
Abstract
The aim of this phase II trial was to estimate the objective response rate (ORR) of two different schedules of ixabepilone [weekly or every 3 weeks (Q3W)] combined with bevacizumab, relative to a reference arm of weekly paclitaxel and bevacizumab. Patients with human epidermal growth factor receptor 2-normal, chemotherapy-naïve metastatic breast cancer (MBC) were randomized 3:3:2 to ixabepilone 16 mg/m2 weekly plus bevacizumab 10 mg/kg Q2W (Arm A: n = 46); ixabepilone 40 mg/m2 Q3W (reduced to 32 mg/m2 after four cycles of treatment) plus bevacizumab 15 mg/kg Q3W (Arm B: n = 45); or paclitaxel 90 mg/m2 weekly plus bevacizumab 10 mg/kg intravenous infusion Q2W (Arm C: n = 32). Of 123 randomized patients, 122 were treated. All were followed for ≥19 months; 5 % of patients remained on study treatment at the time of this analysis. Grade 3 or 4 neutropenia was more common in Arm B (60 %) than Arms A (16 %) or C (22 %); other adverse events were similar. The investigator-assessed ORR was 48, 71, and 63 % for Arms A, B, and C, respectively. Median progression-free survival (randomized patients) was 9.6 months in Arm A, 11.9 months in Arm B, and 13.5 months in Arm C. In conclusion, ixabepilone Q3W plus bevacizumab has clinical activity as first-line therapy for MBC relative to paclitaxel plus bevacizumab, but with significantly greater risk of grade 3 or 4 neutropenia. In addition, these data suggest that weekly dosing of ixabepilone may be less active than Q3W dosing, but with less neutropenia.
Similar content being viewed by others
Introduction
Many improvements have been made in the treatment of metastatic breast cancer (MBC) over the past 5 years; however, few regimens have translated into an incremental gain in overall survival (OS). A number of palliative chemotherapy options exist, with diverse regimens based on class of drug, number of agents, dosage, and schedule. Microtubules are a validated target for anticancer therapy; natural antitubulin agents such as taxanes are active in the treatment of breast cancer. However, development of drug resistance and dose-limiting toxicity are the most critical limitations of taxane therapy. Therefore, agents with the ability to overcome resistance without increasing toxicity are needed.
Vascular endothelial growth factor (VEGF), a key mediator of angiogenesis [1], is over-expressed in many tumor types, including primary breast cancer [2, 3], and has been associated with poor prognosis [4–6]. Bevacizumab is a recombinant humanized monoclonal antibody that blocks binding of human VEGF-A to its receptors. In addition to antiangiogenic effects, bevacizumab may produce clinical benefit through other mechanisms, including direct action against tumor cells [7]. Clinical activity of bevacizumab combinations in advanced breast cancer has been demonstrated in three large, randomized, phase III trials [8–10]; significant improvements have been observed in both response rate (RR) and progression-free survival (PFS), but not OS.
Ixabepilone is a semi-synthetic analog of epothilone B targeting microtubules, but engineered to overcome tumor survival pathways [11]. In preclinical tumor models derived from breast, colon, lung, and kidney cancers, ixabepilone alone and in combination with several targeted antiangiogenic agents (bevacizumab, sunitinib, or brivanib) demonstrated robust synergistic antitumor activity [12], and the synergistic antitumor effect was greater with ixabepilone than paclitaxel [12, 13]. In addition, ixabepilone was more effective than paclitaxel at killing endothelial cells expressing P-glycoprotein in vitro, and inhibiting endothelial cell proliferation and tumor angiogenesis in vivo [13]. In the clinic, ixabepilone has shown efficacy as monotherapy in several phase II trials in patients with MBC [14–16], as well as in combination with capecitabine in two large, phase III trials focusing on patients with chemotherapy-resistant disease [17, 18]. Most trials to date have administered ixabepilone on an every 3 week (Q3W) schedule.
