
Abstract
Cancer cell invasion, intravasation and survival in the bloodstream are early steps of the metastatic process, pivotal to enabling the spread of cancer to distant tissues. Circulating tumor cells (CTCs) represent a highly selected subpopulation of cancer cells that tamed these critical steps, and a better understanding of their biology and driving molecular principles may facilitate the development of novel tools to prevent metastasis. Here, we describe key research advances in this field, aiming at describing early metastasis-related processes such as collective invasion, shedding, and survival of CTCs in the bloodstream, paying particular attention to microenvironmental factors like hypoxia and mechanical stress, considered as important influencers of the metastatic journey.
Graphical abstract

Similar content being viewed by others
Introduction
Metastasis encompasses the spread of cancer cells from the primary tumor to distant sites, via the bloodstream and/or the lymphatic system. Despite consistent advances in cancer treatment, metastatic disease remains largely uncurable and accounts for the vast majority of cancer-associated deaths. Improving our understanding of the molecular and cellular processes that drive metastasis, particularly in its early steps, is crucial for the development of effective treatments to prevent the spread of cancer.
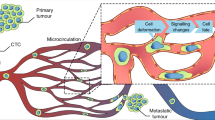
Metastasis of solid tumors is a complex, multi-factorial process involving distinct sequential steps collectively termed as invasion-metastasis cascade. The major steps include cell migration, local invasion through adjacent tissues, intravasation into the bloodstream, arrest and extravasation, dissemination to adjacent or distant sites, survival and adaptation to the foreign microenvironment, establishment of micrometastases, and finally formation of clinically evident macrometastases [1]. Of note, these steps are continuously replicated, even in cells that have already metastasized, fostering metastasis-to-metastasis dissemination [2]. Invasion into the surrounding extracellular matrix (ECM) is one of the essential early steps in the metastatic cascade [3]. Histopathological examination of patient tumor specimens, intravital imaging in mouse cancer models and in vitro experimental systems revealed that tumor cells can display diverse invasive behaviors [4, 5]. Broadly speaking, three major categories can be distinguished: cells can employ either amoeboid or mesenchymal single cell invasion, multicellular streaming (elongated strands of loosely-attached tumor cells moving through a common path) or collective invasion, characterized by sustained cell-cell adhesion [5]. Modes and dynamics of cancer cell invasion are not merely regulated by intrinsic cancer cell factors, but also depend on multiple elements of the tumor microenvironment (TME) [6]. In addition to cancer cells, the TME comprises a broad range of non-malignant cell types, including immune cells, cancer-associated fibroblasts (CAFs), endothelial cells, pericytes, and various other tissue-specific cell types that are all surrounded by vascularized and modified ECM [7].
It is increasingly acknowledged that collective cell invasion, including the formation of cell clusters, is a key mechanism in the progression of solid tumors. Two types of cells can be identified among collectively invading units: leader and follower cells. Leader cells, either tumor-derived or stroma-derived, are in charge of creating low resistance tracks to be exploited for migration, both by using biochemical and biomechanical mechanisms, such as matrix deposition, proteolysis and cytoskeletal remodeling. Leader cells can be classified into four main categories: mesenchymal-like (or hybrid epithelial/mesenchymal) tumor cells, basal epithelial tumor cells, CAFs and tumor-associated macrophages (TAMs) [4]. In contrast to highly invasive leader cells, follower cells are most frequently described as a phenotypically distinct subpopulation of cancer cells, characterized by low invasive potential and migrating along the created invasive paths. The ways by which leader cells collectively coordinate both leader and follower cells, as well as their specific contribution to metastasis remains unclear. Cancer cells that leave the primary tumor and enter the circulation are referred to as circulating tumor cells (CTCs), and act as first-line pioneers of the metastatic cascade [8]. While the primary tumor is thought to shed large numbers of tumor cells, only an extremely small fraction of CTCs will effectively give rise to secondary tumors [3, 9, 10]. CTCs circulate in the blood as single cells or as aggregated cell clusters formed by two or more tumor cells (homotypic CTC clusters) or formed by tumor cells and non-neoplastic cells (heterotypic clusters) [11,12,13,14]. Multicellular CTC clusters have a higher metastatic potential compared to individual CTCs, and their occurrence in patients with various cancer types is linked to a poor prognosis [14, 15].
In this review, we discuss research advances in the field of early metastasis events, with a particular focus on the mechanistic determinants of collective invasion, shedding and survival of CTCs in the bloodstream. Particularly, we will highlight hypoxia and mechanical stress as key regulators of CTC biology and metastatic proclivity.
Hypoxia in the tumor microenvironment
Hypoxia, or low oxygen (O2) tension, is a key factor of the TME that influences the behavior of both tumor and stromal cells. Studies measuring the partial pressure of O2 (pO2) in solid tumors have shown a median pO2 value of ~ 10 mm Hg, as compared with for example 65 and 42 mm Hg in normal human breast and cervix tissue, respectively [16, 17]. pO2 values < 10 mm Hg have been associated with a worse prognosis [17, 18]. Two types of hypoxia are observed in tumors, namely chronic and cycling (intermittent) hypoxia (Fig. 1a). Chronic hypoxia is defined as O2 deficiency over a continuous period of time (at least several hours) and affects cells that are rather distant from blood vessels. In order to grow beyond a certain size, tumors require nourishment in the form of nutrients and O2, as well as an ability to remove metabolic wastes and carbon dioxide. Therefore, during tumor progression, an angiogenic switch is activated that boosts vascular supply by causing the normally quiescent vasculature to continually sprout new blood vessels [19,20,21]. However, the tumor vasculature is not fully functional; due to an improper balance and/or excessive production of angiogenic factors, it is chaotically organized, leaky, and blood often follows different paths through the same vessel [22,23,24,25]. The abnormal tumor vasculature leads to irregular O2 perfusion of the tumor tissue and therefore tumors experience temporal and spatial fluctuations in oxygenation [22, 26, 27]. This type of hypoxia is named cycling hypoxia and affects cells immediately adjacent to inefficiently perfused blood vessels, and when the blood flow is restored, the hypoxia period is followed by a reoxygenation period. Reoxygenation can cause “reoxygenation injury” to the cells, involving free radical formation, reactive oxygen species (ROS) generation, and tissue damage [26, 28].

(a) In solid tumors, HIF-α is stabilized under conditions of low O2 due to reduced vascularization and the establishment of a hypoxic microenvironment. (b) Post-transcriptional regulation of HIF-α subunits. Under normoxic conditions, PHD enzymes utilize oxygen and 2-oxoglutarate as substrates to hydroxylate two proline residues in the HIF-α subunit. These hydroxylation events are required for VHL to bind, ubiquitinate, and target HIF-α for proteasomal degradation. Under hypoxia, hydroxylation is inhibited and HIF-α stabilized. HIF-α heterodimerizes with HIF-1β, interacts with the transcriptional coactivators p300 and CBP, and binds to HRE elements within regulatory regions of target genes. Illustrations were created with BioRender.
Hypoxia-inducible factors (HIFs) are transcription factors that are central to the molecular mechanisms underlying O2 homeostasis and function as master regulators of the adaptive response to hypoxia. HIF-α signaling regulates multiple specific biological processes that are involved in many stages of the metastatic cascade, and the focus of the following sections is on the hypoxia-related steps leading up to the intravasation of cancer cells.
Regulation of hypoxia-inducible factors
HIFs are frequently overexpressed in human cancers because of either intratumoral hypoxia or genetic alterations that lead to HIF-α stabilization (e.g., loss of the von Hippel-Lindau (VHL) tumor suppressor) [29]. HIFs are heterodimers containing an O2-sensitive HIF-α subunit and an O2-independent, constitutively expressed aryl hydrocarbon receptor nuclear translocator (ARNT)/HIF-1β subunit. Three HIF-α subunits have been identified, namely HIF-1α, endothelial PAS domain protein 1 (EPAS1)/HIF-2α, and HIF-3α [29]. HIF-1α is ubiquitously expressed, while HIF-2α is expressed in endothelial cells and in distinct cell populations of kidneys, brain, lungs, liver, gastrointestinal tract, pancreas, and heart [30]. The HIF3A gene gives rise to multiple HIF-3α isoforms by utilizing different promoters, different transcription initiation sites, and alternative splicing [31]. The multiple HIF-3α isoforms have different and even opposite functions, and studies suggest that they negatively regulate the activity of HIF-1α and HIF-2α [31, 32]. Under normoxia, HIF-α subunits are hydroxylated by prolyl hydroxylase domain (PHD)-containing enzymes on two proline residues within the O2-dependent degradation domain (Fig. 1b) [32]. The hydroxylated HIF-α subunits are recognized and targeted for proteasomal degradation by the VHL E3 ubiquitin ligase complex [33]. Hypoxia or oncogenic alterations such as loss of function of VHL, phosphatase and tensin homolog (PTEN), tumor protein 53 (TP53) as well as activation of the phosphoinositide 3-kinase (PI3K)-AKT pathway stabilize HIF-α subunits [34,35,36]. The stabilization of HIF-α subunits under non-hypoxic conditions is a phenomenon termed pseudohypoxia [29]. The HIF-α subunits dimerize with HIF-1β and interact with the transcriptional coactivator p300/cAMP response element-binding protein (CREB)-binding protein (CBP) complex. This transcriptional complex binds to hypoxia-response elements (HREs) in promoters of HIF-α target genes involved in metabolism, nutrient uptake, apoptosis resistance, sustained growth factor signaling, replicative immortality, invasion, metastasis, angiogenesis, and erythropoiesis [29, 37, 38]. HIF-1α and HIF-2α have both overlapping and distinct target genes [32].
In general, HIF signaling reinforces most hallmarks of cancer [20, 21] and confers cancer cells with more aggressive characteristics in hypoxic niches. Increased expression of HIF-1α and/or HIF-2α has been associated with increased tumor aggressiveness and poor prognosis in a broad range of tumor types [39]. Increased levels of HIF-1α, which correlate with poor prognostic outcomes and increased metastasis in patients, have been identified in many solid tumors such as breast, cervical, ovarian, pancreatic, gastric, colorectal, esophageal, lung, liver and prostate cancer [40,41,42,43,44,45]. In breast cancer, survival is significantly decreased in patients with the highest HIF-1α levels, regardless of the lymph node status [46, 47]. HIF-2α expression in primary tumors is associated with distant metastasis and poor outcome in patients with small cell lung cancer, non-small cell lung cancer, clear cell renal cell carcinoma (ccRCCs), neuroblastoma, and breast cancer [48,49,50,51,52]. Intratumoral hypoxia is associated with the invasion and metastasis of HIF-1α-active pancreatic cancer cells [53, 54], and eradication of the HIF-1α-active cells compromises malignant progression. In addition, it has been shown that highly metastatic pancreatic ductal adenocarcinoma (PDAC) subpopulations are enriched for hypoxia-induced genes, and hypoxia-mediated induction of the transcription factor B lymphocyte-induced maturation protein-1 (BLIMP1) contributes to the regulation of a subset of hypoxia-associated gene expression programs [55]. ccRCC is the most common subtype (~ 75%) of renal cancer and complete loss of VHL function occurs in ~ 90% of ccRCCs, leading to constitutive stabilization of the HIF-α subunits and activation of their signaling [51, 52]. HIF-2α is considered to be a driver oncoprotein for ccRCC. Almost a third of all patients with ccRCC show metastatic dissemination at presentation (at which time it has 95% mortality), and ~ 60% have metastases within the initial 2–3 years after diagnosis. Furthermore, it has been shown in experimental models that overexpression of HIFs in several tumor cell types promotes metastasis, whereas inactivation of HIFs decreases the metastatic potential of tumor cells [56].
Several studies have shown that HIF-1α upregulation is more strongly induced by repeated exposures to hypoxia–reoxygenation than by chronic hypoxia [57,58,59,60,61]. The primary consequence of cycling hypoxia is upregulation of HIF-1α activity to a level that supersedes that of chronic hypoxia. The mechanism of enhanced HIF-1α activity is multifactorial, but one factor is the hypoxia-reoxygenation injury-induced production of ROS, which stabilize HIF-1α even in the presence of enhanced oxygenation. In contrast to HIF-1α, the effect of cyclic hypoxia on the stabilization of HIF-2α has been understudied. One study even showed that cyclic hypoxia leads to the degradation of HIF-2α via a calpain-dependent signaling pathway, which in turn results in oxidative stress [62].
