
Abstract
Tricholoma matsutake, a wild edible ectomycorrhizal mushroom, is revered for its distinguished flavor and iconic significance. Here, we test for landscape effects on T. matsutake gene flow and population structure in the Eastern Himalayas. Using single-nucleotide polymorphic (SNP) DNA markers, isolation by distance patterns were tested on eight populations within and between watersheds. We find that high, treeless ridgelines are effective barriers to gene flow, even at distances less than 65 km, whereas populations located within watersheds are structured at greater distances. Mantel tests demonstrated a significant positive correlation between Fst and a “landscape distance” measured as the shortest distance between population pairs below treeline r = 0.574, P = 0.002, whereas strict euclidian distances do not correlate. AMOVA analysis revealed significant partitioning with 91% of the genetic variance found within populations and 7% found between watersheds, indicative of sexually recombining populations with limited gene flow between watersheds. We show that landscape is an important determinant of air-dispersed ectomycorrhizal species population structure in heterogeneous landscapes.
Similar content being viewed by others

Introduction
Gene flow is one of the factors involved in the maintenance of genetic diversity of populations and is among the major determinants of the genetic structure of a species. An increasing body of literature has demonstrated that mushroom-forming basdiomycete fungi vary in the spatial extent of population structure, as would be expected in a group of organisms including species characterized by different life histories and dispersal syndromes. Much less understood are the processes involved in population structuring of these fungi, and at what spatial scales populations begin to diverge. These data are crucial not only to fully understand the evolutionary processes and the biology of these organisms but also to identify the best ways to maintain diverse assemblages of economic and ecologically important symbiotic fungi.
Isolation by distance, (IBD) first proposed by Wright (1943), is the clinal genetic dissimilarity pattern that emerges when gene flow among populations (or gamete dispersal among individuals) is spatially constrained. The spatial scale at which genetic distance and spatial distance correlate, therefore, depends on dispersal distance. In general, ectomycorrhizal mushrooms (EM) with hypogeous fruit bodies producing meiospores that are largely animal-dispersed show population structure at small scales relative to those with epigeous, wind-dispersed meiospores (Roy et al. 2008). Previous studies of epigeous EM systems have shown little to no population structure at scales from 0.045 to 1,090 m (Gherbi et al. 1999; Zhou et al. 1999; Bergemann et al. 2006; Lian et al. 2006). Conversely, studies at a larger geographic scale include examples of structure: Bergemann and Miller (2002) found F st values as high as 0.434 in populations of Russula brevipes separated by over 1,500 km, although they raised the possibility that these populations may represent separate cryptic species. Likewise a study of Tricholoma matsutake by Xu et al. (2008) found F st values ranging from 0.016 to 0.232 in seventeen populations (which partially overlap this study) separated by <50 km–>1,100 km.
Factors other than distance may account for restricted gene flow among populations, however. Landscape genetics, an emerging field which characterizes how landscape features interact with population structure (see Manel et al. 2003; Storfer et al. 2007 for a review of methods and studies), has been used in a diversity of biota and habitats to characterize gene flow patterns. A study by Grubisha et al. (2007) examined populations of two species of the truffle-like ectomycorrhizal genus Rhizopogon in the Channel islands off of Southern California. Their study found that both ocean channels and a non-forested watershed served as barriers to gene-flow. As Rhizopogon spp. are thought to be predominantly mammal-dispersed, it remains unclear whether landscape heterogeneity would have similar effects on air-borne meiospore-producing mushroom population structure. In mushroom species in which gamete dispersal is potentially in the range of thousands of kilometers, is gene flow at distances <500 km less constrained by distance per se than by landscape barriers?
Patchy host distribution may also affect population structure. Gene flow in populations of fungi associated with plant hosts, such as mycorrhizal fungi, is tightly linked to those plants’ distributions, as evidenced by relatively weak structure in fungi without host preference, and relatively strong structure in fungi whose host preference is narrow. In a study of the generalist wood rotting fungus, Fomitopsis pinicola in Europe, Hoegberg et al. (1999) found uniformly high genetic diversity across populations with little evidence for population structure, even across relatively large distances (Russia to Switzerland). This study is consistent with numerous others showing little to no geographic effects on population structure in host-generalist pathogens (i.e. Heterobasidion annosum (Stenlid 1985) and Armillaria gallica (Saville et al. 1996)). These run counter to studies of fungi with narrow host-preference. Thrall et al. (2002), suggest that the host-specificity of Melampsora lini, and the patchy distribution of the host Linum marginale explain restricted gene flow in that system. Similarly, Parrent et al. (2004) found significant structure in populations of the wood decaying fungus Datronia caperata, a mangrove specialist, in fragmented patches of its Laguncularia racemosa host.