Several clinical trials in patients with advanced disease have tried to identify the optimum dose and schedule for administration of paclitaxel and docetaxel. Weekly dosing of paclitaxel was shown to be superior to Q3W dosing in the metastatic setting [19], less toxic than weekly docetaxel, and superior to Q3W paclitaxel or docetaxel in early stage disease [20]. We hypothesized that weekly dosing of ixabepilone might improve efficacy and reduce toxicity compared with Q3W dosing. This clinical trial, CA163-115, was designed to estimate the RR of weekly or Q3W dosing of ixabepilone combined with bevacizumab, with reference treatment of weekly paclitaxel and bevacizumab in patients with chemotherapy-naïve MBC (NCT00370552).
Methods
Patients
Eligible women with human epidermal growth factor receptor 2 (HER2)-normal metastatic or locally advanced breast cancer, previously untreated with chemotherapy for advanced disease, with at least one measurable lesion, Karnofsky performance status score between 80 and 100 %, and life expectancy ≥12 weeks, were enrolled. Patients were permitted to have received prior chemotherapy only in the neoadjuvant/adjuvant setting; any number of prior lines of hormone therapy were allowed.
This trial was conducted in accordance with the Declaration of Helsinki, and in compliance with Good Clinical Practice and local and national regulatory requirements. The protocol was approved by the Institutional Review Board or Independent Ethics Committee at each site before enrollment; all patients provided written informed consent.
Study design
In this multinational, phase II study, patients were randomized in a 3:3:2 ratio to receive ixabepilone weekly or Q3W plus bevacizumab, or weekly paclitaxel plus bevacizumab. The primary endpoint was estimation of RR by Response Evaluation Criteria in Solid Tumors (version 1.0) criteria for each ixabepilone arm relative to the paclitaxel arm. Secondary endpoints included PFS (defined as the time from randomization to disease progression or death), week 24 PFS rate, time to response, duration of response, OS, and safety. Response and progression were determined by the local investigator. Randomization was stratified by disease-free interval from initial diagnosis to first recurrence (≤24 or >24 months), prior taxane therapy (yes or no), and investigative site. Patients initially diagnosed with metastatic disease (or locally advanced disease not amenable to surgery) were included in the >24 month disease-free interval.
For the two ixabepilone experimental arms, patients received either ixabepilone 16 mg/m2 as a 1-h intravenous (IV) continuous infusion on days 1, 8, and 15 in a 28-day cycle, along with bevacizumab as a 10 mg/kg IV infusion every 2 weeks (Q2W; Arm A); or ixabepilone 40 mg/m2, as a 3-h IV infusion on day 1 of a 21-day cycle, along with bevacizumab as a 15 mg/kg IV infusion Q3W (Arm B). After four cycles of treatment on Arm B, all patients remaining on the 40 mg/m2 dose had their ixabepilone dose reduced to 32 mg/m2 (maintenance). For the reference treatment (Arm C), patients received paclitaxel 90 mg/m2 as a 1-h IV infusion on days 1, 8, and 15 of a 28-day cycle, along with bevacizumab administered as a 10 mg/kg IV infusion Q2W.
Treatment for all arms was continued until disease progression or unacceptable toxicity. All patients who received the study drug were evaluated for safety; adverse events (AEs) were assessed according to National Cancer Institute Common Terminology Criteria for Adverse Events (version 3.0).
Statistical analysis
At least 45 patients in each of the experimental arms were required to achieve the maximum width of the exact two-sided 95 % confidence intervals (CIs) for the RRs in these arms to be 31 %, when the corresponding RR was to be 30–50 %. This was a randomized, non-comparative trial. The objective was to estimate endpoints in the three dosing arms, and formal statistical comparisons were not planned due to limitations in sample size.
The primary endpoint, objective RR (ORR), was defined for each arm as the number of patients with best tumor response including complete response (CR) or partial response (PR), divided by the number of randomized patients in the arm. Tumor response analysis consisted of point estimates of the RR and two-sided exact 95 % CIs (Clopper–Pearson method) for each treatment arm. The analyses of PFS and OS were conducted on all randomized patients, and estimated using the Kaplan–Meier method. The two-sided 95 % CIs for the median PFS were reported for each treatment arm. Additional analyses on estimation of time to response and response duration were conducted on response-evaluable patients. The Kaplan–Meier method was used to estimate duration of response, and descriptive statistics (median, minimum, and maximum) were used to summarize time to response for the responders in each arm. Tumor assessments were performed every 8 weeks ± 5 working days until disease progression during the first 12 months from randomization. Thereafter, patients were assessed for tumor response every 3 months until disease progression was documented.