HIF-mediated metabolic reprogramming
In the last few years, several studies have addressed how tumor acidosis participates in cancer progression. In hypoxia, cells shift from O2-dependent mitochondrial adenosine triphosphate (ATP) production to O2-independent production, via glycolysis. In cancer cells, the rate of glucose uptake is dramatically increased and lactate is produced, even in the presence of oxygen and fully functioning mitochondria. This process is known as the Warburg Effect or aerobic glycolysis and required for tumor growth [63]. The rate of glucose metabolism through aerobic glycolysis is higher compared to mitochondrial respiration, and the production of lactate is 10–100 times faster than the complete oxidation of glucose in the mitochondria [63]. In addition, the Warburg effect supports the biosynthetic requirements of proliferating cells by diverting glucose-derived carbon into the multiple branching pathways of glycolysis [64].
HIF signaling enhances glycolysis by inducing genes that encode glucose transporters (e.g., GLUT1/SLC2A1, GLUT3/SLC2A3) and glycolytic enzymes (Fig. 2) [65]. Lactate dehydrogenase A (LDHA), which converts pyruvate to lactate and ensures NAD+ regeneration for glycolysis, and monocarboxylate transporter 4 (MCT4), which transports lactate and H+ out of the cell, are also upregulated by HIF signaling (Fig. 2). Remarkably, lactate produced by hypoxic cancer cells can be taken up by non-hypoxic cancer cells or stromal cells via MCT1 to regenerate pyruvate, used for oxidative phosphorylation [66, 67]. Consequently, glucose freely diffuses through the oxygenated tumor cell sheath to fuel glycolysis of hypoxic tumor cells.

Regulation of glucose and glutamine metabolism and pH by HIF-α. Blue circles indicate proteins encoded by HIF-α target genes. To compensate for the reduced flux of glucose to citrate, reductive glutamine metabolism generates cytosolic citrate for de novo lipid synthesis. α-KG, α-ketoglutarate; Glu, glutamate; OXPHOS, oxidative phosphorylation; NBC, Na+/HCO3− co-transporter; TCA, tricarboxylic acid
Cancer cells have evolved several mechanisms to maintain their intracellular pH [68]. H+ can also be exported by specific H+ transporters, including Na+/H+ exchanger 1 (NHE1/SLC9A1) [68]. Hypoxic cancer cells counteract local acidosis by HIF-dependent induction of the membrane-bound ectoenzyme carbonic anhydrase 9 (CA9), which converts H2O and metabolically generated CO2 to H+ and HCO3− (Fig. 2) [69, 70]. Na+/HCO3− co-transporters facilitate HCO3− flux through the cell membrane to maintain the alkaline intracellular pH. In summary, the activity of MCT4, NHE1, and CA9 leads to intracellular alkalization of cancer cells.
HIF signaling suppresses both the tricarboxylic acid cycle (TCA) and oxidative phosphorylation within mitochondria (Fig. 2). HIF-α induces the expression of pyruvate dehydrogenase kinase 1 (PDK1), which phosphorylates and inhibits the mitochondrial pyruvate dehydrogenase (PDH), thereby inhibiting the conversion of pyruvate to acetyl-CoA for entry into the TCA cycle [71, 72]. A HIF-1α-mediated isoform switch from cytochrome c oxidase subunit 4 isoform 1 (COX4-1) to COX4-2 optimizes the efficiency of mitochondrial respiration under hypoxia [73]. The mitochondrial Lon peptidase 1 (LONP1) is induced by HIF-1α and degrades COX4-1. Attenuation of mitochondrial metabolism and O2 consumption in response to hypoxia is also achieved by HIF-α-mediated suppression of mitochondrial biogenesis and enhancement of selective mitochondrial autophagy (mitophagy) [29].
A common feature of cultured cancer cells is glutamine addiction [74]. Under conditions where HIF-α is stabilized, cells shift from oxidative to reductive glutamine metabolism and reverse TCA cycle flux to compensate for the reduced flux of glucose to citrate (Fig. 2) [29]. Glutamine is metabolized through glutaminolysis to α-ketoglutarate, which is converted to citrate by isocitrate dehydrogenase 1 or 2 and aconitase [75]. Thus, reductive glutamine metabolism is the major source of acetyl-CoA for de novo lipid synthesis when HIF-α is stabilized.
The increased consumption of glucose and glutamine by cancer cells causes metabolic competition for nutrients in the TME between cancer cells, stromal and immune cells, ultimately promoting cancer progression [76]. It can negatively impact the functions of immune cells in the TME such as T cells, TAMs, and myeloid-derived suppressor cells (MDSCs) [77]. TAMs and T cells depend on glucose availability and glucose metabolism. Naïve T cells in a resting state require low amounts of glucose, amino acids, and fatty acids to meet basic energy requirements, but active T cells increase glucose and glutamine catabolism for nucleotide and lipid synthesis as well as ATP production [78]. Tumor cells, macrophages, and tumor-infiltrating lymphocytes (TILs) compete for glucose in the TME, and the high rates of glycolysis in tumor cells limit the availability of glucose to TAMs and TILs, which require sufficient glucose for their effector functions [76, 79]. High lactate concentrations in the TME disturb the metabolism and function of human cytotoxic T lymphocytes and suppress their proliferation and cytokine production [80]. Tumor-derived lactate also suppresses survival and function of TILs and natural killer (NK) cells, leading to immune evasion [81]. The intracellular acidification blocks the interferon-γ production in T and NK cells by preventing the upregulation of the transcription factor nuclear factor of activated T cells (NFAT). High rates of cancer cell glycolysis suppress anti-tumor T cell effector functions by depriving T cells of glucose and the downstream metabolite phosphoenolpyruvate, which regulates Ca2+-NFAT signaling in T cells [79].
TAMs promote tumor cell invasion, intravasation, and induce angiogenesis in primary tumors [82]. The extracellular acidification increases the proteolytic activity of TAMs to enhance cell motility [21, 83]. Cancer cell-derived lactate leads to HIF-1α-dependent polarization of TAMs towards a tumor-permissive M2 phenotype and induces vascular endothelial growth factor A (VEGFA) expression in TAMs [84]. In breast cancer, a subpopulation of perivascular TAMs has been identified that has high levels of the TEK receptor tyrosine kinase and expresses VEGFA, which increases vasculature leakiness and causes tumor cell intravasation [85]. In addition, HIF-1α-mediated proangiogenic signaling in the TME also occurs in breast CAFs [86].
HIF signaling and invasion
Acid-mediated invasion
The increased glucose metabolism in cancer cells decreases the pH in the TME due to lactate secretion, and the acidity results in increased progression and metastasis [87,88,89]. A lactate gradient is formed in the TME with the highest concentration in the most hypoxic regions. In a study using breast and colon cancer cells, invasion and peritumoral pH were monitored using intravital microscopy [87]. The peritumoral pH is acidic and heterogeneous and the regions of highest tumor invasion corresponded to areas of lowest pH, whereas no invasion is evident in regions with normal or near-normal extracellular pH [87]. An examination of core and invasive edges of invasive ductal breast carcinoma showed that tumor edges are characterized by increased staining of HIF-1α, CA9, and GLUT1 [90]. Interestingly, the vascular marker CD34 is co-expressed with these hypoxia markers, suggesting that they might be expressed in well-oxygenated regions. In addition, with the acidosis markers - the pH-(low)-insertion peptides (pHLIP), plasma membrane-localized lysosome-associated membrane protein 2 (Lamp2), and CA9 it has been shown that acidic regions are not restricted to hypoxic areas, yet overlap with highly proliferative, invasive regions at the tumor-stroma interface [91]. These and other studies led to the acid-mediated invasion model [89]. Accordingly, several studies have shown that neutralization of tumor acidosis can inhibit metastasis [89].
The increased motility of cancer cells is due in part to changes in the dynamics of the cytoskeleton. Cancer cells have a slightly higher intracellular pH (pHi >7.4) than normal cells (pHi ~7.2), which leads to de novo assembly of actin filaments through a pH-dependent increase in the activities of several actin-binding proteins [68, 90]. Hypoxia also promotes remodeling of the actin cytoskeleton and cancer cell motility through the activation of the RhoA/Rho-associated protein kinase (ROCK) signaling pathway by HIF-1α, which leads to increased invasion and migration of hepatocellular carcinoma (HCC) cells in vitro and in vivo [92].
Hypoxia-induced ECM remodeling
Cell migration and invasion require the remodeling of ECM structures. For invasion to occur, cancer cells must degrade the surrounding basement membrane (BM). HIF signaling promotes ECM remodeling through the upregulation and secretion of proteolytic enzymes. Matrix metalloproteinases (MMPs) are the main enzymes involved in ECM degradation, and increased levels of many MMPs in both primary tumors and/or metastases are positively associated with tumor progression [93]. HIF signaling induces the expression of MMP1, MMP2, MMP3, MMP9, MMP13, MMP14, and MMP15 [94,95,96,97]. It has been shown that MMP2 and MMP14 are expressed selectively in leader cells during collective invasion [98], and the knockdown of MMP14 inhibited fibrosarcoma and breast cancer cell collective invasion [99]. The HIF-dependent upregulation of the urokinase plasminogen activator surface receptor (PLAUR) also increases the proteolytic activity of cancer cells, thereby altering the interaction between integrins and the ECM, promoting cell invasion [100,101,102].
The tissue inhibitors of metalloproteinases (TIMPs) family consists of four members and controls the enzymatic activity of MMPs [95, 103]. The HIF-dependent downregulation of TIMPs has been shown to increase the invasion ability of cancer cells and enhance metastasis [104,105,106].
In addition to the perforation of the BM by proteolytic enzymes, CAFs assist cancer cells in breaching the BM to promote invasion. CAFs can also facilitate BM breakthrough in a MMP-independent manner, by applying physical forces to the BM and by widening of pre-existing gaps initially created by MMPs or other proteases such as PLAUR [107].
Solid tumors are often characterized by excessive deposition of ECM proteins (referred to as fibrosis), and therefore are often stiffer than the surrounding normal tissue [108,109,110,111]. Changes in the posttranslational modification, deposition, and degradation of the matrix result in changes in the composition, density, and mechanical properties of the ECM [112, 113], which in turn affect ECM stiffness, cell migration, tumor growth, invasion, and metastasis [114,115,116]. Hypoxia has been shown to promote fibrosis in breast cancer [117, 118] and PDAC [119,120,121,122]. Chronic hypoxia is an important determinant of fibrosis and carcinogenesis in the liver [123, 124]. In addition, breast cancer and PDAC aggression associate with a stiffer ECM [113].
The HIF pathway controls the expression of genes encoding collagens and collagen-modifying enzymes [125, 126]. Posttranslational modifications of collagen include the hydroxylation of proline and lysine residues that are catalyzed by prolyl-4-hydroxylase α-subunit isoform 1 (P4HA1) and 2 (P4HA2) and procollagen-lysine 2-oxyglutarate 5-dioxygenase 1 (PLOD1) and 2 (PLOD2) [113, 127, 128]. These enzymes catalyze modifications that are necessary for the production of stiff and aligned collagen fibrils. HIF-1α induces the expression of P4HA1, P4HA2, PLOD1, and PLOD2 in different cancer and non-cancer cell lines [109, 129,130,131,132,133,134,135,136], and enhanced activities of these enzymes are associated with increased collagen deposition in cancer tissue. In sarcoma, increased expression of PLOD2 is associated with a more metastatic phenotype, and loss of HIF-1α and HIF-1α-dependent PLOD2 expression disrupts cell migration and pulmonary metastasis [133]. Hypoxia-induced expression of PLOD2 in sarcomas enhances collagen fiber size and tumor density and thus promotes lung metastasis [137]. The knockdown of PLOD2 also impairs the invasion of breast cancer cells into the adjacent normal tissue of the mammary fat pad [136]. Knockdown of P4HA1, P4HA2, and PLOD2 inhibits the spontaneous metastasis of breast cancer cells to the lungs and lymph nodes of mice [135, 136, 138].