We use Tricholoma matsutake, an edible and medicinal mushroom as our study species. This mushroom has been revered in Japan for centuries for its distinguished flavor, medicinal properties and iconic significance (Hosford et al. 1997; Redhead 1997). The last century saw dramatic declines in Matsutake fruiting bodies in Japan, and by the early 1980’s Matsutake productivity had declined to less than 20% of its pre WWII levels (Redhead 1997). The reasons for this decline may be found in the deforestation of many parts of Japan linked to increasing urbanization and the aggressive attacks on P. densiflora by the pine wood nematode (Hosford et al. 1997). Furthermore, efforts to cultivate this species have not been successful. Scarcity combined with an unflagging demand by Japanese consumers has made Matsutake among the most sought after and expensive mushrooms in the world: in the 1990’s, at the start of the mushroom season, wholesale prices in Japan reached as high as US$1,275/kg (Yun et al. 1997). As the mushroom harvest declined in Japan, imports of T. matsutake soared. By 1986, Yunnan province began to export fresh Matsutake to Japan and is now among the largest exporters in China (He 2003).
Previous studies have examined IBD patterns of this Asian species, at both larger and smaller spatial scales. Chapela and Garbelotto (2004) demonstrate a significant IBD pattern at a continental scale, whereas Xu et al. (2008) find a positive correlation between genetic and geographic distance in populations separated by up to 1,100 km. While the latter study examined possible divergence correlated with elevation, no significant pattern emerged. A study by Lian et al. (2006) of seven T. matsutake fairy rings located within 500 m found no correlation between geographic distance and relatedness.
The main goal of our study was to determine what role landscape features play in T. matsutake population structure at distances not likely to be constrained by meiospore dispersal potential. Specifically, we test whether a high treeless mountain ridgeline is equal or more significant barrier to gene flow as distance. High ridgelines may represent a barrier to gene flow of this host-dependent fungus for several reasons including the lack of any possible tree hosts and adverse environmental conditions, such as increased UV radiation shown to reduce the viability of its air-dispersed propagules (Rotem and Aust 1991; Smits et al. 1996). In order to test our hypothesis, we study the genetic structure of EM populations within and between adjacent watersheds separated by treeless high mountain ridges.
Materials and methods
Site description
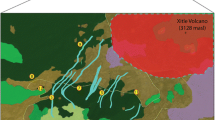
The dramatic topography of northwest Yunnan was caused by the latest and most substantial uplifting event of the Tibetan plateau 1.1–0.6 Ma, concurrent with increased moisture and watercourse diversification (Zhang and Wu 2000). The resulting gorges create habitat islands in low-lying watersheds nestled among Himalayan ridgelines. While this region of China boasts the highest tree line in the Northern hemisphere at 4,900 m (Miehe et al. 2007), tree line within the study area generally ends at 3,900 m with an abrupt shift from Quercus spp. shrubs (which are potential T. matsutake hosts) to alpine meadows and scree (Salick et al. 2004). Short East–West geographic distances are punctuated by substantial physical vicariances and habitat discontinuities as in the mountain slope where Yubeng and Yemen are located (Fig. 1), which rises 4,740 m from river to glacial mountain peak in only 10 km. We sampled two groups of populations, one located within the Mekong River watershed and one located within the Yangtze River watershed: the first one comprises three populations, while five populations were sampled in the latter. Study sites are located in Weixi, Deqin and Shangri-la counties, NW Yunnan province, and were purposely selected for known high productivity of T. matsutake mushrooms.

