
Abstract
Chemotherapy drugs efficiently eradicate rapidly dividing differentiated cells by inducing cell death, but poorly target slowly dividing cells, including cancer stem cells and dormant cancer cells, in the later course of treatment. Prolonged exposure to chemotherapy results in a decrease in the proportion of apoptotic cells in the tumour mass. To investigate and characterize the molecular basis of this phenomenon, microarray-based expression analysis was performed to compare tHcred2-DEVD-EGFP-caspase 3-sensor transfected C-26 tumour cells that were harvested after engraftment into mice treated with or without 5-FU. Peritoneal metastasis was induced by intraperitoneal injection of C-26 cells, which were subsequently reisolated from omental metastatic tumours after the mice were sacrificed by the end of the 10th day after tumour injection. The purity of reisolated tHcred2-DEVD-EGFP-caspase 3-sensor-expressing C-26 cells was confirmed using FLIM, and total RNA was extracted for gene expression profiling. The validation of relative transcript levels was carried out via real-time semiquantitative RT‒PCR assays. Our results demonstrated that chemotherapy induced the differential expression of mediators of cancer cell dormancy and cell survival-related genes and downregulation of both intrinsic and extrinsic apoptotic signalling pathways. Despite the fact that some differentially expressed genes, such as BMP7 and Prss11, have not been thoroughly studied in the context of chemoresistance thus far, they might be potential candidates for future studies on overcoming drug resistance.
Similar content being viewed by others

Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in men and the second most commonly diagnosed cancer in women, with over 1.8 million new cases and nearly 881,000 related deaths reported worldwide in 2018 [1]. Surgical removal of tumours and lymph nodes is considered the first-line treatment for early stage CRC [2, 3]. For patients affected by advanced-stage of CRC, chemotherapy is the most commonly used treatment option in adjuvant and palliative settings for achieving lasting remission and a definitive cure [4]. However, among CRC patients, those with advanced-stage CRC, are most likely to eventually develop resistance to both single and multiple chemotherapeutic agents despite initial positive responses [5].
Cancer cell resistance to chemotherapy is still a major problem. This results in the progression of local or metastatic disease, and metastasis is a leading cause of patient mortality from solid tumours [6]. Chemoresistance has been shown to be caused by numerous genes and multiple complex biological mechanisms, either intrinsic or acquired, including cancer stem cells (CSCs) and dormancy [7], decreased drug accumulation [8], reduced drug-target interactions [9], drug efflux mechanisms [5], alterations in drug targets and signalling transduction molecules [6], enhanced autophagy activity [10], epithelial–mesenchymal transition [11, 12], increased repair of drug-induced DNA damage, and apoptosis evasion [7, 13].
One of the key breakthroughs for advancing colon cancer treatment could be the overcoming drug resistance [5, 14]. Gaining in-depth knowledge on drug resistance mechanisms is of a paramount importance for developing a rational strategy to target resistant cancers [13, 15]. Although various studies have been conducted to clarify the molecular mechanisms of chemoresistance, the underlying mechanisms are still poorly understood. Therefore, to gain a better understanding of factors contributing to chemoresistance, novel targets should be identified.
We have previously reported the syngeneic mouse systems that express a FRET-based caspase-3 sensor that can be employed to analyse the therapeutic effectiveness of chemotherapy-induced apoptosis [16]. This syngeneic system allowed in vitro, in vivo, and ex vivo analysis of chemotherapy-induced apoptosis induction by optically monitoring the caspase-3 sensor state in the tumour cells. Tumour tissue analysis of 5-FU-treated mice revealed the selection of 5-FU-induced apoptosis-resistant tumour cells, which are referred to as cancer-repopulating cells (CRCs). These CRCs are known as CSCs and responsible for posttherapy relapse and metastatic colonization [17], which are the features most closely related to cancer-related death. This pilot study aimed to investigate and characterize the genetic basis of chemoresistance in chemotherapy-resistant cells. The expression analysis was performed by comparing tHcred2-DEVD-EGFP-transfected C-26 tumour cells that were harvested after engraftment into mice treated with or without 5-FU chemotherapy.