ECM stiffening is induced by increased collagen crosslinking, which is mediated by lysyl oxidase (LOX) and LOX-like (LOXL) enzymes [139]. The expression and secretion of LOX, LOXL2, and LOXL4 is induced under hypoxia by HIF-1α [125, 140,141,142,143,144,145]. The ECM remodeling by LOX and LOXL enzymes leads to collagen fiber realignment, bundling, and stiffening with dense collagen bundles that are frequently positioned perpendicular to the tumor, thereby allowing cancer cells to migrate via the fibers and invading the host tissue [146,147,148]. Hypoxic secretion of LOX family members by cancer cells also regulates the formation of premetastatic niches in distant organs, priming these sites for colonization by metastatic cancer cells [56, 109, 111, 139]. Hence, HIF-driven ECM remodeling also has long-distance effects.
An important mediator of cell invasion is the hepatocyte growth factor (HGF) that acts through the MET tyrosine kinase receptor (MET) [149, 150]. HGF is a pleiotropic cytokine and hypoxia sensitizes cells to HGF stimulation by inducing the expression of MET [151, 152]. For example, in non-small-cell lung cancer, it has been shown that prolonged treatment with the MET inhibitor JNJ-605 and the epidermal growth factor receptor inhibitor erlotinib resulted in aggravation of Warburg metabolism in cancer cells with increased lactate release [153].
The gene encoding the C-X-C motif chemokine receptor 4 (CXCR4) is a direct HIF target [154]. CXCR4 is highly expressed in many types of cancer cells, and elevated CXCR4 levels correlate with distant metastases, poor prognosis, and unfavorable outcomes in most solid tumors [155, 156]. HIF signaling also regulates CXCR4 expression in endothelial cells [157]. The upregulation of CXCR4 also aids in the transendothelial migration of cancer cells. A study suggested that hypoxia could drive intravasation via signaling through CXCR4 and its ligand stromal cell derived factor 1 (SDF-1, also known as C-X-C motif chemokine ligand 12 (CXCL12)), which results in adhesion of cancer cells to endothelial cells and trans-endothelial migration of breast cancer cells in in vitro assays [157]. Accordingly, neutralizing the interactions of CXCL12/CXCR4 impaired metastasis of breast cancer cells to regional lymph nodes and lung [158].
Extensive in vitro studies have shown that the migration of cancer cells is associated with an epithelial-to-mesenchymal transition (EMT), whereby cells lose expression of epithelial markers such as E-cadherin and increase expression of mesenchymal markers (e.g., N-cadherin, fibronectin, and vimentin) [159]. This leads to changes in cell-cell and cell-matrix adhesions. Hypoxia and HIF signaling have been linked to EMT-like phenomena by the induction of the transcription factors such as twist family BHLH transcription factor (TWIST) [160], snail family transcriptional repressor 1 (SNAI1) [161,162,163], zinc finger E-box binding homeobox 1 (ZEB1) [164], and snail family transcriptional repressor 2 (SNAI2) [165], and the modulation of cell signaling pathways such as neurogenic locus notch homolog protein (NOTCH), WNT, and AXL [166,167,168,169,170,171,172]. However, the consensus that EMT is required for cancer metastasis has been challenged by studies showing that CTCs are not the homogeneous mesenchymal phenotype described in the EMT-metastasis hypothesis [173]. A study using a mouse lineage-tracing model suggested that cells do not activate EMT in order to metastasize [174]. Along these lines, absence of TWIST1 or SNAI1 did not alter invasion and metastasis in genetically engineered PDAC mouse models [175]. Recent studies suggest that instead of being a binary process, EMT rather occurs through distinct intermediate states, a process referred to as hybrid EMT [176, 177]. Multiple subpopulations of cancer cells have been identified that are associated with different states of EMT, ranging from epithelial to mesenchymal-like states [178]. EMT or mesenchymal-to-epithelial transition (MET) are therefore not “all-or-none” processes, and cells undergoing hybrid EMT may display cellular plasticity associated with distinct invasive and metastatic potential [176, 178]. It has been suggested that the acquisition of a hybrid EMT state is involved in the determination of leader and follower cells [176].
CTCs and hypoxia
In the laboratory setting, a standard O2 level of 20% (160 mm Hg) used for cell cultures is referred to as normoxia. However, the physiological O2 levels (physoxia) found in normal tissues are much lower and average about 5% O2 (38 mm Hg) and range from ~ 3–7% [179]. It has been suggested that physiological hypoxia, the O2 level at which tissues respond to maintain their preferred O2 level, ranges from 2 to 6%, whereas O2 levels < 2% are referred to as pathological hypoxia [179]. O2 levels of 5% mimic more closely in vivo conditions and are therefore commonly utilized for expansion of patient-derived specimens in the laboratory. Along with dedicated media components, physiological hypoxia has been used to expand and maintain patient-derived CTCs ex vivo [180, 181]. While most of the laboratories employ hypoxic conditions, which have been proven critical for certain cancer types [180, 183], some CTC lines have been occasionally cultured under normoxic conditions as well [184].
Clinically, hypoxia and the expression of HIF-α are associated with increased distant metastasis and poor survival in a variety of tumor types [182]. Interestingly, in mouse models of breast cancer, a bimodal distribution of hypoxia has been observed, either restricted within a central core or more scattered throughout the tumor, often in areas with low blood vessel density [180]. Blood vessels are distributed in both normoxic and hypoxic regions of tumors, with a higher presence in normoxic tumor regions. The presence of functional blood vessels in hypoxic tumor areas indicates possible access routes for metastatic cells to the circulatory system. Furthermore, scattered hypoxic areas are due to cycling hypoxia, and cancer cells in these regions might be more prone to intravasation since they are located closer to blood vessels. It has been shown that intra-tumor hypoxia in breast cancer leads to upregulation of cell-cell junction components and intravasation of hypoxic CTC clusters with high metastatic ability, while single CTCs rather derive from normoxic tumor regions [180]. Anti-angiogenic therapies targeting the VEGF pathway have been developed to reduce intratumoral vasculature and consequently starve the tumor from its nutrients. However, in breast cancer patients, VEGF inhibitors lacked efficacy in the long run, even promoting tumor invasiveness and metastasis in some cases [23]. This led to withdrawal of the VEGF-targeting agent Avastin for the treatment of breast cancer patients in 2011 [183]. Accordingly, targeting VEGF in breast cancer mouse models leads to primary tumor shrinkage, but it increases intra-tumor hypoxia and results in a higher CTC cluster shedding rate and metastasis formation [180]. In contrast, pro-angiogenic treatment increases primary tumor size, but it dramatically suppresses the formation of CTC clusters and metastasis [180].
Using a fate-map system for hypoxic cells in vivo, cells exposed to physiological levels of hypoxia in the primary breast tumor were shown to have a four times greater probability of becoming viable CTCs, compared to cells from normoxic regions of the primary tumor [184]. Cells exposed to hypoxia in the primary tumor have the ability to migrate toward a more oxygenated invasive front of tumor regions and intermingle with blood vessels. In addition, post-hypoxic cells have a 6-7-fold greater probability of forming lung metastasis, suggesting that they have an enhanced metastatic potential [184]. Another study showed that oxidative stress prevents the survival of CTCs in the bloodstream and is therefore a limiting step in metastasis [185]. Cells that experience hypoxia in the primary tumor are more resistant to oxidative stress and have lower levels of mitochondrial ROS in both the primary tumor and in the blood [184]. It has been suggested that the enhanced metastatic potential of post-hypoxic cells is in part due to the ROS-resistant phenotype that allows them to survive high levels of ROS in the circulation.
Taken together, these observations highlight a prominent role of hypoxia in the early steps of the metastatic cascade, conferring both advantageous metabolic features as well as invasive traits that result in the intravasation of aggressive precursors of metastasis.
Fluid shear stress
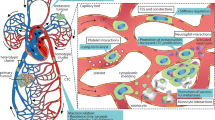
In addition to biochemical signals, biomechanical forces also affect tumor progression (Fig. 3). The effects of mechanical forces on living tissues are in the focus of mechanobiology, a multidisciplinary field at the interface between biology, physics and engineering that investigates how cells sense and respond to physical cues, and how these mechanisms impact their behavior and organization [186]. The field of mechanobiology is growing rapidly thanks to imaging techniques such as atomic force microscopy, technological innovations in force-application methods used to manipulate cells mechanically, microfluidics and advanced computational simulation [186,187,188,189]. Within the field, an extensively studied aspect is mechanotransduction, a process whereby cells convert forces arising from mechanical stimuli into biochemical signals. It involves sensing various physical stimuli through specialized ion channels, integrins and cytoskeletal structures, subsequently triggering alterations in gene expression, protein synthesis and cellular behavior [190]. Several types of solid stresses such as compression, tension, shear and ECM stiffness are applied to tumor cells from their environment [191]. It is widely recognized that these mechanical forces play a significant role in various steps of the metastatic cascade, starting from the growth of the primary tumor, intravasation of cancer cells into the bloodstream, their survival in circulation, infiltration into secondary sites and formation of metastatic outgrowths [192]. In the human body, cancer cells experience two fundamental types of shear stress due to the flow of bodily fluids: shear stress, generated by interstitial fluid flow within the primary TME, and shear stress, generated by the blood flow within the circulatory system [193, 194]. Here, our attention will be directed towards the impact of fluid shear stress on tumor progression both in the primary TME as well as in the bloodstream.

Hypoxia and shear stress regulate dissemination of circulating tumor cells (CTCs). Cancer cells are in interaction with several types of immune cells and experience various biophysical forces both in the primary tumor microenvironment and in the circulation. Primary tumors have normoxic and hypoxic regions with constant interstitial flow, inducing biochemical and biophysical signaling changes in cancer cells. Cancer cells can intravasate as single cells, as a group of cells (homotypic clusters) or together with non-neoplastic cells (heterotypic clusters). When CTCs enter the bloodstream, they are subjected to different levels of fluid shear stress that can lead to cell rupture and apoptosis, and initiate rapid signaling changes. Due to mechanical shielding, CTC clusters are more resistant to shear stress than single cells. In contrast, single cells are more prone to cell membrane blebbing caused by mechanical forces. MDSC, myeloid-derived suppressor cell; CAF, cancer-associated fibroblast; NK, natural killer cell. Illustrations were created with BioRender.
Fluid shear stress exposure in tumor microenvironment
The TME can be considered as a dynamic niche where cells are constantly encountering physical and mechanical cues. Cancer cells in the TME are subjected to shear stress and hydrostatic pressure generated by interstitial fluid flow, tensile and contractile forces [195]. Interstitial fluid flow arises from the movement of fluids through the ECM, commonly occurring between blood and lymphatic vessels [196]. A common characteristic of progressing cancers is an increase in intratumoral pressure and the consequent elevated interstitial flow from the tumor mass to the adjacent healthy stroma [197]. The observed changes are the result of various contributing factors, such as vascular abnormalities, lymphatic co-option, and an increase in cell number and density. Elevated interstitial fluid and blood/lymph flows within the primary tumor produce a constant fluid shear stress, ranging between 0.1 and 1 dyn/cm2, that can affect cell behavior [198]. For example, several studies have shown that physical cues in the primary tumor can promote migration and invasion of cancer cells [199,200,201]. Shear stress can stimulate the formation of circular dorsal ruffles, F-actin rich membrane structures, thereby promoting migration of breast cancer cells [202]. In an in vitro experimental setup, oral squamous cell carcinoma cells were subjected to conditions mimicking various levels of interstitial fluid pressure, by using a humidified pressure chamber with cyclic pressure system connected to an air pump to tightly control atmospheric pressure on cells. This revealed that, as interstitial fluid pressure elevates, cell proliferation, survival and invasion capabilities of cancer cells increase [203]. A study where breast cancer and melanoma cells were seeded into a collagen-Matrigel matrix and placed within a flow chamber has demonstrated that physiological levels of interstitial flow strongly induce their migration through autocrine C-C motif chemokine receptor 7 (CCR7) signaling [204]. Multiple glioma cells lines exposed to pressure-driven flow (mimicking interstitial flow) have increased cell invasion capabilities driven by CXCR4-dependent mechanism [205].
