/mec.12227
DAWE (2022) EPBC Act List of Threatened Fauna. https://www.environment.gov.au/cgi-bin/sprat/public/publicthreatenedlist.pl. Accessed 22 Jun 2022
de Jong MJ, de Jong JF, Hoelzel AR, Janke A (2020) SambaR: an R package for fast, easy and reproducible population-genetic analyses of biallelic SNP datasets. https://doi.org/10.1101/2020.07.23.213793. bioRxiv 2020.07.23.213793
DEHP (2014) National recovery plan for the northern bettong Bettongia tropica. Report to Department of sustainability, Environment, Water, Population and Communities, Canberra. Department of Environment and Heritage Protection, Brisbane
Dennis AJ (2001) Recovery plan for the northern bettong, Bettongia tropica 2000–2004. Queensland Parks and Wildlife Service, Brisbane
Di Fonzo MMI, Pelletier F, Clutton-Brock TH et al (2011) The population growth consequences of variation in individual heterozygosity. PLoS One 6. https://doi.org/10.1371/journal.pone.0019667
Do C, Waples RS, Peel D et al (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (ne) from genetic data. Mol Ecol Resour 14:209–214. https://doi.org/10.1111/1755-0998.12157
Dyer RJ (2017) Applied Population Genetics
Finlayson GR, Finlayson ST, Dickman CR (2010) 22 returning the rat-kangaroos: translocation attempts in the family Potoroidae (superfamily Macropodoidea) and recommendations for conservation. In: Derek M, Eldridge B (eds) Macropods: the Biology of Kangaroos, Wallabies, and rat-kangaroos. CSIRO Publishing
Fischer J, Lindenmayer DB (2000) An assessment of the published results of animal relocations. Biol Conserv 96:1–11. https://doi.org/10.1016/S0006-3207(00)00048-3
Fitzpatrick SW, Bradburd GS, Kremer CT et al (2020) Genomic and fitness consequences of genetic rescue in wild populations. Curr Biol 30:517–522e5. https://doi.org/10.1016/j.cub.2019.11.062
Frankham R (2015) Genetic rescue of small inbred populations: meta-analysis reveals large and consistent benefits of gene flow. Mol Ecol 24:2610–2618. https://doi.org/10.1111/mec.13139
Frankham R, Ballou JD, Eldridge MDB et al (2011) Predicting the Probability of Outbreeding Depression. Conserv Biol 25:465–475. https://doi.org/10.1111/j.1523-1739.2011.01662.x
Frankham R, Ballou JD, Ralls K et al (2017a) Inbreeding reduces reproductive fitness. Genetic management of Fragmented Animal and Plant populations. Oxford University Press, Oxford
Frankham R, Ballou JD, Ralls K et al (2017b) Outbreeding depression is uncommon and predictable. Genetic management of Fragmented Animal and Plant populations. Oxford University, Oxford Press
Frichot E, François O (2015) LEA: an R package for landscape and ecological association studies. Methods Ecol Evol 6:925–929. https://doi.org/10.1111/2041-210X.12382
Frichot E, Mathieu F, Trouillon T et al (2014) Fast and efficient estimation of individual ancestry coefficients. Genetics 196:973–983. https://doi.org/10.1534/genetics.113.160572
Geyle HM, Woinarski JCZ, Baker GB et al (2018) Quantifying extinction risk and forecasting the number of impending australian bird and mammal extinctions. Pac Conserv Biol 24:157–167. https://doi.org/10.1071/PC18006
Gilbert KJ, Whitlock MC (2015) Evaluating methods for estimating local effective population size with and without migration. Evolution 69:2154–2166. https://doi.org/10.1111/evo.12713
Goldberg TL, Grant EC, Inendino KR et al (2005) Increased Infectious Disease susceptibility resulting from Outbreeding Depression. Conserv Biol 19:455–462. https://doi.org/10.1111/j.1523-1739.2005.00091.x
Graham CH, VanDerWal J, Phillips SJ et al (2010) Dynamic refugia and species persistence: tracking spatial shifts in habitat through time. Ecography 33:1062–1069. https://doi.org/10.1111/j.1600-0587.2010.06430.x