Materials and methods
Tumour cell culture maintenance
Wild-type C-26 murine colon carcinoma cells (referred to as C-26 cells), which can be used for construction of syngeneic models in BALB/c mice, obtained from the American Type Culture Collection™ (Manassas, VA). The cells were cultivated in DMEM medium supplemented with 10% (v/v) heat-inactivated foetal bovine serum, 100 U/ml penicillin, 100 lg/ml streptomycin, and 1% (v/v) glutamine in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Stock cultures were stored in liquid nitrogen and used for in vitro experiments within five passages. C-26 cells transfected with tHcred-DEVD-EGFP (referred to as C-26-c3s cells) were maintained under the same conditions as the wild-type cells. For in vivo studies, cells were harvested from subconfluent cultures. Cell viability was determined via trypan blue exclusion assays. The cell number was adjusted to 1 × 106 cells in 0.5 ml of PBS for intraperitoneal injection to induce peritoneal metastases in mice.
Animal experiments and reisolation of tumour cells
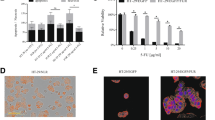
The animal experiments were performed as described in our previous report [16]. Briefly, C26 cells carrying caspase-3 sensors were reisolated from omental metastatic tumours generated by intraperitoneal injection of the cells. C26 cells were reisolated from both omental tumor tissues of untreated controls and 5 days 5-FU-treated mice. We determined the sample size using the permutation method for sample size estimation in R software as previously described for small pilot datasets [18, 19]. The estimated range for group size was 4–6 mice per group. Each group, including both the control and treatment groups, consisted of five mice. To generate omental metastasis, the cells were injected (1 × 106 cells in 0.5 ml of PBS) into the peritoneal cavity of BALB/c mice. The mice in the treatment group received 5-FU (30 mg/kg body mass) injection starting after the fifth day after tumour inoculation. The mice in both groups were sacrificed by the end of the 10th day after tumour injection. The omental tumour tissue specimens were carefully removed from the abdominal cavity. Directly after removal, the tissue samples were dissected into smaller blocks of 1 mm using a scalpel on the culture flask. The tissue pieces were incubated in an atmosphere of 100% humidity and 5% CO2 at 37 °C for 2–3 days in Dulbecco’s modified Eagle´s medium (DMEM). The cells that migrated away from the tissue pieces to the culture dishes, and the remaining necrotic tissue rests was removed during regular medium changes. FLIM Microscopy was used to confirm the purity of the C26 cells via detection of tHcred2-DEVD-EGFP-caspase 3-sensor. Figure 1 shows representative images from an animal experiment, that show the omental metastasis and FLIM features of C26 cells carrying caspase-3 sensor within omental tissue and after their isolation from the tissue and subsequent seeding onto a culture dish.

FLIM of caspase-3 sensor transfected C26 cells in metastatic omental tissue and following their reisolation. The top-left panel depicts a representative image of the normal peritoneal cavity in a mouse. The top-right panel presents an image of peritoneal metastasis generated by the injection of caspase-3 sensor-transfected C26 cells into the peritoneal cavity. The lower-left panel shows a FLIM image of metastatic omental tissue, while the lower-right panel shows a FLIM image after the C26 cells were reisolated and seeded onto a culture dish
Microarray and data analysis
For gene expression profiling, total RNA was purified (Qiagen RNeasy Micro Kit; Qiagen, Crawley, UK) from the reisolated C26 cells of 120 h-5-FU-treated and untreated mice (5 mice in each group; one mouse died during the experiment). Total RNA was used to generate cRNA, which was labelled with biotin according to methods recommended for CodeLink Expression Bioarray System (Amersham, UK). CRNA was then hybridized to DNA oligonucleotide probes attached to a gel matrix followed by secondary labelling and signal detection. The 25-mer microarrays (U133 Plus 2.0; Affymetrix, Santa Clara, CA) were used in our experiment. The microarrays were scanned with GenePix 4000B and analysed using GeneSpring 7.0 software.