Diqing Prefecture in NW. Yunnan Province, China: 27.6°N, 99.5°E. Broken lines are rivers. Major watersheds are (left to right) the Salween, Mekong and Yangtze Rivers. Closed circles denote sites where mushrooms were collected: Jidi,1; Nixi, 2; Xiao Zhongdian (XZD), 3; Puja, 4; Pan Tien Ge (PTG), 5; Yubeng, 6; Yemen, 7; Louji, 8. Dark grey is above the 3,900 elevation contour which roughly corresponds with treeline. Black arrows model the shortest route between populations below treeline (3,900 m): denoting “landscape distance”
Whereas vegetation type and plant community assemblage are profoundly affected by elevation and aspect (Salick et al. 2004), there are no major differences in forest environments between the river watersheds sampled, which are separated by less than 100 km.
Sporocarp samples
Two hundred nine mushrooms, (nine to thirty-nine per site; Table 1) were collected for this study. Sporocarps were found in mixed conifer deciduous forests predominated by Pinus spp. and Quercus spp. in the overstory. Each mushroom was identified based on morphological characters. Species identification for a subset of samples was also verified by sequencing a portion of the nuclear ribosomal operon called the internal transciber spacer (ITS) using primers pair ITS1f and ITS4 (White et al. 1990) and by comparing the resulting DNA sequences with sequences deposited in GenBank through the BLAST algorithm on the NCBI GenBank website (www.ncbi.nlm.nih.gov). Mushrooms were cut into thin strips and dried in silica gel in vacuum chambers. Voucher specimens were deposited with the Kunming Institute of Botany herbarium (KIB). We attempted to sample comparable distances within and between watersheds, and sites were chosen at the Northern and Southern limits of T. matsutake distribution within each watershed.
DNA isolation
Approximately 10 mg of dry mushroom material was removed from the stipe and incubated in 50 μl of Extraction Solution from the Extract-N-Amp Plant PCR Kit (Sigma Aldrich) for 10 min at 65°C followed by 15 min at 95°C. Fifty μl of Dilution Solution was added and vortexed briefly. Genomic extracts were diluted fivefold in 0.1× TE buffer (10 mM Tris pH 8.0, 1 mM EDTA–Na) and stored at −20°C for subsequent PCR.
Genotyping
Six variable single nucleotide polymorphism (SNP) markers were designed based either on the shotgun clone library sequence published by Xu et al. (2007), or on a clone library sequence contig from a microsatellite enrichment described below. TmRC 4 and TmRC 17 are respectively, 455 and 429 bp long (Xu et al. 2007) while TmSNP 8, is 346 base pair fragment located adjacent to a microsatellite sequence identified in this study. TmSNP 8 was amplified using the primer pair TmSNP 8 forward: (TGGGTTGGGTTGTATCCATTTAT) and TmSNP 8 reverse: (AAAGTGCTCAGACTACCCCTCTT) with conditions as follows: 1 μl DNA was added to a PCR reaction containing 1 μM each primer, 1.5 mM MgCl2, 200 μM of each dNTP. 0.75 units of Taq DNA polymerase was added to a total volume of 30 μl for TmRC 4, and 0.25 units of Taq was added to a final volume of 10 μl for TmSNP 8. PCR reactions were initially denatured at 95°C for 30 s followed by 35 cycles of 10 s at 95°C, 30 s at 55°C, 30 s at 72°C and a final extension of 5 min at 72°C. Restriction enzymes complimentary to a single SNP allele and nowhere else on the DNA fragment were selected for PCR-RFLP genotyping assay. TmRC 4 was digested with AciI, AleI, EcoRI and SphI restriction enzymes, and TmSNP 8 was digested with BstBI restriction enzyme (New England Biolabs, Ipswich, USA). Eight μl of PCR product were added to two units of restriction enzyme and 2 μl of 10× reaction buffer, and reactions were incubated for 6 h at 37°C. Reactions were electrophoresed in 2% agarose in 1× TAE and scored as codominant markers.