The safety profile of the combination of ixabepilone/paclitaxel and bevacizumab was assessed through summaries of AEs, serious AEs (SAEs), deaths, AEs leading to discontinuation, and laboratory abnormalities in hematology, liver function, and renal parameters for all treated patients.
Results
Patients
A total of 123 patients with metastatic or locally advanced breast cancer were randomized to receive either ixabepilone in Arm A (n = 46) or Arm B (n = 45), or paclitaxel in Arm C (n = 32), plus bevacizumab, at 23 sites across five countries over 12 months. A total of 122 patients were treated: one patient in Arm A was not treated (Fig. 1). The majority of demographic characteristics were balanced between arms (Table 1) except for sites of metastases; fewer patients had liver and/or lung metastasis in Arm C compared with patients in Arms A and B.

CONSORT diagram. Asterisks key reasons for discontinuation, AE adverse event, Q3W every 3 weeks, QW every week
Exposure
Patients received a median of 6.0 cycles (range 1–14) of chemotherapy in Arm A, 7.0 (range 1–28) in Arm B, and 6.5 (range 2–21) in Arm C. The median number of courses of bevacizumab received by each patient was 6.0 (range 1–24) in Arm A, 10.5 (range 1–37) in Arm B, and 10.5 (range 2–22) in Arm C. Of the 117 patients who had at least two cycles of ixabepilone or paclitaxel chemotherapy, 48 patients (Arm A: 43 %; Arm B: 40 %; Arm C: 41 %) had at least one dose reduction of chemotherapy; of these, peripheral neuropathy was the most common reason for the first dose reduction in all three arms (Arm A: n = 10, 24 %; Arm B: n = 12, 28 %; Arm C: n = 6, 19 %). In Arms A, B and C, 14, 15, and 17 % of patients experienced delay in receiving chemotherapy, and 14, 15, and 16 % of patients experienced delays in receiving bevacizumab, respectively. Dose intensity for ixabepilone was similar between the weekly and Q3W arms (11.5 and 11.2 mg/m2/week), but due to less delivered cycles the cumulative dose was lower on the weekly versus the Q3W arm (231 vs. 257 mg/m2). Ninety-five percent of the patients in the study discontinued treatment at the time of this analysis (Arm A: 98 %; Arm B: 93 %; Arm C 94 %); of these, 50 % discontinued due to disease progression (Arm A: 51 %; Arm B: 51 %; Arm C: 47 %). Eight patients (Arm A: n = 3, 7 %; Arm B: n = 2, 4 %; Arm C: n = 3, 9 %) discontinued bevacizumab and continued treatment on ixabepilone or paclitaxel alone.
Safety
Most patients had treatment-related AEs (Table 2). Treatment-related SAEs were reported in 16 % of patients each in Arms A and C, and 20 % of patients in Arm B. The most common drug-related SAEs, presented as the number of events over the total treated patients in each arm, were neutropenia (7 % of patients in Arm B), leukopenia and hypersensitivity (each, 4 % of patients in Arm B), pyrexia (2 % of patients each in Arms A and B, and 3 % of patients in Arm C), and hypertension (2 % of patients each in Arms A and B). Among the non-hematologic AEs, peripheral neuropathy was most common. Rates were similar between Arms A, B, and C (all grades: 76, 80, and 81 %; grade 3: 18, 24, and 25 %); no grade 4 events were reported. Grade 3 or 4 neutropenia was more common in Arm B (60 %) than in Arm A (16 %) or Arm C (22 %), and febrile neutropenia was only reported for one patient in Arm B. Pyrexia was mostly grade 1 and more common in Arm C (Arm A: 7 %; Arm B: 4 %; Arm C: 19 %); the rate of all-grade hypersensitivity was similar across the three treatment arms.