Validation of relative transcript levels with real-time semiquantitative RT-PCR
To validate the microarray data, semiquantitative real-time PCR was performed on an ABI Prism 7700 Sequence Detection System (PE Applied Biosystems, Foster City, CA, USA). cDNAs were synthesized using the same total RNA samples that were used for the microarray experiments. Total RNA samples (1 µg) were reverse-transcribed using the QuantiTect Reverse Transcription Kit with Integrated Genomic gDNA Wipeout Buffer (Qiagen) according to the manufacturer’s instructions. Primers for quantitative real-time PCR were purchased from Sigma/GenoSys (Steinheim, Germany). The sequences of primers used are listed in Table 1. Real-time PCR was performed with each specific primer pair using SYBR Green PCR Master Mix (PE Applied Biosystems). The data were analysed using the ABI Prism 7000 Sequence Detection System.
Results
Caspase-3-sensor-carrying C26 cells were used to generate peritoneal carcinomatosis models, followed by 5-FU treatment for five days (30 mg/kg body mass). The surviving cells subjected to this therapy were isolated for microarray analysis. Table 2 summarizes genes that are significantly differentially expressed in C26 tumour-bearing mice treated for 120 h with 5-FU in comparison with untreated mice, or that are associated with survival and apoptotic pathways. These genes could potentially contribute to 5-FU chemoresistance. Figure 2 schematically depicts the relative gene expression map for two experimental groups of mice—120 h 5-FU-treated mouse group compared to the untreated mouse group; where differentially expressed genes are shown.

Comparison of mice teated with or without 5-FU for 120 h. The relative expression values for the two mouse experimental groups (mice treated with 5-FU for 120 h compared to untreated mice) are plotted, significantly differentially expressed genes are shown (P < 0.05)
To validate the results of the microarray analysis, several representative genes were analysed using semiquantitative real-time PCR (Table 3). These genes were Bid, Dedd, Dap, Caspase 3, Caspase 8, Caspase 9, Notch1, Prss11, VEGF-A, and Ecm1. Consistent with the microarray data, similar regulatory patterns of these genes were detected.
Using the DAVID Bioinformatics Database, we defined the specific pathways involving the 47 RNA molecules of interest, that contribute to the onset and progression of cancer cell survival and chemoresistance.
In addition to the data collected and processed from various scientific research articles, we decided to include a discussion of our findings from the DAVID Bioinformatics Database, through which we analysed the list of genes that were differentially expressed in cancer cells that survived chemotherapy according to the identifier ENTEREZ_GENE_ID. We used the “Functional Annotation Report” of this program with the primary goal of determining whether the genes overexpressed in mice treated with chemotherapy participate in numerous common pathways leading to cancer cell survival and apoptosis suppression. Among the 47 identified RNAs, 15 were found to be involved in various cancer pathways simultaneously. A total of 14 gene products (Fas, Notch1, Bak1, Bid, Htra1, Bmp7, Casp3, Casp7, Casp8, Casp9, Idb3, Il6, Prkdc, and Tgfbr1) are involved in the positive regulation of apoptosis, with 5 of them (Fas, Bid, Casp3, Casp8, Casp9) and Bcl2 are members of the p53 signalling pathway; 9 of gene products participate in negative regulation of apoptosis (Fas, Bcl2, Casp3, Hsp90ab1, Kitl, Il6, Prkdc, Tgbr1, and Vegfa). We also detected the involvement of 7/47 analysed genes (Bcl2, Jak1, Casp9, Hsp90ab1, Il6, Kitl, and Vegfa) in the PI3K–Akt pathway.