A SNP located on the fragment TmRC 17 (Xu et al. 2007) which was not compatible with PCR RFLP analysis was assayed via Tetra-primer ARMS-PCR (Ye et al. 2001), a protocol in which four primers are used to differentiate codominant genotypes. Two “outer” primers flank a T/C transition mutation site, TMSP17forward: (GCTCGTCTCAAGCTTTGTTGTGATTTAA), and TMSNP17reverse: (CTGCAGTGATTTATCATCCTCCAAGAGA), producing a 429 bp fragment. Each overlapping hemi-nested primer amplifies only in the presence of a specific allele. TMSNP17_C: CTCAGACATTGTCAATCTTGAATTTCCTG produces a 292 bp fragment in the presence of a “C” allele, whereas TMSNP17_T: CAGCTCACTATCCAGACTTCAAACTTCCAT produces a 196 bp fragment in the presence of a “T” allele. One μl genomic DNA was added to a 10 μl PCR reaction containing 1 μM of each inner primer, 0.1 μM of each outer primer, 2.5 mM MgCl2, 200 μM of each dNTP, and 0.25 u Taq DNA polymerase to a total volume of 10 μl. PCR reactions were amplified using the following touchdown thermocycling protocol: initial denature at 95°C for 30 s followed by 10 cycles of 10 s at 95°C, 30 s at 69°C (−1°C each cycle), 30 s at 72°C, 95°C for 30 s followed by 25 cycles of 10 s at 95°C, 30 s at 59°C, 30 s at 72°C and a final extension of 5 min at 72°C. PCR products were electrophoresed and scored as above. Amplicon sequences from a subset of these genotypes were compared with sequence data to confirm that this tetra-primer ARMS-PCR was robust and consistent.
Prior to genotyping using methods outlined above, several different molecular markers were assayed for variation among samples from this study. Ten microsatellite markers described by Lian et al. (2003) for a Japanese population of T. matsutake were tested on a large subset of isolates. Of those loci, only one: TM16, showed variation in a mono-nucleotide repeat motif. These loci were not used for subsequent analyses.
A microsatellite enrichment library was constructed in the Glenn Laboratory (University of Georgia) and screened following the protocols of Glenn and Schable (2005). Thirty-five primer pairs were used to amplify genomic DNA from one mushroom isolate from the eight sampling locations. None of these microsatellites showed polymorphism, and were not used in subsequent analyses.
Data analysis
Multilocus genotypes were compiled, and chi-square tests comparing observed and expected heterozygosities were conducted in GenAleX 6 (Peakall and Smouse 2006) (Table 1) to determine conformance to Hardy Weinberg equilibrium.
The analysis of molecular variance (AMOVA) was used to determine where molecular variance is partitioned within a hierarchical framework. Because we were interested in determining the role of topography on population substructure, our hierarchy was constructed to test variance among populations as well as among watersheds. This series of analyses was conducted in GenAleX 6 with 9,999 permutations.
Statistical independence of loci was examined by testing linkage disequilibrium between pairs of SNP markers in GENEPOP on the Web (Rousset and Raymond 1995). A second test of linkage disequilibrium: the index of association (I A) was calculated using the program MULTILOCUS (Agapow and Burt 2001). The I A tests the independence of alleles across multiple loci within individuals, with confidence statistics computed by comparing observed frequencies against a randomized data set. The I A statistic may be used to infer reproductive strategies, since significant multi-locus linkage may be the result of clonal populations.
Pairwise population differentiation was calculated using two methods. Fisher’s exact test for genotypic differentiation compares multilocus allele frequencies between populations using a chi-square test of significance. Because this test accounts for allele identities in the analysis, we prefer it for SNP analyses where only two alleles are possible. The second test of pairwise population differentiation used was F st, which uses homozygosity levels to infer rates of dissimilarity between pairs of populations. This test is more common, and was used here to enable direct comparisons with other studies.
A topographical model of hypothetical dispersal paths within Matsutake habitat was calculated as the “landscape distance” between populations below the 3,900 m treeline (Fig. 1). Distances were calculated using a 30 m digital elevation model in ArcGIS 9.2 (ESRI, Redlands, CA, USA). Paths between populations at elevations higher than 3,900 m were excluded using Cost Distance in ArcGIS 9.2 Spatial Analyst. Resulting distances, in these cases, followed the 3,900 m elevation contour around high ridges (see Fig. 1 for simplified landscape distance routes, and Table 2a for distances). Euclidian distance was measured as horizontal distance between populations.
We examine the correlation between these distance measurements and population genetic dissimilarity via a Mantel’s test. These were calculated with 10,000 permutations using the online version of GENEPOP with log-transformed pairwise F st (Rousset 1997), with 100 batches, 1,000 iterations per batch as described in Goudet et al. (1996).