Haouchar D, Pacioni C, Haile J et al (2016) Ancient DNA reveals complexity in the evolutionary history and taxonomy of the endangered australian brush-tailed bettongs (Bettongia: Marsupialia: Macropodidae: Potoroinae). Biodivers Conserv 25:2907–2927. https://doi.org/10.1007/s10531-016-1210-y
Harmon LJ, Braude S (2010) 12. Conservation of small populations: Effective Population Sizes, Inbreeding, and the 50/500 rule. In: Braude S, Low BS (eds) An introduction to methods and models in Ecology, Evolution, and Conservation Biology. Princeton University Press, Princeton, pp 125–138
Hoffmann AA, Miller AD, Weeks AR (2020) Genetic mixing for population management: from genetic rescue to provenancing. Evol Appl 14:634–652. https://doi.org/10.1111/eva.13154
Houde ALS, Garner SR, Neff BD (2015) Restoring species through reintroductions: strategies for source population selection. Restor Ecol 23:746–753. https://doi.org/10.1111/rec.12280
IUCN (2013) Guidelines for reintroductions and other conservation translocations
Jamieson IG, Allendorf FW (2012) How does the 50/500 rule apply to MVPs? Trends Ecol Evol 27:578–584. https://doi.org/10.1016/j.tree.2012.07.001
Jolliffe IT (2003) Principal component analysis. Technometrics 45:276
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kanowski J, Smith M, Fleming A, Roshier D (2018) Effective conservation of critical weight range mammals: reintroduction projects of the Australian Wildlife Conservancy. pp 269–279
Kitada S, Nakamichi R, Kishino H (2021) Understanding population structure in an evolutionary context: population-specific FST and pairwise FST. G3 (Bethesda) 11:jkab316. https://doi.org/10.1093/g3journal/jkab316
Kjeldsen SR, Zenger KR, Leigh K et al (2016) Genome-wide SNP loci reveal novel insights into koala (Phascolarctos cinereus) population variability across its range. Conserv Genet 17:337–353. https://doi.org/10.1007/s10592-015-0784-3
Kyriazis CC, Wayne RK, Lohmueller KE (2021) Strongly deleterious mutations are a primary determinant of extinction risk due to inbreeding depression. Evol Lett 5:33–47. https://doi.org/10.1002/evl3.209
Laidlaw MJ, Butler DW (2012) Queensland Herbarium Potential habitat modelling methodology fro Queensland’s threatened flora and fauna
Legge S, Woinarski JCZ, Burbidge AA et al (2018) Havens for threatened australian mammals: the contributions of fenced areas and offshore islands to the protection of mammal species susceptible to introduced predators. Wildl Res 45:627–644. https://doi.org/10.1071/WR17172
Liddell E, Sunnucks P, Cook CN (2021) To mix or not to mix gene pools for threatened species management? Few studies use genetic data to examine the risks of both actions, but failing to do so leads disproportionately to recommendations for separate management. Biol Conserv 256:109072. https://doi.org/10.1016/j.biocon.2021.109072
Lott MJ, Wright BR, Kemp LF et al (2020) Genetic management of captive and reintroduced bilby populations. J Wildl Manag 84:20–32. https://doi.org/10.1002/jwmg.21777
MacFarlane AET (2019) Long-term consequences of genetic rescue on island populations of South Island robins. 201
Marshall TC, Spalton JA (2000) Simultaneous inbreeding and outbreeding depression in reintroduced arabian oryx. Anim Conserv forum 3:241–248. https://doi.org/10.1111/j.1469-1795.2000.tb00109.x
Martin S, Ball S, Peeters P (2006) Reintroduction of the brush tailed Bettong. Bettongia penicillata ogilbyi) into Lincoln National Park
Maschinski J, Wright SJ, Koptur S, Pinto-Torres EC (2013) When is local the best paradigm? Breeding history influences conservation reintroduction survival and population trajectories in times of extreme climate events. Biol Conserv 159:277–284. https://doi.org/10.1016/j.biocon.2012.10.022