Furthermore, increased interstitial fluid causes a decrease in transcapillary transport, leading to decreased anti-cancer drug uptake [206]. A study investigating interstitial fluid pressure in patients with metastatic melanoma has shown that interstitial fluid pressure levels significantly increases over time in patients that are not responding to immunotherapy, whereas they decreases in responders [207]. Moreover, clinical data on cervical cancer has shown that patients with higher interstitial fluid pressure are significantly more prone to develop distant metastasis and recurrence [208].
Fluid shear stress exposure in vascular microenvironment
Not only in the primary tumor, but also during their hematogenous dissemination, cancer cells are notably exposed to circulatory fluid shear stress, the most prominent mechanical force in the bloodstream [209]. Circulatory fluid shear stress arises due to tangential frictional forces generated by blood flow acting on the surface of cells. Upon intravasation, cancer cells experience various levels of shear stress (ranging 1–30 dyn/cm2) [210], depending on the vessel type. Higher levels of shear stress are imposed in arteries (15–30 dyn/cm2) and in capillaries (10–20 dyn/cm2), whereas in aorta, vena cava and veins shear stress levels are lower (< 10 dyn/cm2) [211]. However, in the vicinity of vessel bifurcations at the heart, shear stress levels can raise up to ~ 3350 dyn/cm2 [212]. Fluid shear stress can be studied with various technologies both experimentally and computationally, including parallel plate flow chambers, microfluidic devices, syringe/peristaltic pumps and computational fluid dynamics modelling [213,214,215,216]. For instance, a recent study investigated the effect of brief pulses of high-level shear stress on prostate cancer cells by using a syringe pump to mimic high shear stress values in the turbulent flow region around the heart. They demonstrated that cancer cells activate RhoA-myosin II and stiffen in response to shear stress. Moreover, shear stress triggers the elevation of cortical F-actin, which further contributes to cell stiffness [214]. In another report, using a peristaltic pump to imitate arterial levels of shear stress, including high levels achieved during exercise, it has been shown that high shear stress (60 dyn/cm2) destroys over 90% of CTCs derived from multiple types of cancer within 4 h of circulation [213]. In a different study, computational fluid dynamics simulations were performed to mathematically predict the behavior of CTC clusters under various levels of shear stress [215]. Tumor cell intravasation is more likely to occur in areas with reduced fluid flow, as low shear stress is less detrimental for tumor cells during intravasation [217]. Recently, it has been reported that human cancer cells avoid high shear stress regions in the process of intravasation through the activation of transient receptor potential cation channel, subfamily M, member 7, also known as TRPM7 channel, a key shear-stress sensor, which promotes calcium influx into the cell followed by RhoA/myosin-II and calmodulin/IQGAP1/ Cdc42 pathway activation, subsequently reversing migration [217]. Even though carcinoma cells have higher mechanical resistance compared to non-transformed epithelial cells, shear stress in the blood circulation can readily destroy many of the intravasated cancer cells, explaining the relative inefficiency of the metastatic process [198]. Arterial levels of shear stress (15–30 dyn/cm2) could also increase ROS, which impair mitochondria activity and lead to further cellular death [218]. While shear forces are thought to drastically reduce the viability and number of CTCs in the circulation, a small fraction of these are mechanically robust and develop mechano-adaptive resistance. Fluid shear stress also involves human Piezo type mechanosensitive ion channel component 1 (PIEZO1) activation, which leads to the influx of extracellular calcium [219, 220]. Among other signals, PIEZO1 activates Akt/mTOR (mammalian target of rapamycin) pathway in breast cancer, and may subsequently positively regulate cell motility and survival [221, 222]. Although regulation of cytoskeletal organization and certain mechanosensitive ion channels such as PIEZO1 and TRPM7 have been shown to play a role in shear stress related responses, further mechanisms are likely to be involved.
Along with the mechanical trapping, blood flow is also a regulator of intravascular arrest of CTCs. It has been shown that early arrest on the vascular endothelium is mediated by integrin αvβ3 and CD44. Moreover, during the stronger adhesion to endothelium, cancer cells utilize integrin α5β1-dependent adhesions, which facilitate the adherence of CTCs to low shear stress regions of the vascular wall [223]. Due to the vessel diameter and numerous bifurcations, capillary beds seem to be one of the most common locations of CTC entrapment. When CTCs come across a capillary, they either become trapped and exit the vessel (in a process called extravasation) or they pass through the capillary without getting trapped. While CTCs are transiting or arrested in capillaries, shear forces can induce morphological changes and cause severe cell deformation and, hence, affect cell fate [224]. During CTC squeezing through tight capillary constrictions, cytoplasmic deformation activates several mechanotransduction signaling pathways, including yes-associated protein 1 / transcriptional co-activator with PDZ-binding motif (YAP/TAZ) and GTPase RhoA. Upon activation of Rho-family GTPase RhoA, several downstream effectors are triggered, including ROCK. As a result, ROCK promotes the activation of myosin II molecular motor, responsible for cytoskeletal rearrangement via contraction of actomyosin networks and can further enhance cancer cell survival and metastatic ability [225, 226].
Resistance to shear stress
To resist detrimental effects of shear stress, cancer cells developed various resistance mechanisms, including travelling with companions. Although both single CTCs and CTC clusters are accountable for metastasis formation, they display different survival abilities in the circulation due to their physical characteristics [11]. A study performed using a microfluidic system to mimic capillary constrictions has revealed that CTC clusters may migrate through the capillary bed by rapidly unfolding into single-file chain-like geometry. This structural strategy facilitates their migration and substantially reduces the hydrodynamic resistance [227], however, fabricated glass capillaries may not fully recapitulate the properties that are ascribable to endothelial walls. Moreover, a recent study has revealed that shear stress disassociates the CTC clusters due to strong forces and disaggregates most of the CTC clusters in breast cancer [215]. Along the same line, it has been shown that majority of the CTC events that have been captured from the peripheral blood of patients and experimental models are single CTCs, with clusters being considered as a rarer event [11, 13]. While rare in numbers, CTC clusters have higher metastatic potential compared to single cells [11]. Compared to single CTCs, CTC clusters have upregulated expression of cell junction components, such as plakoglobin. Knockdown of plakoglobin halts the formation of CTC clusters and suppresses lung metastasis. Therefore, plakoglobin dependent intercellular adhesion within CTC clusters contributes to the multicellular nature of clusters as well as to their higher metastasis efficiency [11]. Increasing evidence points toward a clear CTC cluster survival advantage within the bloodstream, possibly due to their enhanced resistance to shear stress [224], among other factors. Various non-neoplastic elements have been suggested to facilitate CTC survival in circulation, likely impacting on shear stress resistance, among other aspects. For instance, through cytokines crosstalk within individual CTC-neutrophil clusters, neutrophils promote proliferation of CTCs while in circulation, thereby accelerating metastasis seeding [13]. Accordingly, breast cancer patients with detectable CTC-neutrophil clusters are characterized by a poor prognosis [13]. In another study employing an in vitro model of shear induced damage, investigators have shown that in the absence of platelets, ovarian cancer cells display a higher shear-induced membrane damage, suggesting that platelet adhesion may be protective in this context [228]. CAFs can also accompany CTCs on their metastatic journey in the blood, as observed in patients and experimental models [12]. An in vitro study addressed the role of CAFs in cancer cell survival under extremely high shear stress (5920 dyn/cm2) using a 3-D cell co-culture system. When co-cultured with prostate cancer cells, CAFs provide resistance to high shear, support tumor cell survival and promote proliferation via intercellular contact and soluble derived factors such as CCL2, CCL7 and CXCL5 [229]. In contrast to healthy individuals, circulating cancer-associated macrophages-like cells were isolated from the peripheral blood of breast, pancreatic and prostate cancer patients. A subset of these disseminated macrophages, so called circulating giant macrophages, is able to bind CTCs [230]. Recently, it has been discovered that bacteria also contribute to cancer cell survival in the bloodstream. Intracellular microbiota enhances the resistance of breast cancer cells to fluid shear stress by cytoskeletal rearrangements in CTCs, promoting cell viability. Particularly, bacteria are able to trigger the fluid shear stress-related pathways and regulate stress response within the circulation. Mechanistically, cytoplasmic bacteria inhibit the activation of RhoA and ROCK and, therefore, contractile forces triggered by mechanical forces can be alleviated [231]. Interestingly, circadian clock-regulated hemodynamics could also affect the survival of CTCs in bloodstream [232]. Given that the metastatic spread of breast cancer in mouse models and patients has been reported to occur mostly during the rest phase [233], other circadian clock-regulated parameters such as blood pressure, heartbeat and cardiac output could be contributing to these processes [234].
Overall, CTCs utilize various survival strategies, such as interactions with immune and stromal cells, allowing them to withstand shear stress. So far, most of the studies focused on molecular pathways related to mechanosensitive ion channels and cytoskeletal rearrangements, which have been widely acknowledged for their role in mechanotransduction. However, further unbiased mechanistic studies, such as genomic and transcriptomic analysis, may provide a broader overview of fluid-shear regulated genes and their impact on the metastatic cascade, possibly highlighting novel opportunities for intervention.
Conclusions
We have provided an overview of the literature that impacts the ability of cancer cells to intravasate and travel in the bloodstream, particularly concerning the role of hypoxia and shear stress in these early stages of metastasis. Although the discussed large volume of experimental evidence highlights the relevance of these processes for the metastatic abilities of CTCs, several open questions remain. Considering that hypoxia is one of the triggers of CTC cluster shedding, it would be interesting to mechanistically investigate which hypoxia-related signals are critical in this context. Also, while the mechanical forces associated with fluid shear stress trigger several downstream biological events that affect the fate of cancer cells, the biological effects and consequences of shear stress exposure remain to be fully understood, particularly in physiological systems. Along these lines, the development of sophisticated microfluidics systems combined with computational simulations and in vivo experiments may provide new insights in understanding how flow dynamics and shear stress alter cell behavior and regulate metastasis. Deep insights into the mechanistic drivers of CTC intravasation and survival in circulation may highlight new opportunities for therapeutic interventions aimed at metastasis suppression.
Data Availability
Not applicable.