Fifty-five percent of patients (Arm A: 53 %; Arm B: 51 %; Arm C: 63 %) in the study discontinued treatment of either one or both study drug combinations due to treatment-related AEs. Grade 3 peripheral sensory neuropathy led to the discontinuation of treatment in 19 patients (Arm A: 13.3 %; Arm B: 15.6 %; Arm C: 18.8 %). A total of 32 (26 %) randomized patients (Arm A: 26 %; Arm B: 33 %; Arm C: 16 %) died due to their underlying disease. None died within 30 days of the last dosing date and there were no drug-related deaths.
Primary efficacy measure (ORR)
The ORR (assessed by investigators among all randomized patients) was 48 % (22/46; 95 % CI 32.9–63.1) for Arm A, 71 % (32/45; 95 % CI 55.7–83.6) for Arm B, and 63 % (20/32; 95 % CI 43.7–78.9) for Arm C (Table 3). Two patients (4 %) each in Arms A and B, and four patients (13 %) in Arm C, had a CR; 20 patients (43 %) in Arm A, 30 patients (67 %) in Arm B, and 16 patients (50 %) in Arm C had a PR; 18 patients (39 %) in Arm A, nine patients (20 %) in Arm B, and 11 patients (34 %) in Arm C had stable disease. Five patients (11 %) in Arm A, three patients (7 %) in Arm B, and no patients in Arm C, had progressive disease as the best response.
A sensitivity analysis (defined retrospectively) computing ORR by excluding those patients assumed as non-responders for this analysis, who received non-protocol therapy (any systemic therapy, surgery, or radiation) prior to achieving a CR or PR, resulted in an ORR of 46 % in Arm A, 69 % in Arm B, and 59 % in Arm C.
Other outcome measures
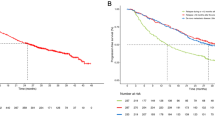
At the time of the final analysis, 102 randomized patients (83 %) had progressed or died, giving an estimated median PFS of 9.6 months (95 % CI 6.1–11.7) for patients in Arm A, 11.9 months (95 % CI 8.7–14.7) for patients in Arm B, and 13.5 months (95 % CI 10.0–18.2) for patients in Arm C (Table 4; Fig. 2). A sensitivity analysis of PFS censoring patients who received non-protocol therapy prior to disease progression or last tumor assessment (in case no progression was observed) at the earliest start date of this subsequent therapy, showed a median PFS of 9.7 months in Arm A, 13.8 months in Arm B, and 13.7 months in Arm C. At week 24, the estimated PFS rates were 75 % in Arm A, 86 % in Arm B, and 94 % in Arm C (Table 4). Similar results were also reported for the estimated week 24 PFS rates with censoring for non-protocol therapy administered prior to progressive disease: 82 % in Arm A, 88 % in Arm B, and 94 % in Arm C.

PFS. CI confidence interval, Bev bevacizumab, Ixa ixabepilone, Pac paclitaxel, PFS progression-free survival, QW every week, Q3W every 3 weeks
The 1-year OS rates were 91 % in Arm A, 89 % in Arm B, and 91 % in Arm C (Table 4); however, the median OS could not be determined based on the number of deaths at the time of the final analysis (12 in Arm A, 15 in Arm B, and five in Arm C). Median time to response (for randomized patients with a response of CR or PR) was similar among the three study arms (Table 4; Arm A: 8.2 weeks; Arm B: 8.3 weeks; Arm C: 8.1 weeks). Median duration of response for randomized patients with a response of CR or PR was 10.1 months in Arm A, 10.3 months in Arm B, and 13.1 months in Arm C (Table 4).