Discussion
It is well known that chemotherapy drugs efficiently eradicate rapidly dividing differentiated cells by inducing cell death at earlier stages but poorly target slowly dividing cells, including CSCs and dormant cancer cells [20]. Our previously published work demonstrated this phenomenon in a mouse model of peritoneal carcinomatosis of colon carcinoma, where under longer chemotherapy exposure, a lower proportion of apoptotic cells in the tumour mass was detected [16]. To gain a better understanding of the mechanisms involving reversible genetic alterations that could lead to chemoresistance, we analysed of genes of interest in 5-FU-treated mice under chemotherapy treatment to determine their relation to chemoresistance development by determining whether the corresponding gene products promote the survival of cancer cells. The results revealed the downregulation of both intrinsic and extrinsic apoptotic signalling pathways and the upregulation of some mediators of cancer cell dormancy and survival-related genes. The critical mediators of intrinsic and extrinsic apoptotic signaling pathways, such as Bak1, Casp7, Bid, Bcl2, was significantly downregulated in cancer cells exposed to 5-FU. Moreover, some of the important genes of these pathways, such as Casp9 and Casp3, were not regulated.
One of the most likely mechanisms underlying the acquired resistance to 5-FU-induced apoptosis is the differential expression of Fas pathway members. Several genes in this pathway, such as Bid, DEDD, Caspase 7, and DAP were significantly downregulated. Fas (also known as Tnfrsf6) is a death domain-containing member of the TNF receptor superfamily that plays an important role in regulating apoptosis, the pathogenesis of several malignancies, and immune system disorders [21]. Crosslinking of the Fas receptor through Fas ligands or agonistic antibodies results in the formation of death-inducing signal complexes, which include the adaptor proteins FADD/MORT-1 and Caspase 8 [22]. Interestingly, the Caspase 8 and the Tnfrsf6 gene, which are essential for apoptosis execution, were not differentially expressed in C26 cells that survived the chemotherapy. Cleavage of BID (Bcl-2 family proapoptotic protein required for death receptor-mediated apoptosis) by Caspase 8 induces its strong proapoptotic activity, which eventually causes mitochondrial damage and, in due course, cell shrinkage and nuclear condensation [23]. DEDD (DEFT), known as a death effector domain-containing protein, accelerates Fas-induced apoptosis by interacting with FAS-associated death domain-containing protein (FADD) and caspase-8 [24, 25]. Another differentially expressed proapoptotic factor, DAP, is still being studied in terms of its functional role in pathways leading to apoptosis [26]. DAP is a negative regulator of autophagy; that is, it can prevent or suppress authophagy [27]. DAP-kinase was discovered to have potent tumour-suppressive properties, linking the control of apoptosis to metastasis [28]. Caspase 7, which was also downregulated in our study, has been demonstrated to be activated during Fas-induced apoptosis [29]. The downregulation of the aforementioned members of the Fas pathway is consistent with the fact that this pathway is known to promote apoptosis; therefore, its downregulation might contribute to the progression of chemotherapy resistance through apoptosis evasion. However,there is a preliminary study that seems to be contradictory; the authors proposed that Fas signalling induces epithelial–mesenchymal transition (EMT), which promotes motility and metastasis, in gastrointestinal cancers [30]. Efforts are being made to develop cancer therapies based on Fas signalling, but these agents need to be administered cautiously because activation of this pathway can not only induce apoptosis, but also induce resistance to chemotherapy [31]. This finding is of significance importance in our research because it might explain why, as mentioned earlier, Fas (Tnfrsf6) is involved in both the negative and positive regulation of apoptosis according to functional clustering based on the DAVID Bioinformatics Database; hence, more comprehensive studies are needed to clarify the role of the Fas pathway, and eventually advance chemotherapy-based treatments.
So-called survival signalling pathways counter apoptosis signalling pathways. In this context, several genes in the MAPK1/MAPK3 signalling pathway (Kit, Il6, Jak1, and Kitl) were significantly downregulated after 5-FU treatment. Because of its intrinsic complexity and diverse crosstalk with other signalling pathways, the regulation of this pathway remains unclear, as does its involvement in chemoresistance [32].