References
Vanharanta S, Massagué J (2013) Origins of metastatic traits. Cancer Cell 24:410–421. https://doi.org/10.1016/j.ccr.2013.09.007
Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E et al (2015) The evolutionary history of lethal metastatic prostate cancer. Nature 520:353–357. https://doi.org/10.1038/nature14347
Fidler IJ (2003) The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat Rev Cancer 3:453–458. https://doi.org/10.1038/nrc1098
Vilchez Mercedes SA, Bocci F, Levine H, Onuchic JN, Jolly MK, Wong PK (2021) Decoding leader cells in collective cancer invasion. Nat Rev Cancer 21:592–604. https://doi.org/10.1038/s41568-021-00376-8
Friedl P, Locker J, Sahai E, Segall JE (2012) Classifying collective cancer cell invasion. Nat Cell Biol 14:777–783. https://doi.org/10.1038/ncb2548
Clark AG, Vignjevic DM (2015) Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol 36:13–22. https://doi.org/10.1016/j.ceb.2015.06.004
de Visser KE, Joyce JA (2023) The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 41:374–403. https://doi.org/10.1016/j.ccell.2023.02.016
Pantel K, Speicher MR (2016) The biology of circulating tumor cells. Oncogene 35:1216–1224. https://doi.org/10.1038/onc.2015.192
Gupta GP, Massagué J (2006) Cancer metastasis: building a framework. Cell 127:679–695. https://doi.org/10.1016/j.cell.2006.11.001
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2:563–572. https://doi.org/10.1038/nrc865
Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA et al (2014) Circulating Tumor cell clusters are oligoclonal precursors of breast Cancer metastasis. Cell 158:1110–1122. https://doi.org/10.1016/j.cell.2014.07.013
Duda DG, Duyverman AMMJ, Kohno M, Snuderl M, Steller EJA, Fukumura D et al (2010) Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A 107:21677–21682. https://doi.org/10.1073/pnas.1016234107
Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J et al (2019) Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566:553–557. https://doi.org/10.1038/s41586-019-0915-y
Schuster E, Taftaf R, Reduzzi C, Albert MK, Romero-Calvo I, Liu H (2021) Better together: circulating tumor cell clustering in metastatic cancer. Trends in Cancer 7:1020–1032. https://doi.org/10.1016/j.trecan.2021.07.001
Ring A, Nguyen-Sträuli BD, Wicki A, Aceto N (2023) Biology, vulnerabilities and clinical applications of circulating tumour cells. Nat Rev Cancer 23:95–111. https://doi.org/10.1038/s41568-022-00536-4
Vaupel P, Höckel M, Mayer A (2007) Detection and characterization of Tumor Hypoxia using pO2 histography. Antioxid Redox Signal 9:1221–1236. https://doi.org/10.1089/ars.2007.1628
Walsh JC, Lebedev A, Aten E, Madsen K, Marciano L, Kolb HC (2014) The clinical importance of assessing Tumor Hypoxia: relationship of Tumor Hypoxia to Prognosis and Therapeutic Opportunities. Antioxid Redox Signal 21:1516–1554. https://doi.org/10.1089/ars.2013.5378
Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26:225–239. https://doi.org/10.1007/s10555-007-9055-1
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during Tumorigenesis. Cell 86:353–364. https://doi.org/10.1016/S0092-8674(00)80108-7
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70. https://doi.org/10.1016/S0092-8674(00)81683-9
Hanahan D, Weinberg RA (2011) Hallmarks of Cancer: the Next Generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hardee ME, Dewhirst MW, Agarwal N, Sorg BS (2009) Novel imaging provides new insights into mechanisms of oxygen transport in tumors. Curr Mol Med 9:435–441. https://doi.org/10.2174/156652409788167122
Loges S, Mazzone M, Hohensinner P, Carmeliet P (2009) Silencing or fueling metastasis with VEGF inhibitors: Antiangiogenesis revisited. Cancer Cell 15:167–170. https://doi.org/10.1016/j.ccr.2009.02.007
Carmeliet P, Jain RK (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discovery 10:417–427. https://doi.org/10.1038/nrd3455
Nussenbaum F, Herman IM (2010) Tumor angiogenesis: insights and innovations. J Oncol. https://doi.org/10.1155/2010/132641
Dewhirst MW, Cao Y, Moeller B (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8:425–437. http://www.nature.com/nrc/journal/v8/n6/suppinfo/nrc2397_S1.html
Michiels C, Tellier C, Feron O (2016) Cycling hypoxia: a key feature of the tumor microenvironment. Biochimica et Biophysica Acta (BBA) -. Reviews on Cancer 1866:76–86. https://doi.org/10.1016/j.bbcan.2016.06.004
Granger DN, Kvietys PR (2015) Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol 6:524–551. https://doi.org/10.1016/j.redox.2015.08.020
Schönenberger MJ, Kovacs WJ (2015) Hypoxia signaling pathways: modulators of oxygen-related organelles. Front Cell Dev Biol 3:42. https://doi.org/10.3389/fcell.2015.00042
Wiesener MS, Jürgensen JS, Rosenberger C, Scholze C, Hörstrup JH, Warnecke C et al (2003) Widespread, hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J 17:271–273. https://doi.org/10.1096/fj.02-0445fje
Duan C (2016) Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am J Physiology-Cell Physiol 310:C260–C269. https://doi.org/10.1152/ajpcell.00315.2015
Keith B, Johnson RS, Simon MC (2012) HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12:9–22. https://doi.org/10.1038/nrc3183
Kaelin WG (2017) The VHL tumor suppressor gene: insights into Oxygen Sensing and Cancer. Trans Am Clin Climatol Assoc 128:298–307
Bárdos JI, Ashcroft M (2004) Hypoxia-inducible factor-1 and oncogenic signalling. BioEssays 26:262–269. https://doi.org/10.1002/bies.20002
Maynard MA, Ohh M (2007) The role of hypoxia-inducible factors in cancer. Cell Mol Life Sci 64:2170–2180. https://doi.org/10.1007/s00018-007-7082-2
Kaelin WG Von Hippel–Lindau disease: insights into oxygen sensing, protein degradation, and cancer. J Clin Investig 2022;132. https://doi.org/10.1172/JCI162480
Nakazawa MS, Keith B, Simon MC (2016) Oxygen availability and metabolic adaptations. Nat Rev Cancer 16:663–673. https://doi.org/10.1038/nrc.2016.84
Missiaen R, Lesner NP, Simon MC (2023) HIF: a master regulator of nutrient availability and metabolic cross-talk in the tumor microenvironment. EMBO J 42:e112067. https://doi.org/10.15252/embj.2022112067
Araos J, Sleeman JP, Garvalov BK (2018) The role of hypoxic signalling in metastasis: towards translating knowledge of basic biology into novel anti-tumour strategies. Clin Exp Metastasis 35:563–599. https://doi.org/10.1007/s10585-018-9930-x
Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D et al (1999) Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res 59:5830–5835
Liao D, Corle C, Seagroves TN, Johnson RS (2007) Hypoxia-inducible Factor-1α is a Key Regulator of Metastasis in a transgenic model of Cancer initiation and progression. Cancer Res 67:563–572. https://doi.org/10.1158/0008-5472.CAN-06-2701
Baba Y, Nosho K, Shima K, Irahara N, Chan AT, Meyerhardt JA et al (2010) HIF1A overexpression is Associated with Poor Prognosis in a cohort of 731 colorectal cancers. Am J Pathol 176:2292–2301. https://doi.org/10.2353/ajpath.2010.090972
Zheng S-S, Chen X-H, Yin X, Zhang B-H (2013) Prognostic significance of HIF-1α expression in Hepatocellular Carcinoma: a Meta-analysis. PLoS ONE 8:e65753. https://doi.org/10.1371/journal.pone.0065753
Chen L, Shi Y, Yuan J, Han Y, Qin R, Wu Q et al (2014) HIF-1 alpha overexpression correlates with poor overall survival and Disease-Free Survival in Gastric Cancer Patients Post-Gastrectomy. PLoS ONE 9:e90678. https://doi.org/10.1371/journal.pone.0090678
Méndez-Blanco C, Fernández-Palanca P, Fondevila F, González-Gallego J, Mauriz JL (2021) Prognostic and clinicopathological significance of hypoxia-inducible factors 1α and 2α in hepatocellular carcinoma: a systematic review with meta-analysis. Ther Adv Med Oncol 13:1758835920987071. https://doi.org/10.1177/1758835920987071
Schindl M, Schoppmann SF, Samonigg H, Hausmaninger H, Kwasny W, Gnant M et al (2002) Overexpression of Hypoxia-inducible factor 1α is Associated with an unfavorable prognosis in Lymph Node-positive breast Cancer. Clin Cancer Res 8:1831–1837
Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM et al (2003) Levels of hypoxia-inducible factor-1α independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 97:1573–1581. https://doi.org/10.1002/cncr.11246
Giatromanolaki A, Koukourakis MI, Sivridis E, Turley H, Talks K, Pezzella F et al (2001) Relation of hypoxia inducible factor 1α and 2α in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br J Cancer 85:881–890. https://doi.org/10.1054/bjoc.2001.2018
Holmquist-Mengelbier L, Fredlund E, Löfstedt T, Noguera R, Navarro S, Nilsson H et al (2006) Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 10:413–423. https://doi.org/10.1016/j.ccr.2006.08.026
Helczynska K, Larsson A-M, Holmquist Mengelbier L, Bridges E, Fredlund E, Borgquist S et al (2008) Hypoxia-inducible Factor-2α correlates to distant recurrence and poor outcome in invasive breast Cancer. Cancer Res 68:9212–9220. https://doi.org/10.1158/0008-5472.CAN-08-1135
Hakimi AA, Pham CG, Hsieh JJ (2013) A clear picture of renal cell carcinoma. Nat Genet 45:849–850. https://doi.org/10.1038/ng.2708
Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E et al (2018) The Cancer Genome Atlas Comprehensive Molecular characterization of renal cell carcinoma. Cell Rep 23:313–326e5. https://doi.org/10.1016/j.celrep.2018.03.075
Kizaka-Kondoh S, Itasaka S, Zeng L, Tanaka S, Zhao T, Takahashi Y et al (2009) Selective killing of Hypoxia-Inducible Factor-1–Active cells improves survival in a mouse model of invasive and metastatic pancreatic Cancer. Clin Cancer Res 15:3433–3441. https://doi.org/10.1158/1078-0432.CCR-08-2267
Tao J, Yang G, Zhou W, Qiu J, Chen G, Luo W et al (2021) Targeting hypoxic tumor microenvironment in pancreatic cancer. J Hematol Oncol 14:14. https://doi.org/10.1186/s13045-020-01030-w
Chiou S-H, Risca VI, Wang GX, Yang D, Grüner BM, Kathiria AS et al (2017) BLIMP1 induces transient metastatic heterogeneity in pancreatic Cancer. Cancer Discov 7:1184–1199. https://doi.org/10.1158/2159-8290.CD-17-0250
Rankin EB, Giaccia AJ (2016) Hypoxic control of metastasis. Science 352:175–180. https://doi.org/10.1126/science.aaf4405
Martinive P, Defresne F, Bouzin C, Saliez J, Lair F, Grégoire V et al (2006) Preconditioning of the Tumor vasculature and tumor cells by intermittent hypoxia: implications for Anticancer Therapies. Cancer Res 66:11736–11744. https://doi.org/10.1158/0008-5472.CAN-06-2056
Hsieh C-H, Lee C-H, Liang J-A, Yu C-Y, Shyu W-C (2010) Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol Rep 24:1629–1636. https://doi.org/10.3892/or_00001027
Malec V, Gottschald OR, Li S, Rose F, Seeger W, Hänze J (2010) HIF-1α signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic Biol Med 48:1626–1635. https://doi.org/10.1016/j.freeradbiomed.2010.03.008
Chen W-L, Wang C-C, Lin Y-J, Wu C-P, Hsieh C-H (2015) Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J Translational Med 13:389. https://doi.org/10.1186/s12967-015-0758-8
Pressley M, Gallaher JA, Brown JS, Tomaszewski MR, Borad P, Damaghi M et al (2021) Cycling hypoxia selects for constitutive HIF stabilization. Sci Rep 11:5777. https://doi.org/10.1038/s41598-021-85184-8
Nanduri J, Wang N, Yuan G, Khan SA, Souvannakitti D, Peng Y-J et al (2009) Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities.Proc Natl Acad Sci 106:1199–204. https://doi.org/10.1073/pnas.0811018106
Liberti MV, Locasale JW (2016) The Warburg Effect: how does it Benefit Cancer cells? Trends Biochem Sci 41:211–218. https://doi.org/10.1016/j.tibs.2015.12.001
Pavlova NN, Zhu J, Thompson CB (2022) The hallmarks of cancer metabolism: still emerging. Cell Metabol 34:355–377. https://doi.org/10.1016/j.cmet.2022.01.007
Semenza GL (2013) HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 123:3664–3671. https://doi.org/10.1172/JCI67230
Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E (2006) Comparison of metabolic pathways between Cancer cells and stromal cells in Colorectal Carcinomas: a metabolic survival role for Tumor-Associated Stroma. Cancer Res 66:632–637. https://doi.org/10.1158/0008-5472.CAN-05-3260
Sonveaux P, Végran F, Schroeder T, Wergin MC et al (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. JCI 118:3930–42. https://doi.org/10.1172/jci36843
Corbet C, Feron O (2017) Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer 17(10):577–593. https://doi.org/10.1038/nrc.2017.77
Supuran CT (2008) Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discovery 7:168–181. https://doi.org/10.1038/nrd2467
Noor SI, Jamali S, Ames S, Langer S, Deitmer JW, Becker HM (2018) A surface proton antenna in carbonic anhydrase II supports lactate transport in cancer cells. ELife 7:e35176. https://doi.org/10.7554/eLife.35176
Kim J, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metabol 3:177–185. https://doi.org/10.1016/j.cmet.2006.02.002
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metabol 3:187–197
Fukuda R, Zhang H, Kim J, Shimoda L, Dang CV, Semenza GL (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129:111–122. https://doi.org/10.1016/j.cell.2007.01.047
Wise DR, Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35:427–433. https://doi.org/10.1016/j.tibs.2010.05.003
Anastasiou D, Cantley LC (2012) Breathless cancer cells get fat on glutamine. Cell Res 22:443–446. https://doi.org/10.1038/cr.2012.5
Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD et al (2015) Metabolic competition in the Tumor Microenvironment is a driver of Cancer Progression. Cell 162:1229–1241. https://doi.org/10.1016/j.cell.2015.08.016
Sormendi S, Wielockx B (2018) Hypoxia pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Frontiers in Immunology 2018;9. https://doi.org/10.3389/fimmu.2018.00040
Tao J-H, Barbi J, Pan F (2015) Hypoxia-inducible factors in T lymphocyte differentiation and function. A review in the theme: Cellular responses to Hypoxia. Am J Physiology-Cell Physiol 309:C580–C589. https://doi.org/10.1152/ajpcell.00204.2015
Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R et al (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162:1217–1228. https://doi.org/10.1016/j.cell.2015.08.012
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M et al (2007) Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 109:3812–3819. https://doi.org/10.1182/blood-2006-07-035972
Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A et al (2016) LDHA-Associated Lactic Acid Production blunts Tumor Immunosurveillance by T and NK cells. Cell Metabol 24:657–671. https://doi.org/10.1016/j.cmet.2016.08.011
Cassetta L, Pollard JW (2023) A timeline of tumour-associated macrophage biology. Nat Rev Cancer 23:238–257. https://doi.org/10.1038/s41568-022-00547-1
Mohamed MM, Sloane BF (2006) Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 6:764–775. https://doi.org/10.1038/nrc1949
Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V et al (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513:559–563. https://doi.org/10.1038/nature13490
Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian B-Z et al (2015) Real-time imaging reveals local, transient vascular permeability, and Tumor Cell Intravasation stimulated by TIE2hi macrophage–derived VEGFA. Cancer Discov 5:932–943. https://doi.org/10.1158/2159-8290.CD-15-0012
De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A, Maggiolini M (2013) HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res 15:R64. https://doi.org/10.1186/bcr3458
Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A et al (2013) Acidity generated by the Tumor Microenvironment drives local Invasion. Cancer Res 73:1524–1535. https://doi.org/10.1158/0008-5472.CAN-12-2796
Keenan MM, Chi J-T (2015) Alternative fuels for cancer cells. Cancer J 21. https://doi.org/10.1097/ppo.0000000000000104
Gillies RJ (2022) Cancer heterogeneity and metastasis: life at the edge. Clin Exp Metastasis 39:15–19. https://doi.org/10.1007/s10585-021-10101-2
Webb BA, Chimenti M, Jacobson MP, Barber DL (2011) Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer 11:671–677. https://doi.org/10.1038/nrc3110
Rohani N, Hao L, Alexis MS, Joughin BA, Krismer K, Moufarrej MN et al (2019) Acidification of Tumor at Stromal Boundaries drives transcriptome alterations Associated with aggressive phenotypes. Cancer Res 79:1952–1966. https://doi.org/10.1158/0008-5472.CAN-18-1604
Huang D, Cao L, Xiao L, Song J, Zhang Y, Zheng P et al (2019) Hypoxia induces actin cytoskeleton remodeling by regulating the binding of CAPZA1 to F-actin via PIP2 to drive EMT in hepatocellular carcinoma. Cancer Lett 448:117–127. https://doi.org/10.1016/j.canlet.2019.01.042
Deryugina EI, Quigley JP (2006) Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 25:9–34. https://doi.org/10.1007/s10555-006-7886-9
Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G et al (2003) Regulation of Colon Carcinoma Cell Invasion by Hypoxia-Inducible factor 11. Cancer Res 63:1138–1143
Gonzalez-Avila G, Sommer B, García-Hernández AA, Ramos C (2020) Matrix Metalloproteinases’ role in Tumor Microenvironment. In: Birbrair A (ed) Tumor Microenvironment: Extracellular Matrix Components – Part A. Springer International Publishing, Cham, pp 97–131. https://doi.org/10.1007/978-3-030-40146-7_5.
Sun H, Zhang D, Yao Z, Lin X, Liu J, Gu Q et al (2017) Anti-angiogenic treatment promotes triple-negative breast cancer invasion via vasculogenic mimicry. Cancer Biol Ther 18:205–213. https://doi.org/10.1080/15384047.2017.1294288
Petrella BL, Lohi J, Brinckerhoff CE (2005) Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2α in von Hippel–Lindau renal cell carcinoma. Oncogene 24:1043–1052. https://doi.org/10.1038/sj.onc.1208305
Nabeshima K, Inoue T, Shimao Y, Okada Y, Itoh Y, Seiki M et al (2000) Front-cell-specific expression of membrane-type 1 matrix metalloproteinase and gelatinase a during cohort migration of colon carcinoma cells induced by hepatocyte growth factor/scatter factor. Cancer Res 60:3364–3369
Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C et al (2007) Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9:893–904. https://doi.org/10.1038/ncb1616
Graham CH, Forsdike J, Fitzgerald CJ, Macdonald-Goodfellow S (1999) Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int J Cancer 80:617–623. https://doi.org/10.1002/(SICI)1097-0215(19990209)80:4<617::AID-IJC22>3.0.CO;2-C
Büchler P, Reber HA, Tomlinson JS, Hankinson O, Kallifatidis G, Friess H et al (2009) Transcriptional regulation of urokinase-type plasminogen activator receptor by Hypoxia-Inducible factor 1 is crucial for Invasion of pancreatic and Liver Cancer. Neoplasia 11:196–IN12. https://doi.org/10.1593/neo.08734
Nishi H, Sasaki T, Nagamitsu Y, Terauchi F, Nagai T, Nagao T et al (2016) Hypoxia inducible factor-1 mediates upregulation of urokinase-type plasminogen activator receptor gene transcription during hypoxia in cervical cancer cells. Oncol Rep 35:992–998. https://doi.org/10.3892/or.2015.4449
Arpino V, Brock M, Gill SE (2015) The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol 44–46:247–254. https://doi.org/10.1016/j.matbio.2015.03.005
Choi JY, Jang YS, Min SY, Song JY (2011) Overexpression of MMP-9 and HIF-1α in breast Cancer cells under hypoxic conditions. J Breast Cancer 14:88–95
Kai AK, Chan LK, Lo RC, Lee JM, Wong CC, Wong JC et al (2016) Down-regulation of TIMP2 by HIF-1alpha/miR-210/HIF-3alpha regulatory feedback circuit enhances cancer metastasis in hepatocellular carcinoma. Hepatology 64:473–487. https://doi.org/10.1002/hep.28577
Liu L, Sun L, Zhao P, Yao L, Jin H, Liang S et al (2010) Hypoxia promotes metastasis in human gastric cancer by up-regulating the 67-kDa laminin receptor. Cancer Sci 101:1653–1660. https://doi.org/10.1111/j.1349-7006.2010.01592.x
Glentis A, Oertle P, Mariani P, Chikina A, El Marjou F, Attieh Y et al (2017) Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat Commun 8:924. https://doi.org/10.1038/s41467-017-00985-8
Frantz C, Stewart KM, Weaver VM (2010) The extracellular matrix at a glance. J Cell Sci 123:4195–4200. https://doi.org/10.1242/jcs.023820
Gilkes DM, Semenza GL, Wirtz D (2014) Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer 14:430–439. https://doi.org/10.1038/nrc3726
Xiong G-F, Xu R (2016) Function of cancer cell-derived extracellular matrix in tumor progression. J Cancer Metastasis Treat 2:357–364. https://doi.org/10.20517/2394-4722.2016.08
Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I (2018) The hypoxic tumour microenvironment. Oncogenesis 7:10. https://doi.org/10.1038/s41389-017-0011-9
Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196:395–406. https://doi.org/10.1083/jcb.201102147
Piersma B, Hayward M-K, Weaver VM (2020) Fibrosis and cancer: a strained relationship. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1873:188356. https://doi.org/10.1016/j.bbcan.2020.188356
Gkretsi V, Stylianopoulos T (2018) Cell adhesion and matrix stiffness: coordinating cancer cell invasion and metastasis. Frontiers in Oncology 2018;8. https://doi.org/10.3389/fonc.2018.00145
Li X, Wang J (2020) Mechanical tumor microenvironment and transduction: cytoskeleton mediates cancer cell invasion and metastasis. Int J Biol Sci 16:2014–2028. https://doi.org/10.7150/ijbs.44943
Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z (2020) Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun 11:5120. https://doi.org/10.1038/s41467-020-18794-x
Colpaert CG, Vermeulen PB, Fox SB, Harris AL, Dirix LY, Van Marck EA (2003) The Presence of a fibrotic focus in invasive breast carcinoma correlates with the expression of Carbonic anhydrase IX and is a marker of Hypoxia and Poor Prognosis. Breast Cancer Res Treat 81:137–147. https://doi.org/10.1023/A:1025702330207
Trastour C, Benizri E, Ettore F, Ramaioli A, Chamorey E, Pouysségur J et al (2007) HIF-1α and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer 120:1451–1458. https://doi.org/10.1002/ijc.22436
Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T (2008) Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiology-Gastrointestinal Liver Physiol 295:G709–G717. https://doi.org/10.1152/ajpgi.90356.2008
Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S et al (2009) Cancer-Stellate cell interactions perpetuate the Hypoxia-Fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 11:497–508. https://doi.org/10.1593/neo.81618
Spivak-Kroizman TR, Hostetter G, Posner R, Aziz M, Hu C, Demeure MJ et al (2013) Hypoxia triggers hedgehog-mediated tumor–stromal interactions in pancreatic Cancer. Cancer Res 73:3235–3247. https://doi.org/10.1158/0008-5472.CAN-11-1433
Mello AM, Ngodup T, Lee Y, Donahue KL, Li J, Rao A et al (2022) Hypoxia promotes an inflammatory phenotype of fibroblasts in pancreatic cancer. Oncogenesis 11:56. https://doi.org/10.1038/s41389-022-00434-2
Nath B, Szabo G (2012) Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology 55:622–633. https://doi.org/10.1002/hep.25497
Roth KJ, Copple BL (2015) Role of Hypoxia-Inducible factors in the development of liver fibrosis. Cell Mol Gastroenterol Hepatol 1:589–597. https://doi.org/10.1016/j.jcmgh.2015.09.005
Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ et al (2005) Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105:659–669. https://doi.org/10.1182/blood-2004-07-2958
Kabei K, Tateishi Y, Nozaki M, Tanaka M, Shiota M, Osada-Oka M et al (2018) Role of hypoxia-inducible factor-1 in the development of renal fibrosis in mouse obstructed kidney: special references to HIF-1 dependent gene expression of profibrogenic molecules. J Pharmacol Sci 136:31–38. https://doi.org/10.1016/j.jphs.2017.12.004
Myllyharju J, Kivirikko KI (2004) Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet 20:33–43. https://doi.org/10.1016/j.tig.2003.11.004