Mcgill R, Tukey JW, Larsen WA (1978) Variations of Box Plots. Am Stat 32:12–16. https://doi.org/10.1080/00031305.1978.10479236
Mijangos JL, Gruber B, Berry O et al (2022) dartR v2: an accessible genetic analysis platform for conservation, ecology and agriculture. Methods Ecol Evol 13:2150–2158. https://doi.org/10.1111/2041-210X.13918
Mims MC, Day CC, Burkhart JJ et al (2019) Simulating demography, genetics, and spatially explicit processes to inform reintroduction of a threatened char. Ecosphere 10:e02589. https://doi.org/10.1002/ecs2.2589
Moritz C, Hoskin C, MacKenzie j et al (2009) J b, Identification and dynamics of a cryptic suture zone in tropical rainforest. Proceedings of the Royal Society B: Biological Sciences 276:1235–1244. https://doi.org/10.1098/rspb.2008.1622
Moseby KE, Read JL, Paton DC et al (2011) Predation determines the outcome of 10 reintroduction attempts in arid South Australia. Biol Conserv 144:2863–2872. https://doi.org/10.1016/j.biocon.2011.08.003
Moseby KE, Lollback GW, Lynch CE (2018) Too much of a good thing; successful reintroduction leads to overpopulation in a threatened mammal. Biol Conserv 219:78–88. https://doi.org/10.1016/j.biocon.2018.01.006
Nei M (1986) Definition and estimation of fixation indices. Evolution 40:643–645. https://doi.org/10.2307/2408586
Nei M, Chesser RK (1983) Estimation of fixation indices and gene diversities. Ann Hum Genet 47:253–259. https://doi.org/10.1111/j.1469-1809.1983.tb00993.x
Noble JC, Gillen J, Jacobson G et al (2001) The potential for degradation of Landscape function and cultural values following the extinction of Mitika (Bettongia Lesueur) in Central Australia. In: Conacher AJ (ed) Land Degradation: Papers selected from contributions to the Sixth Meeting of the International Geographical Union’s commission on Land Degradation and Desertification, Perth, Western Australia, 20–28 September 1999. Springer Netherlands, Dordrecht, pp 71–89
Pacioni C, Wayne AF, Spencer PBS (2013) Genetic outcomes from the translocations of the critically endangered woylie. Curr Zool 59:294–310. https://doi.org/10.1093/czoolo/59.3.294
Pacioni C, Williams MR, Lacy RC et al (2017) Predators and genetic fitness: key threatening factors for the conservation of a bettong species. Pac Conserv Biol 23:200–212. https://doi.org/10.1071/PC17002
Pearson K (1901) LIII. On lines and planes of closest fit to systems of points in space. The London, Edinburgh, and Dublin Philosophical Magazine and. J Sci 2:559–572. https://doi.org/10.1080/14786440109462720
Pope LC, Estoup A, Moritz C (2000) Phylogeography and population structure of an ecotonal marsupial, Bettongia tropica, determined using mtDNA and microsatellites. Mol Ecol 9:2041–2053. https://doi.org/10.1046/j.1365-294X.2000.01110.x
Portas TJ, Cunningham RB, Spratt D et al (2016) Beyond morbidity and mortality in reintroduction programmes: changing health parameters in reintroduced eastern bettongs Bettongia gaimardi. Oryx 50:674–683. https://doi.org/10.1017/S0030605315001283
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rick K, Ottewell K, Lohr C et al (2019) Population Genomics of Bettongia lesueur: Admixing increases genetic diversity with no evidence of Outbreeding Depression. Genes 10:851. https://doi.org/10.3390/genes10110851
Sagvik J, Uller T, Olsson M (2005) Outbreeding depression in the common frog, Rana temporaria. Conserv Genet 6:205–211. https://doi.org/10.1007/s10592-004-7829-3
Schmidt TL, Jasper M, Weeks AR, Hoffmann AA (2020) Unbiased population heterozygosity estimates from genome-wide sequence data. https://doi.org/10.1101/2020.12.20.423694. bioRxiv 2020.12.20.423694