We also showed the upregulation of several genes in PI3K-Akt pathway (Bcl2, Jak1, Casp9, Hsp90ab1, Il6, Kitl, VEGF A), which is an intracellular signal transduction pathway, a so-called cell survival pathway, that promotes proliferation, cell survival, metabolism, growth, and angiogenesis in response to extracellular inputs [33, 34]. A wide range of human cancers, including breast, colon, gastric, lung, and prostate cancers have been shown to be associated with PI3K activity [35, 36]. Further evidence has shown that Akt (protein kinase B), a downstream kinase of PI3K, is also involved in malignant transformation [37]. Inhibition of the PI3K-Akt pathway could be an advantageous strategy for developing state-of-the-art chemotherapeutic treatment methods and is currently being intensively investigated as a potential cancer treatment strategy [36, 38].
BMP7 gene (bone morphogenetic protein-7), which belongs to the transforming growth factor-β superfamily and is associated with the dormancy of cancer cells, including CSCs, was significantly upregulated in tumour cells treated with 5-FU in a manner dependent on the activation of p21, p38 MAPK, and N-myc downstream-regulated gene 1 via BMP receptor-2 [39]. BMP7 can positively regulate EMT, a process that increases the rate, frequency, or extent of epithelial-to-mesenchymal transition and negatively regulates cell death [40]. EMT, in turn, plays a pivotal role in predicting cancer cell growth into macrometastases [12]. Verschi et al. showed that BMP7 is highly expressed in low-grade CRC patients with both colon adenoma and adenocarcinoma, suggesting that this phenomenon is an early event in CRC [41]. This group demonstrated that BMP7 exerts potent antitumour activity by inducing the differentiation of PIK3CA wild-type CRC stem cells (wt CR-CSCs) and suggested that BMP7-based combination therapies may represent potential novel treatment options for CRC.
Additionally, vascular endothelial growth factor (VEGF-A) and Notch1, which are both key factors in angiogenesis, were considerably upregulated in chemotherapy-treated cells. As chemotherapy augments nutrient and oxygen dependency [42], it can be presumed that tumour cells could benefit from the promotion of angiogenesis. VEGF-A is a critical stimulator of angiogenesis because its binding to VEGF receptors stimulates endothelial cell migration and proliferation, both of which serve as keys in the development of new blood vessels [43]. Furthermore, VEGF-A controls vessel sprouting and branching by inducing the expansion of endothelial tip cells, and increasing vascular permeability, which, in turn, might also contribute to angiogenesis and tumour progression [44]. ESM-1 overexpression could be caused by an analogous mechanism. Hhex-mediated suppression of ESM-1 is required for normal vascular endothelial function, tumour vasculogenesis, and cancer progression [45]. In this context, Kang et al. suggested that ESM-1 may be a useful therapeutic target for CRC [46].
Notch1 is also a negative regulator of cell death, and ligands of Notch1 play an important roles in cell fate determination [47]. Although the expression of Notch1 and its ligand in the vascular endothelium and defects in the vascular phenotypes of targeted mutants in the Notch pathway have already been described [48], the specific signalling pathways controlling their expression remain unknown [49].
The HtrA1 gene was significantly upregulated in chemotherapy-selected cells. HtrA1 has already been hypothesized to function as a tumour suppressor [50]. The first clinical study on melanoma was carried out by Baldi and colleagues [51], who reported significant HtrA1 upregulation in primary tumours compared with that in metastases, and suggested that HtrA1 expression could be an indicator of disease progression. Downregulation of HtrA1 protein is associated with poor survival in mesothelioma [52], hepatocellular carcinoma [53], and breast cancer [54]; in the latter study, nodepositivity was associated with shorter survival. HtrA1 downregulation has also been connected to a poor chemotherapy response in patients with gastric cancer [55]. These findings suggest a possible prognostic role for HtrA1 expression.
Conclusion
Based on our results and discussion, it can be concluded that understanding the exact mechanism of chemoresistance is crucial for overcoming this challenge. An auspicious and powerful approach for the treatment of resistant and recurrent neoplastic diseases could be provided by the reprogramming tumour cells to undergo drug-induced apoptosis by means of novel targeted agents. This can be achieved by downregulating the involved dysregulated antiapoptotic factors or activation of proapoptotic factors in tumor cells. Deeper research works on the intrinsic cell kinetics and mechanisms that promote and sustai cancer cells in a dormant state and the long-term consequences of dormancy, are critical for improving current therapeutic treatment outcomes.