Results
Within-population genetic data
All loci were found to be polymorphic across populations. The locus at EcoRI was located on fragment TmRC 4 within 200 bp of loci at AleI and AciI and found to be in linkage disequilibrium with those two loci in the majority of the populations, therefore subsequent population genetic analyses used only the TmRC 4 fragment SNP loci located at the SphI, AleI, and AciI restriction sites. Sample size per population and within-population statistics are presented in Table 1.
Population structure
Significant pairwise population genotypic differentiation (Fisher’s exact test) was detected among some populations located in different watersheds (Table 2b). Populations within watersheds showed no differentiation regardless of distance except in one pairwise comparison of the two most distant populations in the Mekong river watershed: Pan Tien Ge and Yubeng. The two sites were 125.8 km apart, characterized by an F st of 0.03 and significant genotypic differentiation (χ2 23.76, DF 10, P = 0.01). Between all populations, F st values ranged from 0.001 to a maximum of 0.112.
Landscape distances were comparable to geographic distances within watersheds, but were higher for between-watershed comparisons (maximum 256 km, Fig. 1; Table 2a). Mantel test showed significant positive correlation between genetic dissimilarity (F st/(1−F st)) and landscape distance (Log km) between populations r = 0.57, P = 0.002, slope = 0.19 (Fig. 2). Euclidian distance, however, did not significantly correlate with genetic distance r = 0.41, P = 0.12, slope = 0.18 (Fig. 2).

Mantel tests demonstrate a positive correlation between geographic distance (X axis Log distance km) and population dissimilarity F st/(1 − F st) values (Y axis). The slope of the correlation between population comparisons measured using landscape distance (a, see text for details) was significant, P = 0.002, and had a higher correlation coefficient: r = 0.57 than did Euclidian geographic distances between populations r = 0.41, which was not significant P = 0.12
Variance partitioning as determined by AMOVA analysis determined that most (91%) genetic variation was found within populations. Less, though significant, variation (7%) was also found among watersheds. A small amount (2%) of molecular variance was detected among populations in general (Table 3).
Discussion
In this study, we show that the relationship between landscape and genetic affinity is an important determinant of ectomycorrhizal (EM) population structure in heterogeneous landscapes. Low F st values, insignificant Fisher tests of genotypic differentiation, and “among-populations” AMOVA results provide evidence of close genetic affinity among populations within watersheds. Only slight variance (2%) was partitioned among populations within watersheds, a relatively small value compared to measures of variance among the other hierarchical categories. Pairwise genotypic differentiation was only found between two populations within the Mekong watershed located 125 km apart. These findings, in conjunction with positive (though statistically insignificant) Mantel’s correlations suggest detectable isolation-by-distance patterns manifest in the absence of topographical boundaries, albeit at greater distances. It is probable that had we increased either our sample size or the maximum pair-wise distance within watersheds we may have found such a relationship.
This study is among the first to specifically address the role played by topographic features in determining the genetic structure of populations of a fungal species. Pairwise F st values as high as 0.112 as well as significant tests of genotypic differentiation among pairwise comparisons of populations in different watersheds provides evidence that a high ridgeline (elevation > 4,500 m) may be an effective barrier to gene flow among populations. This conclusion is also supported by results of the AMOVA analysis indicating that a significant, percentage of molecular variance was partitioned between watersheds. A relatively low differentiation between watersheds would be expected of populations, such as the ones studied here, that would have parted from a common founding population only recently, as post ice-age warming allowed for the recolonization of high altitude watersheds from lower areas (Hewitt 1996). This would also result in relatively low levels of genetic diversity within Yunnan province. Our difficulty obtaining polymorphic microsatellite markers for this study, and the low allelic variation of ribosomal-coding sequences in Yunnan T. matsutake reported by Sha et al. (2007) might fit this model. These results, however, are somewhat contradictory to the relatively abundant SNP diversity found in this study and in that of Xu et al. (2008). This discrepancy might be further investigated as more is understood of the T. matsutake and related genomes.