Shafer ABA, Wolf JBW, Alves PC et al (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30:78–87. https://doi.org/10.1016/j.tree.2014.11.009
Sharman G, Murtagh C, Johnson P, Weaver C (1980) The chromosomes of a rat-kangaroo attributable to Bettongia tropica (Marsupialia:Macropodidae). Aust J Zool 28:59. https://doi.org/10.1071/ZO9800059
Short J (2009) The characteristics and success of vertebrate translocations within Australia: a progress report to Department of Agriculture, Fisheries and Forestry. June 2009. 103
Short J (2016) Predation by feral cats key to the failure of a long-term reintroduction of the western barred bandicoot (Perameles bougainville). Wildl Res 43:38–50. https://doi.org/10.1071/WR15070
Singhal S, Moritz C (2013) Reproductive isolation between phylogeographic lineages scales with divergence. Proceedings of the Royal Society B: Biological Sciences 280:20132246. https://doi.org/10.1098/rspb.2013.2246
Sunnucks P, Hales DF (1996) Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol Biol Evol 13:510–524. https://doi.org/10.1093/oxfordjournals.molbev.a025612
Taylor G, Canessa S, Clarke RH et al (2017) Is Reintroduction Biology an Effective Applied Science? Trends Ecol Evol 32:873–880. https://doi.org/10.1016/j.tree.2017.08.002
Thavornkanlapachai R, Mills HR, Ottewell K et al (2019) Mixing genetically and morphologically distinct populations in translocations: asymmetrical introgression in a newly established Population of the Boodie (Bettongia lesueur). Genes 10:729. https://doi.org/10.3390/genes10090729
Tracy LN, Wallis GP, Efford MG, Jamieson IG (2011) Preserving genetic diversity in threatened species reintroductions: how many individuals should be released? Anim Conserv 14:439–446. https://doi.org/10.1111/j.1469-1795.2011.00448.x
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci*. Conserv Genet 7:167. https://doi.org/10.1007/s10592-005-9100-y
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262. https://doi.org/10.1111/j.1752-4571.2009.00104.x
Waples RS, England PR (2011) Estimating Contemporary Effective Population size on the basis of linkage disequilibrium in the Face of Migration. Genetics 189:633–644. https://doi.org/10.1534/genetics.111.132233
Weeks AR, Sgro CM, Young AG et al (2011) Assessing the benefits and risks of translocations in changing environments: a genetic perspective. Evol Appl 4:709–725. https://doi.org/10.1111/j.1752-4571.2011.00192.x
Weeks AR, Stoklosa J, Hoffmann AA (2016) Conservation of genetic uniqueness of populations may increase extinction likelihood of endangered species: the case of australian mammals. Front Zool 13:31. https://doi.org/10.1186/s12983-016-0163-z
Weeks AR, Heinze D, Perrin L et al (2017) Genetic rescue increases fitness and aids rapid recovery of an endangered marsupial population. Nat Commun 8:1071. https://doi.org/10.1038/s41467-017-01182-3
Weir BS, Goudet J (2017) A unified characterization of Population structure and relatedness. Genetics 206:2085–2103. https://doi.org/10.1534/genetics.116.198424
White LC, Moseby KE, Thomson VA et al (2018) Long-term genetic consequences of mammal reintroductions into an Australian conservation reserve. Biol Conserv 219:1–11. https://doi.org/10.1016/j.biocon.2017.12.038
Wigginton JE, Cutler DJ, Abecasis GR (2005) A note on exact tests of Hardy-Weinberg equilibrium. Am J Hum Genet 76:887–893. https://doi.org/10.1086/429864
Wilder AP, Navarro AY, King SND et al (2020) Fitness costs associated with ancestry to isolated populations of an endangered species. Conserv Genet 21:589–601. https://doi.org/10.1007/s10592-020-01272-8