Discussion
Despite recent advances in treatment, MBC remains incurable with a median survival of just over 2 years [9]. For patients with newly diagnosed, hormone resistant HER2-normal metastatic disease, taxanes remain a current standard of care option. Several clinical trials have tried to identify the optimum dose and schedule for administration of paclitaxel and docetaxel. Preclinical and clinical evidence suggested that docetaxel was more effective than Q3W paclitaxel, and that weekly paclitaxel was more effective than dosing Q3W [19, 21]. In the large, randomized Cancer and Leukemia Group B (CALGB) 9840 trial, assessing more than 700 patients by preplanned analysis, weekly scheduling of paclitaxel improved RR (40 vs. 28 %; P = 0.0017) and median time to disease progression (9 vs. 5 months; P = 0.0008) compared with Q3W [19]. In the adjuvant setting, the Eastern Cooperative Oncology Group (ECOG) 1199 trial further supported this schedule of paclitaxel as efficacious and tolerable [20]. Exposure to lower, more frequent doses of paclitaxel may potentially exploit antiangiogenic effects; one rationale for combining weekly paclitaxel with bevacizumab in the metastatic setting (ECOG 2100) [9, 22]. Despite the recent withdrawal of accelerated approval of bevacizumab for the treatment of MBC, the combination with weekly paclitaxel remains the most impressive data for bevacizumab to date, with an almost doubling of PFS compared with paclitaxel alone.
In this open-label, randomized, phase II trial, the combination of ixabepilone and bevacizumab was found to be safe and active as first-line therapy in patients with HER2-normal MBC. Imbalances in baseline characteristics may have favored the paclitaxel plus bevacizumab arm (Arm C); Arm C had fewer patients with liver metastasis, and patients had a longer time from initial diagnosis to randomization. An ORR of 71 % in the Q3W ixabepilone arm suggests similar clinical activity for this combination relative to paclitaxel and bevacizumab (63 %), and median PFS was similar at 13.8 and 13.7 months for Arms B and C when adjusted for non-protocol treatment. Interestingly, a similar ORR of 49.2 % and a median PFS of 11.8 months were reported in ECOG 2100 [9]. The CIRG/TORI 010 randomized phase II study of first-line chemotherapy for MBC (weekly paclitaxel vs. weekly paclitaxel plus bevacizumab vs. weekly paclitaxel plus motesanib) also reported a median PFS of 11.5 months and an ORR rate of 52 % for the paclitaxel/bevacizumab arm (n = 97) [23]. Both the ORR (48 %) and PFS (9.6 months) for the weekly ixabepilone arm were inferior to both Q3W ixabepilone and weekly paclitaxel, although median dose intensity was similar, suggesting that the improved efficacy of the Q3W schedule is due to a higher delivered dose of ixabepilone at each infusion. Estimation of median OS was not possible in the current trial, but 1-year OS rates were 91, 89, and 91 % for Arms A, B, and C, respectively.
The safety data from this study demonstrated that ixabepilone plus bevacizumab (Arms A and B) was reasonably well tolerated, with comparable discontinuation rates for toxicity to paclitaxel plus bevacizumab (Arm C). In particular, the incidence of grade 3 peripheral sensory neuropathy (no grade 4 reported) was similar among the three study arms. However, grade 3 or 4 neutropenia was more common in Arm B (60 %) than Arm A (16 %) or Arm C (22 %), but was not associated with an increase in rates of febrile neutropenia (reported in one patient in Arm B), consistent with that observed in monotherapy studies with this dose and schedule of ixabepilone. Bevacizumab-associated toxicities (grade 3 or 4 hypertension and proteinuria) were within the expected range in all three arms relative to phase III studies of chemotherapy plus bevacizumab in first-line MBC [8–10]. The addition of bevacizumab had no meaningful impact on the frequency or severity of ixabepilone or paclitaxel-related toxicities.
These data demonstrated acceptable efficacy and safety with weekly ixabepilone compared with Q3W dosing, and supported the design and dosing of the phase III cooperative group trial CALGB 40502, comparing weekly ixabepilone or weekly nab-paclitaxel with weekly paclitaxel, given in combination with bevacizumab (NCT00785291) [24]. While no definitive conclusions can be made from this trial about the relative differences in efficacy between the two ixabepilone arms, these data suggest that the Q3W schedule of ixabepilone plus bevacizumab (Arm B) might be more active than the weekly schedule (Arm A). Indeed, the results of CALGB 40502 confirmed inferior efficacy with weekly ixabepilone compared with weekly paclitaxel [24]. Q3W ixabepilone was associated with a greater rate of neutropenia but not more febrile neutropenia; otherwise toxicity was manageable and similar between the three arms. Notably, the toxicity associated with Q3W ixabepilone was more favorable than that reported in heavily pre-treated patients, with rates of grade 3 peripheral neuropathy similar to weekly paclitaxel. These data, taken in combination with the recently reported data from CALGB 40502, suggest that ixabepilone should be administered in the Q3W schedule. Weekly paclitaxel may be as efficacious in the first-line setting, with less neutropenia.