Using Mantel correlations, we tested two dispersal scenarios: a strict IBD model in which spore dispersal is not constrained by landscape features, and an isolation by landscape model in which Matsutake spore dispersal is constrained to stepwise dispersal within the geographic range of its host. Although the slopes of these correlations do not differ dramatically, we found that landscape distances better predict genetic dissimilarity than do euclidian distance measurements. It should be noted that despite our best efforts, replication between and within watersheds was imperfect. Our results are consistent with previous studies of host-dependent microbes that have found correlations between habitat discontinuities and population structure where host gaps may restrict gene flow (Thrall et al. 2002; Parrent et al. 2004). Although basidiospore production in mushrooms is both prolific and potentially far-ranging, most spore dispersal studies demonstrate steeply declining densities over short distances (Morkkynen et al. 1997; James and Vilgalys 2001). The rarity of long-distance dispersal is exacerbated by low probability of subsequent establishment. Because habitat and elevation are inextricably correlated in NW Yunnan, we were unable to isolate the biological from the environmental determinants. It is equally possible that landscape distance correlates better with genetic dissimilarity because viable spores are less likely to disperse across high topographical features regardless of whether or not suitable habitat is present. Mycophagous mamalls (including Rhinopithecus bieti, the Yunnan snub-nosed monkey, acording to local observers) and human harvesters may also play some role in medium and long distance T. matsutake dispersal, respectively.
High rates of heterozygozity and recombination in T. matsutake at small spatial scales, and the fact that 91% of molecular variance was partitioned within populations are consistent with dispersal via sexually-produced basidiospores. This dispersal scenario concurs with the results of Murata et al. (2005), who found no correlation between T. matsutake genetic similarity and distance at spatial scales <500 m using inter-retrotransposon amplified polymorphism analysis. Furthermore, our I A analysis (Table 1) showed lack of any significant linkage between loci, a pattern consistent with a mode of reproduction favoring sexual recombination. Five populations did have at least one locus with an excess of homozygotes, however. This pattern could be explained by markers that are not selectively neutral or non-random mating including inbreeding.
Recognizing patterns of gene flow is essential to preserving biodiversity at many levels. We use population structure measurements as a proxy for measuring gene flow, although population structure may result from several other factors aside from gene flow per se. Our results indicate that watersheds separated by treeless ridge tops in NW Yunnan province may contain genetically distinct populations of Matsutake, each of which contributes novel genetic variation to a larger metapopulation. Furthermore, our understanding of disperal in mycorrhizal fungi is fundamental to understanding spatial-temporal patterns in recolonization of degraded habitats and tree plantations. This study identifies a scale at which fungal recolonization may occur by propagules originating from different locations: a scale correlated with the dispersal ability of the studied organism, and with the topographic features of an area. We find population genetic affinity is better predicted by distances within watersheds compared to distances across ridgetops, suggesting gene-flow is less restricted among fungal populations with intervening and continuous host plant range. Although the within-watershed results need to be replicated, this study showed that the IBD model may apply to T. matsutake for distances over 125 km, and provided some of the first evidence that the isolation by landscape model applies to fungi. Whether high ridges are a barrier to fungal dispersal because of lack of the host or because of unfavorable environmental conditions could not be studied here and warrants further consideration.
Finally, our results also enrich the literature on what constitutes an effective gene-flow barrier for a microbe. It appears that for a host-dependant microbial symbiont like T. matsutake, barriers for host dispersal may also represent barriers for the microbe. This finding has important implications for the understanding of evolutionary processes in microbes like the fungi, and provides some fodder for the ongoing debate as to whether or not “Everything is everywhere” in microbial biogeography (Baas-Becking 1934).