References
Bray F et al (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin 68(6):394–424
Mutch MG (2016) The Surgical Management of Colon Cancer. The ASCRS textbook of colon and rectal surgery. Springer, Berlin, pp 443–470
Vogel JD et al (2017) The American society of colon and rectal surgeons clinical practice guidelines for the treatment of colon cancer. Dis Colon Rectum 60(10):999–1017
Meyers BM et al (2016) Adjuvant systemic chemotherapy for stages II and III colon cancer after complete resection: a clinical practice guideline. Curr Oncol 23(6):418–424
Hu T et al (2016) Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J Gastroenterol 22(30):6876–6889
Pan ST et al (2016) Molecular mechanisms for tumour resistance to chemotherapy. Clin Exp Pharmacol Physiol 43(8):723–737
Recasens A, Munoz L (2019) Targeting cancer cell dormancy. Trends Pharmacol Sci 40(2):128–141
Wang X, Zhang H, Chen X (2019) Drug resistance and combating drug resistance in cancer. Cancer Drug Resist 2:141–160
Maji S et al (2018) Bcl-2 antiapoptotic family proteins and chemoresistance in cancer. Advances in cancer research. Elsevier, Amsterdam, pp 37–75
Huang Z et al (2016) Stress management by autophagy: implications for chemoresistance. Int J Cancer 139(1):23–32
Oh JH, Deasy JO (2016) A literature mining-based approach for identification of cellular pathways associated with chemoresistance in cancer. Brief Bioinform 17(3):468–478
Talukdar S et al (2019) Dormancy and cancer stem cells: an enigma for cancer therapeutic targeting. Elsevier, Amsterdam, pp 43–84
Gimenez-Bonafe P, Tortosa A, Perez-Tomas R (2009) Overcoming drug resistance by enhancing apoptosis of tumor cells. Curr Cancer Drug Targets 9(3):320–340
Longley DB, Johnston PG (2005) Molecular mechanisms of drug resistance. J Pathol 205(2):275–292
Fujita K et al (2017) Cancer therapy due to apoptosis: galectin-9. Int J Mol Sci 18:1
Keese M et al (2010) Fluorescence lifetime imaging microscopy of chemotherapy-induced apoptosis resistance in a syngenic mouse tumor model. Int J Cancer 126(1):104–113
Crea F et al (2015) The epigenetic/noncoding origin of tumor dormancy. Trends Mol Med 21(4):206–211
Lin W-J, Hsueh H-M, Chen JJ (2010) Power and sample size estimation in microarray studies. BMC Bioinform 11(1):1–9
Tibshirani R (2006) A simple method for assessing sample sizes in microarray experiments. BMC Bioinform 7:1–6
Linde N, Fluegen G, Aguirre-Ghiso J (2016) The relationship between dormant cancer cells and their microenvironment. Elsevier, Amsterdam, pp 45–71
Wajant H (2002) The Fas signaling pathway: more than a paradigm. Science 296(5573):1635–1636
Kischkel FC et al (1995) Cytotoxicity-dependent APO‐1 (Fas/CD95)‐associated proteins form a death‐inducing signaling complex (DISC) with the receptor. EMBO J 14(22):5579–5588
Li H et al (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94(4):491–501
Reed JC (2000) Mechanisms of apoptosis. Am J Pathol 157(5):1415–1430
Stegh AH et al (1998) DEDD, a novel death effector domain-containing protein, targeted to the nucleolus. EMBO J 17(20):5974–5986
Gozuacik D, Kimchi A (2006) DAPk protein family and cancer. Autophagy 2(2):74–79
Liang C (2010) Negative regulation of autophagy. Cell Death Differ 17(12):1807–1815
Levy-Strumpf N, Kimchi A (1998) Death associated proteins (DAPs): from gene identification to the analysis of their apoptotic and tumor suppressive functions. Oncogene 17(25):3331–3340
Lamkanfi M, Kanneganti T-D (2010) Caspase-7: a protease involved in apoptosis and inflammation. Int J Biochem Cell Biol 42(1):21–24
Zheng H et al (2013) Fas signaling promotes motility and metastasis through epithelial–mesenchymal transition in gastrointestinal cancer. Oncogene 32(9):1183–1192
Zheng H et al (2014) Fas signaling promotes chemoresistance in gastrointestinal cancer by up-regulating P-glycoprotein. Oncotarget 5(21):10763