Woinarski JCZ (2015) Critical-weight-range marsupials in northern Australia are declining: a commentary on Fisher (2014) ‘The current decline of tropical marsupials in Australia: is history repeating?’ Global Ecology and Biogeography 24:118–122. https://doi.org/10.1111/geb.12165
Woinarski J (2018) Mammal Red Hot List. The Australian mammals most at risk of extinction
Woinarski JCZ, Legge S, Fitzsimons JA et al (2011) The disappearing mammal fauna of northern Australia: context, cause, and response. Conserv Lett 4:192–201. https://doi.org/10.1111/j.1755-263X.2011.00164.x
Woinarski JCZ, Burbidge AA, Harrison PL (2015) Ongoing unraveling of a continental fauna: decline and extinction of australian mammals since european settlement. PNAS 112:4531–4540. https://doi.org/10.1073/pnas.1417301112
Woinarski JCZ, Braby MF, Burbidge AA et al (2019) Reading the black book: the number, timing, distribution and causes of listed extinctions in Australia. Biol Conserv 239:108261. https://doi.org/10.1016/j.biocon.2019.108261
WWF (2020) Living planet report 2020: bending the curve of biodiversity loss. WWF, Gland
WWF-Australia (2018) Bettongia tropica population status, distribution, habitat use and impact of fire. Final Report prepared by WWF-Australia, Sydney NSW
Zeisset I, Beebee TJC (2013) Donor population size rather than local adaptation can be a key determinant of amphibian translocation success. Anim Conserv 16:359–366. https://doi.org/10.1111/acv.12003
Acknowledgements
We acknowledge Western Yalanji, Buluwai and Tableands Yidinji Peoples upon whose Traditional lands this research was undertaken. We would like to thank the people who collected samples for this study, especially AWC staff, QPWS rangers, Chris Pocknee, Western Yalanji and Buluwai Rangers, JCU staff and students and the numerous volunteers who made it possible. We are grateful to Peter Cooper for his warm hospitality. Thanks to Andrew Weeks for his assistance with analysis and interpretation, and to Lin Schwarzkopf for proofreading drafts. Use of the data is made possible through a joint partnership between James Cook University, Australian Wildlife Conservancy and the State of Queensland, Australia through the Department of Environment and Science, Queensland Parks and Wildlife Service.
Funding
This work was funded by Australian Government Caring for our Country (TAG14-00542), Wet Tropics Management Authority (WTMA/2017/947 & WTMA/2018/947), Skyrail Rainforest Foundation, and Community Sustainability Action Grants (CSAT 113 & CSAP 030). Sequencing was additionally supported by the Department of Environment and Science.
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Contributions
S.J.T. collected the samples, extracted the DNA, analysed the data and wrote the manuscript. S.A., J.K. and M.F. designed the sample locations, coordinated field work and collected the samples. B.C., D.T.M. and J.P. provided advice on the data analysis and interpretation of results. All authors contributed to the editing process and read the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This work was carried out under JCU Animal Ethics Committee approval (A2019 and A2221) and Australian Wildlife Conservancy ethics approval (CA 2020/08/1404 and CA 2020-06-1388).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Todd, S.J., McKnight, D.T., Congdon, B.C. et al. Diversity and structure of Bettongia tropica: using population genetics to guide reintroduction and help prevent the extinction of an endangered Australian marsupial. Conserv Genet 24, 739–754 (2023). https://doi.org/10.1007/s10592-023-01533-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01533-2