References
Agapow PM, Burt A (2001) Indices of multilocus linkage disequilibrium. Mol Ecol Notes 1:101–102. doi:10.1046/j.1471-8278.2000.00014.x
Baas-Becking LGM (1934) Geobiologie; of inleiding tot de milieukunde. WP Van Stockum and Zoon NV, The Netherlands
Bergemann SE, Miller SL (2002) Size, distribution, and persistence of genets in local populations of the late-stage ectomycorrhizal basidiomycete, Russula brevipes. New Phytol 156:313–320. doi:10.1046/j.1469-8137.2002.00507.x
Bergemann SE, Douhan GW, Garbelotto M, Miller SL (2006) No evidence of population structure across three isolated subpopulations of Russula brevipes in an oak/pine woodland. New Phytol 170:177–184. doi:10.1111/j.1469-8137.2006.01654.x
Chapela IH, Garbelotto M (2004) Phylogeography and evolution in matsutake and close allies inferred by analyses of ITS sequences and AFLPs. Mycologia 96:730–741. doi:10.2307/3762107
Gherbi H, Delaruelle C, Selosse MA, Martin F (1999) High genetic diversity in a population of the ectomycorrhizal basidiomycete Laccaria amethystina in a 150-year-old beech forest. Mol Ecol 8:2003–2013. doi:10.1046/j.1365-294x.1999.00801.x
Glenn TC, Schable NA (2005) Isolating Microsatellite DNA Loci. Methods Enzymol 395:202–222. doi:10.1016/S0076-6879(05)95013-1
Goudet J, Raymond M, de-Meeus T, Rousset F (1996) Testing differentiation in diploid populations. Genetics 144:1933–1940
Grubisha LC, Bergemann SE, Bruns TD (2007) Host islands within the California Northern Channel Islands create fine-scale genetic structure in two sympatric species of the symbiotic ectomycorrhizal fungus Rhizopogon. Mol Ecol 16:1811–1822. doi:10.1111/j.1365-294X.2007.03264.x
He J (2003) Cross-scale institutional linkages of commercial matsutake mushroom management and marketing: a preliminary study of an NTFP in Zhongdian County, Yunnan. Yunnan Science and Technology Press, China
Hewitt GM (1996) Some genetic consequences of ice ages, and their role, in divergence and speciation. Biol J Linn Soc Lond 58:247–276
Hoegberg N, Holdenrieder O, Stenlid J (1999) Population structure of the wood decay fungus Fomitopsis pinicola. Heredity 83:354–360. doi:10.1038/sj.hdy.6885970
Hosford D, Pilz D, Molina R, Amaranthus R (1997) Ecology and management of the commercially harvested american matsutake mushroom. In: [USDA general technical report No. PNW-GTR 412]. USDA, USA
James TY, Vilgalys R (2001) Abundance and diversity of Schizophyllum commune spore clouds in the Caribbean detected by selective sampling. Mol Ecol 10:471–479. doi:10.1046/j.1365-294x.2001.01224.x
Lian C, Hogetsu T, Matsushita N, Guerin-Laguette A, Suzuki K, Yamada A (2003) Development of microsatellite markers from an ectomycorrhizal fungus, Tricholoma matsutake, by an ISSR-suppression-PCR method. Mycorrhiza 13:27–31
Lian C, Narimatsu M, Nara K, Hogetsu T (2006) Tricholoma matsutake in a natural Pinus densiflora forest: correspondence between above-and below-ground genets, association with multiple host trees and alteration of existing ectomycorrhizal communities. New Phytol 171:825–836. doi:10.1111/j.1469-8137.2006.01801.x
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197. doi:10.1016/S0169-5347(03)00008-9
Miehe G, Miehe S, Vogel J, Co S, La D (2007) Highest treeline in the northern hemisphere found in southern Tibet. Mt Res Dev 27:169–173. doi:10.1659/mrd.0792
Morkkynen T, Weissenberg KV, Pappine A (1997) Estimation of dispersal gradients of S-and P-type basidiospores of Heterobasidion Annosum. Eur J Forest Pathol 27:291–300. doi:10.1111/j.1439-0329.1997.tb01083.x
Murata H, Ohta A, Yamada A, Narimatsu M, Futamura N (2005) Genetic mosaics in the massive persisting rhizosphere colony “shiro” of the ectomycorrhizal basidiomycete Tricholoma matsutake. Mycorrhiza 15:505–512. doi:10.1007/s00572-005-0358-1
Parrent JL, Garbelotto M, Gilbert GS (2004) Population genetic structure of the polypore Datronia caperata in fragmented mangrove forests. Mycol Res 108:403–410. doi:10.1017/S0953756204009773
Peakall ROD, Smouse PE (2006) Genalex 6: genetic analysis in excel population genetic software for teaching and research. Mol Ecol Notes 6:288–295. doi:10.1111/j.1471-8286.2005.01155.x
Redhead S (1997) The pine mushroom industry in Canada and the United States: Why it Exists and Where it is Going. In: Palm MA, Chapela IH, (eds) Mycology in Sustainable Development: Expanding Concepts and Vanishing Borders, pp 11–46