References
Marty M, Pivot X (2008) The potential of anti-vascular endothelial growth factor therapy in metastatic breast cancer: clinical experience with anti-angiogenic agents, focusing on bevacizumab. Eur J Cancer 44:912–920
Brown LF, Berse B, Jackman RW, Tognazzi K, Guidi AJ, Dvorak HF, Senger DR, Connolly JL, Schnitt SJ (1995) Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in breast cancer. Hum Pathol 26:86–91
Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, Moghaddam A, Whitehouse R, Bicknell R, Harris AL (1997) Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res 57:963–969
Gasparini G, Toi M, Gion M, Verderio P, Dittadi R, Hanatani M, Matsubara I, Vinante O, Bonoldi E, Boracchi P et al (1997) Prognostic significance of vascular endothelial growth factor protein in node-negative breast carcinoma. J Natl Cancer Inst 89:139–147
Linderholm BK, Lindahl T, Holmberg L, Klaar S, Lennerstrand J, Henriksson R, Bergh J (2001) The expression of vascular endothelial growth factor correlates with mutant p53 and poor prognosis in human breast cancer. Cancer Res 61:2256–2260
Toi M, Inada K, Suzuki H, Tominaga T (1995) Tumor angiogenesis in breast cancer: its importance as a prognostic indicator and the association with vascular endothelial growth factor expression. Breast Cancer Res Treat 36:193–204
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8:579–591
Miles DW, Chan A, Dirix LY, Cortés J, Pivot X, Tomczak P, Delozier T, Sohn JH, Provencher L, Puglisi F et al (2010) Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol 28:3239–3247. doi:10.1200/JCO.2008.21.6457
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE (2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357:2666–2676
Robert NJ, Diéras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON, Perez EA, Yardley DA, Chan SY, Zhou X et al (2011) RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol 29:1252–1260. doi:10.1200/JCO.2010.28.0982
Lee FYF, Wen ML, Shen H, She Y, Lenhart R, Covello K, McGlinchey K, Menard K, Wiebeiek A, Raventos-Suarez C et al (2008) Ixabepilone overcomes multiple mechanisms of drug resistance including overexpression of class III ß tubulin and breast cancer resistance protein. Eur J Cancer 6: abstract 219
Lee F, Lewin A, Wen M, Ryseck R, Fargnoli J, Poruchynsky M, Fojo T, Mudenda B, Rugo H (2009) Antiangiogenic (AG) synergy with ixabepilone (ixa): translation of preclinical studies to the clinical setting. Cancer Res 69:206
Lee FY, Covello KL, Castaneda S, Hawken DR, Kan D, Lewin A, Wen ML, Ryseck RP, Fairchild CR, Fargnoli J et al (2008) Synergistic antitumor efficacy of ixabepilone (BMS-247550) plus bevacizumab in multiple in vivo tumor models. Clin Cancer Res 14:8123–8131
Perez EA, Lerzo G, Pivot X, Thomas E, Vahdat L, Bosserman L, Viens P, Cai C, Mullaney B, Peck R et al (2007) Efficacy and safety of ixabepilone (BMS-247550) in a phase II study of patients with advanced breast cancer resistant to an anthracycline, a taxane, and capecitabine. J Clin Oncol 25:3407–3414
Roche H, Yelle L, Cognetti F, Mauriac L, Bunnell C, Sparano J, Kerbrat P, Delord JP, Vahdat L, Peck R et al (2007) Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, as first-line therapy in patients with metastatic breast cancer previously treated with anthracycline chemotherapy. J Clin Oncol 25:3415–3420
Thomas E, Tabernero J, Fornier M, Conté P, Fumoleau P, Lluch A, Vahdat LT, Bunnell CA, Burris HA, Viens P et al (2007) Phase II clinical trial of ixabepilone (BMS-247550), an epothilone B analog, in patients with taxane-resistant metastatic breast cancer. J Clin Oncol 25:3399–3406
Sparano JA, Vrdoljak E, Rixe O, Xu B, Manikhas A, Medina C, Da Costa SC, Ro J, Rubio G, Rondinon M et al (2010) Randomized phase III trial of ixabepilone plus capecitabine versus capecitabine in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol 28:3256–3263