Yuan J et al (2020) The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol 13(1):1–19
Xie Y et al (2019) PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia. Mol Med Rep 19(2):783–791
Hers I, Vincent EE, Tavaré JM (2011) Akt signalling in health and disease. Cell Signal 23(10):1515–1527
Steelman LS et al (2008) Akt as a therapeutic target in cancer. Expert Opin Ther Targets 12(9):1139–1165
Yang J et al (2019) Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer 18(1):1–28
McCubrey JA et al (2006) Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul 46(1):249–279
Castel P et al (2021) The present and future of PI3K inhibitors for cancer therapy. Nat cancer 2(6):587–597
Gao X-l et al (2017) Cancer cell dormancy: mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther 10:5219
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Investig 119(6):1420–1428
Veschi V et al (2020) Targeting chemoresistant colorectal cancer via systemic administration of a BMP7 variant. Oncogene 39(5):987–1003
Ma J, Waxman DJ (2008) Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther 7(12):3670–3684
Shibuya M (2011) Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti-and pro-angiogenic therapies. Genes cancer 2(12):1097–1105
Gerhardt H et al (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol 161(6):1163–1177
Cong R et al (2006) Hhex is a direct repressor of endothelial cell-specific molecule 1 (ESM-1). Biochem Biophys Res Commun 346(2):535–545
Kang YH et al (2012) ESM-1 regulates cell growth and metastatic process through activation of NF-κB in colorectal cancer. Cell Signal 24(10):1940–1949
Kolev V et al (2008) EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat Cell Biol 10(8):902–911
Limbourg FP et al (2005) Essential role of endothelial Notch1 in angiogenesis. Circulation 111(14):1826–1832
Liu Z-J et al (2003) Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol Cell Biol 23(1):14–25
Altobelli E et al (2015) HtrA1: its future potential as a novel biomarker for cancer. Oncol Rep 34(2):555–566
Baldi A et al (2002) The HtrA1 serine protease is down-regulated during human melanoma progression and represses growth of metastatic melanoma cells. Oncogene 21(43):6684–6688
Baldi A et al (2008) The serine protease HtrA1 is a novel prognostic factor for human mesothelioma. Future Med. https://doi.org/10.2217/14622416.9.8.1069
Zhu F et al (2010) Serine protease HtrA1 expression in human hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int 9(5):508–512
Lehner A et al (2013) Downregulation of serine protease HTRA1 is associated with poor survival in breast cancer. PLoS One 8(4):e60359
Catalano V et al (2011) HtrA1, a potential predictor of response to cisplatin-based combination chemotherapy in gastric cancer. Histopathology 58(5):669–678
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: Vugar Yagublu and Michael Keese; Methodology: Vugar Yagublu and Bayram Bayramov; Formal analysis and investigation: Bayram Bayramov and Javahir Hajibabazade; Writing—original draft preparation: Vugar Yagublu and Shalala Abdulrahimli; Writing—review and editing: Christoph Reissfelder; Supervision: Christoph Reissfelder and Michael Keese. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare no competing interests.
Ethical approval
Experiments were performed in accordance with German legislation on the protection of animals and the Guide for the Care and Use of Laboratory Animals.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yagublu, V., Bayramov, B., Reissfelder, C. et al. Microarray-based detection and expression analysis of drug resistance in an animal model of peritoneal metastasis from colon cancer. Clin Exp Metastasis (2024). https://doi.org/10.1007/s10585-024-10283-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10585-024-10283-5