Rotem J, Aust HJ (1991) The effect of ultraviolet and solar radiation and temperature on survival of fungal propagules. J Phytopathol 133:76–84. doi:10.1111/j.1439-0434.1991.tb00139.x
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rousset F, Raymond M (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Roy M, Dubois M-P, Proffit M, Vincenot L, Desmarais E, Selosse M-A (2008) Evidence from population genetics that the ectomycorrhizal basidiomycete Laccaria amethystina is an actual multihost symbiont. Mol Ecol 17:2825–2838. doi:10.1111/j.1365-294X.2008.03790.x
Salick J, Anderson JD, Woo J, Sherman RE, Cili N, Yin XZ, Na A, Dorje S (2004) Tibetan ethnobotany and gradient analysis: menri (Medicine Mountains), Eastern Himalayas. In: The millennium ecosystem assessment. Bridging scales and epistemologies: linking local knowledge and global science in multi-scale assessments. Alexandria, Egypt
Saville BJ, Yoell H, Anderson JB (1996) Genetic exchange and recombination in populations of the root-infecting fungus Armillaria gallica. Mol Ecol 5:485–497. doi:10.1111/j.1365-294X.1996.tb00341.x
Sha T, Zhang HB, Ding HS, Li ZJ, Cheng LZ, Zhao ZW, Zhang YP (2007) Genetic diversity of Tricholoma matsutake in Yunnan Province. Chin Sci Bull 52:1212–1216. doi:10.1007/s11434-007-0194-0
Smits N, Fargues J, Rougier M, Goujet R, Itier B (1996) Effects of temperature and solar radiation interactions on the survival of quiescent conidia of the entomopathogenic hyphomycete Paecilomyces fumosoroseus (Wize) Brown and Smith. Mycopathologia 135:163–170. doi:10.1007/BF00632338
Stenlid J (1985) Population structure of Heterobasidion annosum as determined by somatic incompatibility, sexual incompatibility, and isoenzyme patterns. Can J Bot 63:2268–2273
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP (2007) Putting the ‘landscape’ in landscape genetics. Heredity 98:128–142. doi:10.1038/sj.hdy.6800917
Thrall PH, Burdon JJ, Bever JD (2002) Local adaptation in the Linum marginale–Melampsora lini host-pathogen interaction. Evol Int J Org Evol 56:1340–1351
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR protocols: a guide to methods and applications 18:315–322
Wright S (1943) Isolation by distance. Genetics 28:114–138
Xu J, Guo H, Yang ZL (2007) Single nucleotide polymorphisms in the ectomycorrhizal mushroom Tricholoma matsutake. Microbiology 153:2002. doi:10.1099/mic.0.2006/005686-0
Xu J, Sha TAO, Li Y, Zhao Z, Yang Z (2008) Recombination and genetic differentiation among natural populations of the ectomycorrhizal mushroom Tricholoma matsutake from southwestern China. Mol Ecol 17:1238–1247. doi:10.1111/j.1365-294X.2007.03665.x
Ye S, Dhillon S, Ke X, Collins AR, Day INM (2001) An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res 29:e88. doi:10.1093/nar/29.17.e88
Yun W, Hall IR, Evans LA (1997) Ectomycorrhizal fungi with edible fruiting bodies. 1. Tricholoma matsutake and related fungi. Econ Bot 51:311–327
Zhang Q, Wu S (2000) Mountain geoecology and sustainable development of the Tibetan Plateau. Kluwer Academic, Norwell
Zhou Z, Miwa M, Hogetsu T (1999) Analysis of genetic structure of a Suillus grevillei population in a Larix kaempferi stand by polymorphism of inter-simple sequence repeat (ISSR). New Phytol 144:55–63. doi:10.1046/j.1469-8137.1999.00504.x
Acknowledgments
This work was supported by grants from the BP Conservation Programme, the Richard E. Schultes award from the Society for Economic Botany, and NSF Grant No. DGE-0232016 to Dr. Kenneth Y. Kaneshiro of the Center for Conservation Research and Training at the University of Hawaii at Manoa. We are grateful to Cui Yi, Cina Wongji and Cilin Qipin for their assistance in the field, and the people and municipal governments of the villages in which these studies were conducted. We are also grateful for the consideration and input of two anonymous reviewers.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Amend, A., Garbelotto, M., Fang, Z. et al. Isolation by landscape in populations of a prized edible mushroom Tricholoma matsutake . Conserv Genet 11, 795–802 (2010). https://doi.org/10.1007/s10592-009-9894-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-009-9894-0