Gjaltema RAF, Bank RA (2017) Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit Rev Biochem Mol Biol 52:74–95. https://doi.org/10.1080/10409238.2016.1269716
Hofbauer K-H, Gess B, Lohaus C, Meyer HE, Katschinski D, Kurtz A (2003) Oxygen tension regulates the expression of a group of procollagen hydroxylases. Eur J Biochem 270:4515–4522. https://doi.org/10.1046/j.1432-1033.2003.03846.x
Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM (2006) Concordant regulation of Gene expression by Hypoxia and 2-Oxoglutarate-dependent dioxygenase inhibition: THE ROLE OF HIF-1α, HIF-2α, AND OTHER PATHWAYS*. J Biol Chem 281:15215–15226. https://doi.org/10.1074/jbc.M511408200
Aro E, Khatri R, Gerard-O’Riley R, Mangiavini L, Myllyharju J, Schipani E (2012) Hypoxia-inducible Factor-1 (HIF-1) but not HIF-2 is essential for hypoxic induction of collagen prolyl 4-Hydroxylases in primary newborn mouse Epiphyseal Growth plate Chondrocytes*. J Biol Chem 287:37134–37144. https://doi.org/10.1074/jbc.M112.352872
Bentovim L, Amarilio R, Zelzer E (2012) HIF1α is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development 139:4473–4483. https://doi.org/10.1242/dev.083881
Eisinger-Mathason TSK, Zhang M, Qiu Q, Skuli N, Nakazawa MS, Karakasheva T et al (2013) Hypoxia-dependent modification of collagen networks promotes Sarcoma Metastasis. Cancer Discov 3:1190–1205. https://doi.org/10.1158/2159-8290.Cd-13-0118
Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL (2013) Hypoxia-inducible factor 1 (HIF-1) promotes Extracellular Matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in Fibroblasts*. J Biol Chem 288:10819–10829. https://doi.org/10.1074/jbc.M112.442939
Gilkes DM, Chaturvedi P, Bajpai S, Wong CC, Wei H, Pitcairn S et al (2013) Collagen Prolyl Hydroxylases are essential for breast Cancer metastasis. Cancer Res 73:3285–3296. https://doi.org/10.1158/0008-5472.CAN-12-3963
Gilkes DM, Bajpai S, Wong CC, Chaturvedi P, Hubbi ME, Wirtz D et al (2013) Procollagen Lysyl hydroxylase 2 is essential for Hypoxia-Induced breast Cancer Metastasis. Mol Cancer Res 11:456–466. https://doi.org/10.1158/1541-7786.MCR-12-0629
Lewis DM, Pruitt H, Jain N, Ciccaglione M, McCaffery JM, Xia Z et al (2019) A Feedback Loop between Hypoxia and Matrix stress relaxation increases Oxygen-Axis Migration and Metastasis in Sarcoma. Cancer Res 79:1981–1995. https://doi.org/10.1158/0008-5472.CAN-18-1984
Xiong G, Deng L, Zhu J, Rychahou PG, Xu R (2014) Prolyl-4-hydroxylase α subunit 2 promotes breast cancer progression and metastasis by regulating collagen deposition. BMC Cancer 14:1. https://doi.org/10.1186/1471-2407-14-1
Liburkin-Dan T, Toledano S, Neufeld G Lysyl Oxidase Family enzymes and their role in Tumor Progression. Int J Mol Sci 2022;23. https://doi.org/10.3390/ijms23116249
Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A et al (2009) Hypoxia-Induced Lysyl oxidase is a critical mediator of bone marrow cell recruitment to Form the Premetastatic Niche. Cancer Cell 15:35–44. https://doi.org/10.1016/j.ccr.2008.11.012
Schietke R, Warnecke C, Wacker I, Schödel J, Mole DR, Campean V et al (2010) The Lysyl Oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in Hypoxia: Insights into cellular transformation processes mediated by HIF-1*. J Biol Chem 285:6658–6669. https://doi.org/10.1074/jbc.M109.042424
Wong CC-L, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P et al (2011) Hypoxia-inducible factor 1 is a master regulator of breast cancer metastatic niche formation. Proc Natl Acad Sci 108:16369–74. https://doi.org/10.1073/pnas.1113483108
Wong CC-L, Zhang H, Gilkes DM, Chen J, Wei H, Chaturvedi P et al (2012) Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J Mol Med 90:803–815. https://doi.org/10.1007/s00109-011-0855-y
Barker HE, Cox TR, Erler JT (2012) The rationale for targeting the LOX family in cancer. Nat Rev Cancer 12:540–552. https://doi.org/10.1038/nrc3319
Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E et al (2015) Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med 7:1063–1076. https://doi.org/10.15252/emmm.201404827
Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ (2006) Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med 4:38. https://doi.org/10.1186/1741-7015-4-38
Wolf K, Friedl P (2011) Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol 21:736–744. https://doi.org/10.1016/j.tcb.2011.09.006
Dekker Y, Le Dévédec SE, Danen EHJ, Liu Q (2022) Crosstalk between hypoxia and extracellular matrix in the tumor microenvironment in breast cancer. Genes 2022;13. https://doi.org/10.3390/genes13091585
Bottaro DP, Rubin JS, Faletto DL, Chan AM-L, Kmiecik TE, Vande Woude GF et al (1991) Identification of the hepatocyte growth factor receptor as the c-met Proto-Oncogene product. Science 251:802–804. https://doi.org/10.1126/science.1846706
Yang X, Liao H-Y, Zhang H-H (2022) Roles of MET in human cancer. Clin Chim Acta 525:69–83. https://doi.org/10.1016/j.cca.2021.12.017
Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM (2003) Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 3:347–361. https://doi.org/10.1016/S1535-6108(03)00085-0
Scarpino S, Cancellario d’Alena F, Di Napoli A, Pasquini A, Marzullo A, Ruco LP (2004) Increased expression of Met protein is associated with up-regulation of hypoxia inducible factor-1 (HIF-1) in tumour cells in papillary carcinoma of the thyroid. J Pathol 202:352–358. https://doi.org/10.1002/path.1522
Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernández-Pérez D, Isella C et al (2018) Increased lactate secretion by Cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted Therapies. Cell Metabol 28:848–865e6. https://doi.org/10.1016/j.cmet.2018.08.006
Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W (2003) Chemokine receptor CXCR4 downregulated by von hippel-lindau tumour suppressor pVHL. Nature 425:307–311. https://doi.org/10.1038/nature01874
Nengroo MA, Khan MA, Verma A, Datta D (2022) Demystifying the CXCR4 conundrum in cancer biology: beyond the surface signaling paradigm. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1877:188790. https://doi.org/10.1016/j.bbcan.2022.188790
Korbecki J, Kojder K, Kapczuk P, Kupnicka P, Gawrońska-Szklarz B, Gutowska I et al (2021) The Effect of Hypoxia on the expression of CXC Chemokines and CXC Chemokine Receptors—A review of literature. Int J Mol Sci 22. https://doi.org/10.3390/ijms22020843
Jin F, Brockmeier U, Otterbach F, Metzen E (2012) New Insight into the SDF-1/CXCR4 Axis in a breast carcinoma model: Hypoxia-Induced endothelial SDF-1 and Tumor Cell CXCR4 are required for Tumor Cell Intravasation. Mol Cancer Res 10:1021–1031. https://doi.org/10.1158/1541-7786.MCR-11-0498
Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME et al (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–56. https://doi.org/10.1038/35065016
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 15:178–196. https://doi.org/10.1038/nrm3758
Yang M-H, Wu M-Z, Chiou S-H, Chen P-M, Chang S-Y, Liu C-J et al (2008) Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol 10:295–305. https://doi.org/10.1038/ncb1691
Imai T, Horiuchi A, Wang C, Oka K, Ohira S, Nikaido T et al (2003) Hypoxia attenuates the expression of E-Cadherin via Up-Regulation of SNAIL in ovarian carcinoma cells. Am J Pathol 163:1437–1447. https://doi.org/10.1016/S0002-9440(10)63501-8
Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA et al (2010) ERβ impedes prostate Cancer EMT by destabilizing HIF-1α and inhibiting VEGF-Mediated snail nuclear localization: implications for Gleason Grading. Cancer Cell 17:319–332. https://doi.org/10.1016/j.ccr.2010.02.030
Luo D, Wang J, Li J, Post M (2011) Mouse snail is a target gene for HIF. Mol Cancer Res 9:234–245. https://doi.org/10.1158/1541-7786.MCR-10-0214
Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R et al (2015) HIF-1α promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in Colorectal Cancer. PLoS ONE 10:e0129603. https://doi.org/10.1371/journal.pone.0129603
Storci G, Sansone P, Mari S, D’Uva G, Tavolari S, Guarnieri T et al (2010) TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol 225:682–691. https://doi.org/10.1002/jcp.22264
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U (2008);105:6392–7. https://doi.org/10.1073/pnas.0802047105
Chen J, Imanaka N, Chen J, Griffin JD (2010) Hypoxia potentiates notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br J Cancer 102:351–360. https://doi.org/10.1038/sj.bjc.6605486
Rankin EB, Giaccia AJ (2016) The receptor tyrosine kinase AXL in cancer progression. Cancers 2016;8. https://doi.org/10.3390/cancers8110103
Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN et al (2014) Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci 111:13373–13378. https://doi.org/10.1073/pnas.1404848111
Clevers H (2006) Wnt/β-Catenin signaling in Development and Disease. Cell 127:469–480. https://doi.org/10.1016/j.cell.2006.10.018
MacDonald BT, Tamai K, He X (2009) Wnt/β-Catenin signaling: components, mechanisms, and Diseases. Dev Cell 17:9–26. https://doi.org/10.1016/j.devcel.2009.06.016
Hong C-F, Chen W-Y, Wu C-W (2017) Upregulation of wnt signaling under hypoxia promotes lung cancer progression. Oncol Rep 38:1706–1714. https://doi.org/10.3892/or.2017.5807
Aceto N, Toner M, Maheswaran S, Haber DA (2015) En Route to Metastasis: circulating Tumor cell clusters and epithelial-to-mesenchymal transition. Trends in Cancer 1:44–52. https://doi.org/10.1016/j.trecan.2015.07.006
Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC et al (2015) Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527:472–476. https://doi.org/10.1038/nature15748
Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H et al (2015) Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527:525–530. https://doi.org/10.1038/nature16064
Nagai T, Ishikawa T, Minami Y, Nishita M (2020) Tactics of cancer invasion: solitary and collective invasion. J Biochem 167:347–355. https://doi.org/10.1093/jb/mvaa003
Williams ED, Gao D, Redfern A, Thompson EW (2019) Controversies around epithelial–mesenchymal plasticity in cancer metastasis. Nat Rev Cancer 19:716–732. https://doi.org/10.1038/s41568-019-0213-x
Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S et al (2018) Identification of the tumour transition states occurring during EMT. Nature 556:463–468. https://doi.org/10.1038/s41586-018-0040-3
McKeown SR (2014) Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br J Radiol 87:20130676. https://doi.org/10.1259/bjr.20130676
Donato C, Kunz L, Castro-Giner F, Paasinen-Sohns A, Strittmatter K, Szczerba BM et al (2020) Hypoxia triggers the intravasation of clustered circulating Tumor cells. Cell Rep 32:108105. https://doi.org/10.1016/j.celrep.2020.108105
Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R et al (2019) Circulating Tumor Cell Clustering Shapes DNA methylation to Enable Metastasis Seeding. Cell 176:98–112e14. https://doi.org/10.1016/j.cell.2018.11.046
Rankin EB, Nam J-M, Giaccia AJ, Hypoxia (2016) Signaling the Metastatic Cascade. Trends in Cancer 2:295–304. https://doi.org/10.1016/j.trecan.2016.05.006
FDA Pulls Approval for Avastin in Breast Cancer (2011) Cancer Discov 1:OF1–2. https://doi.org/10.1158/2159-8290.CD-ND112311OL-08
Godet I, Shin YJ, Ju JA, Ye IC, Wang G, Gilkes DM (2019) Fate-mapping post-hypoxic tumor cells reveals a ROS-resistant phenotype that promotes metastasis. Nat Commun 10:4862. https://doi.org/10.1038/s41467-019-12412-1
Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z et al (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527:186–191. https://doi.org/10.1038/nature15726
Lim CT, Bershadsky A, Sheetz MP, Mechanobiology (2010) J R Soc Interface 7. https://doi.org/10.1098/rsif.2010.0150.focus
Nakanishi J, Uto K (2022) An introduction to material-based Mechanobiology. In: Nakanishi J, Uto K (eds) Material-based Mechanobiology. The Royal Society of Chemistry, pp 1–20. https://doi.org/10.1039/9781839165375-00001.