Thomas ES, Gomez HL, Li RK, Chung HC, Fein LE, Chan VF, Jassem J, Pivot XB, Klimovsky JV, de Mendoza FH et al (2007) Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J Clin Oncol 25:5210–5217
Seidman AD, Berry D, Cirrincione C, Harris L, Muss H, Marcom PK, Gipson G, Burstein H, Lake D, Shapiro CL et al (2009) Randomized phase III trial of weekly compared with every-3-weeks paclitaxel for metastatic breast cancer, with trastuzumab for all HER-2 overexpressors and random assignment to trastuzumab or not in HER-2 nonoverexpressors: final results of Cancer and Leukemia Group B protocol 9840. J Clin Oncol 26:1642–1649
Sparano JA, Wang M, Martino S, Jones V, Perez EA, Saphner T, Wolff AC, Sledge GW Jr, Wood WC, Davidson NE (2008) Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med 358:1663–1671
Sparano JA (2000) Taxanes for breast cancer: an evidence-based review of randomized phase II and phase III trials. Clin Breast Cancer 1:32–40
Gray R, Bhattacharya S, Bowden C, Miller K, Comis RL (2009) Independent review of E2100: a phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol 27:4966–4972. doi:10.1200/JCO.2008.21.6630
Martin M, Roche H, Pinter T, Crown J, Kennedy MJ, Provencher L, Priou F, Eiermann W, Adrover E, Lang I et al (2011) Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Oncol 12:369–376. doi:10.1016/S1470-2045(11)70037-7
Rugo HS, Barry WT, Moreno-Aspitia A, Lyss AP, Cirrincione C, Mayer EL, Naughton M, Layman RM, Carey LA, Somer RA et al (2012) CALGB 40502/NCCTG N063H: Randomized phase III trial of weekly paclitaxel (P) compared to weekly nanoparticle albumin bound nab-paclitaxel (NP) or ixabepilone (Ix) with or without bevacizumab (B) as first-line therapy for locally recurrent or metastatic breast cancer (MBC). J Clin Oncol 30: abstract CRA1002
Acknowledgments
The authors take full responsibility for the content of this publication and confirm that it reflects their viewpoint and scientific expertise. They wish to acknowledge StemScientific, funded by Bristol-Myers Squibb, for providing writing and editorial support. The authors would also like to acknowledge the contribution of Boyd Mudenda to this study during his time at Bristol-Myers Squibb.
Conflict of interest
Dino Amadori, Daniela Aldrighetti, PierFranco Conte, Cristian Villanueva, Michelle Melisko and Xavier Pivot declare that they have no conflicts of interest. Hope S. Rugo has received research funding from Bristol-Myers Squibb, Genentech and Eisai. Mario Campone has received remuneration from Novartis, and has performed a consultant/advisory role for Novartis and Servier. Andrew Wardley has performed a consultant/advisory role for Roche. M. Brent McHenry, David Liu and Francis Lee are employees and stock owners of Bristol-Myers Squibb.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper was earlier presented at the annual meeting of the American Society of Clinical Oncology: Rugo HS, Campone M, Amadori D, Wardley AM, Aldrighetti D, Conte PF, Liu D, Mudenda B, McHenry MB, Pivot XB (2009) Randomized phase II study of weekly versus every 3-week ixabepilone plus bevacizumab (ixa/bev) versus paclitaxel plus bev (pac/bev) as first-line therapy for metastatic breast cancer (MBC) (suppl; abstr 1029).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Rugo, H.S., Campone, M., Amadori, D. et al. A randomized, phase II, three-arm study of two schedules of ixabepilone or paclitaxel plus bevacizumab as first-line therapy for metastatic breast cancer. Breast Cancer Res Treat 139, 411–419 (2013). https://doi.org/10.1007/s10549-013-2552-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2552-8