Kim AA, Nekimken AL, Fechner S, O’Brien LE, Pruitt BL (2018) Microfluidics for mechanobiology of model organisms. Methods Cell Biol 146:217–259. https://doi.org/10.1016/bs.mcb.2018.05.010
Krieg M, Fläschner G, Alsteens D, Gaub BM, Roos WH, Wuite GJL et al (2018) Atomic force microscopy-based mechanobiology. Nat Rev Phys 1:41–57. https://doi.org/10.1038/s42254-018-0001-7
Cardozo CP (2020) Mechanotransduction: Overview. encyclopedia of bone biology, elsevier; 2020: p. 217. https://doi.org/10.1016/B978-0-12-801238-3.62233-X
Wang JH, Li B (2010) Mechanics rules cell biology. BMC Sports Sci Med Rehabil 2:16. https://doi.org/10.1186/1758-2555-2-16
Montagner M, Dupont S (2020) Mechanical forces as determinants of disseminated metastatic cell fate. Cells 9:250. https://doi.org/10.3390/cells9010250
Michor F, Liphardt J, Ferrari M, Widom J (2011) What does physics have to do with cancer? Nat Rev Cancer 11:657–670. https://doi.org/10.1038/nrc3092
Swartz MA, Fleury ME (2007) Interstitial Flow and its Effects in Soft tissues. Annu Rev Biomed Eng 9:229–256. https://doi.org/10.1146/annurev.bioeng.9.060906.151850
Vasilaki D, Bakopoulou A, Tsouknidas A, Johnstone E, Michalakis K (2021) Biophysical interactions between components of the tumor microenvironment promote metastasis. Biophys Rev 13:339–357. https://doi.org/10.1007/s12551-021-00811-y
Yao W, Li Y, Ding G (2012) Interstitial fluid Flow: the mechanical environment of cells and Foundation of Meridians. Evidence-Based Complement Altern Med 2012:1–9. https://doi.org/10.1155/2012/853516
Munson JM, Shieh AC (2014) Interstitial fluid flow in cancer: implications for disease progression and treatment. Cancer Manag Res 6:317–328. https://doi.org/10.2147/CMAR.S65444
Barnes JM, Nauseef JT, Henry MD (2012) Resistance to Fluid Shear stress is a conserved Biophysical Property of Malignant cells. PLoS ONE 7:e50973. https://doi.org/10.1371/journal.pone.0050973
Mierke CT (2020) Mechanical Cues affect Migration and Invasion of cells from three different directions. Front Cell Dev Biol 8:583226. https://doi.org/10.3389/fcell.2020.583226
Mierke CT (2019) The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep Prog Phys 82:064602. https://doi.org/10.1088/1361-6633/ab1628
Fischer T, Wilharm N, Hayn A, Mierke CT (2017) Matrix and cellular mechanical properties are the driving factors for facilitating human cancer cell motility into 3D engineered matrices. Converg Sci Phys Oncol 3:044003. https://doi.org/10.1088/2057-1739/aa8bbb
Qin X, Zhang Y, He Y, Chen K, Zhang Y, Li P et al (2021) Shear stress triggered circular dorsal ruffles formation to facilitate cancer cell migration. Arch Biochem Biophys 709:108967. https://doi.org/10.1016/j.abb.2021.108967
Yu T, Liu K, Wu Y, Fan J, Chen J, Li C et al (2013) High interstitial fluid pressure promotes tumor cell proliferation and invasion in oral squamous cell carcinoma. Int J Mol Med 32:1093–1100. https://doi.org/10.3892/ijmm.2013.1496
Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA (2007) Autologous Chemotaxis as a mechanism of Tumor Cell Homing to Lymphatics via interstitial Flow and Autocrine CCR7 Signaling. Cancer Cell 11:526–538. https://doi.org/10.1016/j.ccr.2007.04.020
Munson JM, Bellamkonda RV, Swartz MA (2013) Interstitial Flow in a 3D Microenvironment increases Glioma Invasion by a CXCR4-Dependent mechanism. Cancer Res 73:1536–1546. https://doi.org/10.1158/0008-5472.CAN-12-2838
Heldin C-H, Rubin K, Pietras K, Östman A (2004) High interstitial fluid pressure — an obstacle in cancer therapy. Nat Rev Cancer 4:806–813. https://doi.org/10.1038/nrc1456
Curti BD, Urba WJ, Alvord WG, Janik JE, Smith JW, Madara K et al (1993) Interstitial pressure of subcutaneous nodules in melanoma and lymphoma patients: changes during treatment. Cancer Res 53:2204–2207
Fyles A, Milosevic M, Pintilie M, Syed A, Levin W, Manchul L et al (2006) Long-term performance of interstial fluid pressure and hypoxia as prognostic factors in cervix cancer. Radiother Oncol 80:132–137. https://doi.org/10.1016/j.radonc.2006.07.014
Gensbittel V, Kräter M, Harlepp S, Busnelli I, Guck J, Goetz JG (2021) Mechanical adaptability of Tumor cells in Metastasis. Dev Cell 56:164–179. https://doi.org/10.1016/j.devcel.2020.10.011
Fernandes DC, Araujo TLS, Laurindo FRM, Tanaka LY (2018) Hemodynamic Forces in the Endothelium: From Mechanotransduction to Implications on Development of Atherosclerosis. Endothelium and Cardiovascular Diseases, Elsevier; p. 85–95. https://doi.org/10.1016/B978-0-12-812348-5.00007-6
Chatterjee S (2018) Endothelial mechanotransduction, Redox Signaling and the regulation of vascular inflammatory pathways. Front Physiol 9:524. https://doi.org/10.3389/fphys.2018.00524
Strony J, Beaudoin A, Brands D, Adelman B (1993) Analysis of shear stress and hemodynamic factors in a model of coronary artery stenosis and thrombosis. Am J Physiol Heart Circ Physiol 265:H1787–H1796. https://doi.org/10.1152/ajpheart.1993.265.5.H1787
Regmi S, Fu A, Luo KQ (2017) High Shear stresses under Exercise Condition destroy circulating Tumor cells in a Microfluidic System. Sci Rep 7:39975. https://doi.org/10.1038/srep39975
Moose DL, Krog BL, Kim T-H, Zhao L, Williams-Perez S, Burke G et al (2020) Cancer cells resist Mechanical Destruction in circulation via RhoA/Actomyosin-Dependent mechano-adaptation. Cell Rep 30:3864–3874e6. https://doi.org/10.1016/j.celrep.2020.02.080
Marrella A, Fedi A, Varani G, Vaccari I, Fato M, Firpo G et al (2021) High blood flow shear stress values are associated with circulating tumor cells cluster disaggregation in a multi-channel microfluidic device. PLoS ONE 16:e0245536. https://doi.org/10.1371/journal.pone.0245536
Mitchell M, King M (2013) Computational and experimental models of cancer cell response to fluid shear stress. Front Oncol 3:44. https://doi.org/10.3389/fonc.2013.00044
Yankaskas CL, Bera K, Stoletov K, Serra SA, Carrillo-Garcia J, Tuntithavornwat S et al (2021) The fluid shear stress sensor TRPM7 regulates tumor cell intravasation. Sci Adv 7:eabh3457. https://doi.org/10.1126/sciadv.abh3457
Regmi S, Fung TS, Lim S, Luo KQ (2018) Fluidic shear stress increases the anti-cancer effects of ROS-generating drugs in circulating tumor cells. Breast Cancer Res Treat 172:297–312. https://doi.org/10.1007/s10549-018-4922-8
Liu H, Hu J, Zheng Q, Feng X, Zhan F, Wang X et al (2022) Piezo1 channels as Force Sensors in Mechanical Force-Related chronic inflammation. Front Immunol 13:816149. https://doi.org/10.3389/fimmu.2022.816149
Kim O-H, Choi YW, Park JH, Hong SA, Hong M, Chang IH et al (2022) Fluid shear stress facilitates prostate cancer metastasis through Piezo1-Src-YAP axis. Life Sci 308:120936. https://doi.org/10.1016/j.lfs.2022.120936
Dombroski JA, Hope JM, Sarna NS, King MR (2021) Channeling the force: Piezo1 mechanotransduction in Cancer Metastasis. Cells 10:2815. https://doi.org/10.3390/cells10112815
Zhou H, Huang S (2011) Role of mtor signaling in tumor cell motility, invasion and metastasis. Curr Protein Pept Sci 12:30–42. https://doi.org/10.2174/138920311795659407
Osmani N, Follain G, García León MJ, Lefebvre O, Busnelli I, Larnicol A et al (2019) Metastatic tumor cells exploit their adhesion repertoire to Counteract Shear Forces during intravascular arrest. Cell Rep 28:2491–2500e5. https://doi.org/10.1016/j.celrep.2019.07.102
Perea Paizal J, Au SH, Bakal C (2021) Squeezing through the microcirculation: survival adaptations of circulating tumour cells to seed metastasis. Br J Cancer 124:58–65. https://doi.org/10.1038/s41416-020-01176-x
Huang X, Yang Y, Zhao Y, Cao D, Ai X, Zeng A et al (2018) RhoA-stimulated intra‐capillary morphology switch facilitates the arrest of individual circulating tumor cells. Int J Cancer 142:2094–2105. https://doi.org/10.1002/ijc.31238
Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M et al (1996) Regulation of myosin phosphatase by rho and Rho-Associated kinase (Rho-Kinase). Science 273:245–248. https://doi.org/10.1126/science.273.5272.245
Au SH, Storey BD, Moore JC, Tang Q, Chen Y-L, Javaid S et al (2016) Clusters of circulating tumor cells traverse capillary-sized vessels. Proc Natl Acad Sci USA 113:4947–4952. https://doi.org/10.1073/pnas.1524448113
Egan K, Cooke N, Kenny D (2014) Living in shear: platelets protect cancer cells from shear induced damage. Clin Exp Metastasis 31:697–704. https://doi.org/10.1007/s10585-014-9660-7
Ortiz-Otero N, Clinch AB, Hope J, Wang W, Reinhart-King CA, King MR (2020) Cancer associated fibroblasts confer shear resistance to circulating tumor cells during prostate cancer metastatic progression. Oncotarget 11:1037–1050. https://doi.org/10.18632/oncotarget.27510
Adams DL, Martin SS, Alpaugh RK, Charpentier M, Tsai S, Bergan RC et al (2014) Circulating giant macrophages as a potential biomarker of solid tumors. Proc Natl Acad Sci USA 111:3514–3519. https://doi.org/10.1073/pnas.1320198111
Fu A, Yao B, Dong T, Chen Y, Yao J, Liu Y et al (2022) Tumor-resident intracellular microbiota promotes metastatic colonization in breast cancer. Cell 185:1356–1372e26. https://doi.org/10.1016/j.cell.2022.02.027
Diamantopoulou Z, Gvozdenovic A, Aceto N (2023) A new time dimension in the fight against metastasis. Trends Cell Biol S0962892423000211. https://doi.org/10.1016/j.tcb.2023.02.002
Diamantopoulou Z, Castro-Giner F, Schwab FD, Foerster C, Saini M, Budinjas S et al (2022) The metastatic spread of breast cancer accelerates during sleep. Nature 607:156–162. https://doi.org/10.1038/s41586-022-04875-y
Veerman DP, Imholz BPM, Wieling W, Wesseling KH, van Montfrans GA (1995) Circadian Profile of systemic hemodynamics. Hypertension 26:55–59. https://doi.org/10.1161/01.HYP.26.1.55
Acknowledgements
We thank members and collaborators of the Aceto laboratory for scientific feedback and discussions.
Funding
Open access funding provided by Swiss Federal Institute of Technology Zurich. The Aceto laboratory is supported by the European Research Council (101001652), the strategic focus area of Personalized Health and Related Technologies at ETH Zurich (PHRT-541), the Future and Emerging Technologies programme of the European Commission (801159-B2B), the Swiss National Science Foundation (212183), the Swiss Cancer League (KLS-4834-08-2019), the Basel Cancer League (KLbB-4763-02-2019), the ETH Lymphoma Challenge (LC-02-22) and the ETH Zürich. WJK is supported by the Swiss Cancer League (KFS-4735-02-2019) and the Swiss National Science Foundation (192614).
Author information
Authors and Affiliations
Contributions
ESI, AG, WJK, and NA wrote the article. ESI, AG and WJK performed literature search. ESI and WJK designed the figures.
Corresponding author
Ethics declarations
Competing interests
NA is co-founder and member of the board of PAGE Therapeutics AG, Switzerland, listed as inventor in patent applications related to CTCs, paid consultant for companies with an interest in liquid biopsy, and a Novartis shareholder.
Ethics approval and consent to participate
Not applicable.
Consent to publish
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ildiz, E.S., Gvozdenovic, A., Kovacs, W.J. et al. Travelling under pressure - hypoxia and shear stress in the metastatic journey. Clin Exp Metastasis 40, 375–394 (2023). https://doi.org/10.1007/s10585-023-10224-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-023-10224-8