
Abstract
Pockmarks are formed as a result of gas (methane) or/and groundwater outflow from the sea bottom. Methane, the second most important (after CO2) greenhouse gas, has a significant impact on biogeochemical processes in the bottom sediments by affecting the cycling of some elements, e.g. C, Fe, and S. Active pockmarks may also lead to changes in water column conditions by causing nutrients release from sediments. In the present study, we have focused on the impact of biogeochemical processes in pockmarks (methanogenesis, anaerobic methane oxidation, and groundwater seepage) on the transformation of iron (Fe) and the mineral composition of the sediment. In pore water, concentrations of hydrogen sulfide, phosphate, ammonia, sulfate, chloride, dissolved inorganic carbon, iron, and methane were analyzed. In the sediment, Fe speciation was performed using sequential extraction. The mineral composition was determined using powder X-Ray diffraction and scanning electron microscopy. The results from two pockmarks (with active gas seepage and groundwater infiltration) and two reference stations in the southern Baltic Sea show that geochemical conditions in pockmark sediments are significantly different from those in the typical muddy sea bottom. Pore water in pockmarks is characterized by lower sulfate and higher dissolved carbon concentrations as compared to areas of the seafloor where such structures are absent. This is due to the outflow of groundwater, which was confirmed by lower chloride concentration. In addition, sulfate is used to oxidize methane diffusing from deeper layers. Sediments in pockmarks are enriched in Fe(II) carbonates and depleted in Fe(III) (oxy)hydroxides, resulting from the anaerobic oxidation of methane with Fe(III) (Fe-AOM). Ferrous iron produced in large quantities during Fe-AOM is precipitated with carbonates.
Similar content being viewed by others


Introduction
Environmental conditions in shelf seas are strongly influenced by human activities, e.g. an increased supply of nutrients. The consequence is eutrophication and increased demand for O2 to decompose large amounts of organic matter (Díaz and Rosenberg 2008; Rabalais et al. 2014). It can be expected that the extent of O2-deficient seabed zones in coastal seas will increase in the coming years due to progressive eutrophication. The process will intensify under the influence of climate change, e.g. for the Baltic Sea, we can expect lower oxygen solubility at higher temperatures and an increased inflow of nutrients from land due to higher precipitation (Kabel et al. 2012; Carstensen et al. 2014). In addition, the increased influx of river waters in the northeastern part of the Baltic Sea may cause the freshening of seawater (Graham 2004; Meier et al. 2022) and decrease the concentration of sulfate, the most crucial electron acceptor in the anaerobic decomposition of organic matter in marine sediments. Due to high demand for oxidants for the decomposition of organic matter and the limited availability of sulfates, an increase in the intensity of methanogenesis can be expected.
Accumulation of methane produced by the microbial communities of archaea in anoxic sediments where a large amount of organic matter is decomposed is one of the reasons for the formation of pockmarks on the seabed (Hovland and Judd 1988). When the sediment pore water becomes supersaturated with methane, gas bubbles are formed, which may be released into the water (Idczak et al. 2020). Methane often escapes from the sediment along with groundwater, which seeps toward the surface and is enriched with this gas (Bussmann and Suess 1998). Groundwater seepage dilutes the pore water and reduces the availability of sulfates to mineralize organic matter and to oxidize methane. This, in turn, modifies the microbial composition of surface sediments (Haverkamp et al. 2014; Giovanelli et al. 2016) and the course of early diagenesis, e.g., higher rates of methanogenesis and anaerobic methane oxidation (AOM) as well as dissolved carbon production (Hung et al. 2016). The presence of groundwater outflow (Whiticar 1981; Judd and Hovland 2009), oxidation of methane (Werner 1981; Söderberg et al. 1992) and higher concentrations of organic matter with its anaerobic decomposition (Kelley et al. 1994; Kruglyakova et al. 2002) make the chemical composition of pore waters in pockmarks different from typical marine sediments. This, in turn, may be reflected in the mineral composition of the sediment, as the degree of saturation with specific mineral phases affects the possibility of precipitation (Peckmann et al. 2001; Judd and Hovland 2009). In addition, gas and groundwater outflow may increase nutrient concentrations in the water column above pockmarks, further supporting biological productivity (Hovland et al. 1988; Zeppilli et al. 2012; O’Reilly et al. 2021).
The processes associated with the occurrence of pockmarks on the seabed have wide-ranging consequences for sulfur and iron cycling in the marine environment. Anaerobic methane oxidation coupled with sulfate reduction (SO42−-AOM), mediated by a consortium of anaerobic methanotrophic archaea and sulfate reducing bacteria, is the main mechanism preventing the emission of this gas into the water column (Hinrichs et al. 1999; Knittel and Boetius 2009). The by-product of SO42−AOM is H2S, which reductively dissolves Fe (oxy)hydroxides (Raiswell and Canfield 2012). When large amounts of hydrogen sulfide are produced, high concentrations of this compound provide conditions suitable for abundant production by chemoautotrophic bacteria that support the growth of benthic communities (Levin 2005).
In addition to sulfate, ferric iron, mainly in the form of Fe(III) oxyhydroxides, can act as an oxidizing agent during AOM (Hinrichs et al. 1999; Beal et al. 2009; Knittel and Boetius 2009; Egger et al. 2018). Fe(III) can also be an electron acceptor in the process of mineralization of organic matter by dissimilatory reduction of Fe (Froelich et al. 1979). These processes lead to a reduction in the content of Fe(III) oxyhydroxides in the sediments and an increase in ferrous Fe concentration in pore water (Sørensen 1982; Brocławik et al. 2020). Fe(II) can be readily released into the water column (Hyacinthe and Van Capellen 2004) and used by phytoplankton (Rooze 2016), or it can be immobilized in the sediment for an extended period by the formation of authigenic minerals (Raiswell and Canfield 2012). The increasing concentration of Fe(II) in pore water, in combination with the high concentration of dissolved inorganic carbon associated with intensive mineralization of organic matter and AOM, can lead to supersaturation of pore waters with Fe carbonate and precipitation of authigenic carbonate minerals. The most actively precipitated minerals in methane-bearing sediments are Mg-calcite and aragonite (CaCO3), dolomite (CaMg(CO3)2), magnesite (MgCO3), as well as Fe- and Mn-carbonates (Peckmann et al. 2001; Greinert et al. 2001; Teichert et al. 2005; Mavromatis et al. 2012; Brocławik et al. 2020; Łukawska-Matuszewska et al. 2022). Ferrous Fe can also be removed from pore water during reaction with hydrogen sulfide present in anaerobic sediment and form Fe monosulfides, which can be converted into pyrite and permanently buried in sediments (Raiswell and Canfield 2012). Therefore, biogeochemical processes in sediments determine the form of occurrence and, consequently, the bioavailability of Fe in pockmark areas. Since Fe is a micro-nutrient necessary for the phytoplankton growth (Martin 1990; Archer and Johnson 2000), its excessive accumulation in shelf sediments may limit the supply of this element to the open ocean and, also, limit the primary production. On the other hand, pockmarks are suggested to provide significant amounts of dissolved iron to the water due to the reduction and dissolution of solid Fe (III) oxides and hydroxides (Lamaitre et al. 2014). It is suggested that pockmarks and this type of cold, seabed structures mobilize approx. 0.47 Mt of iron every year (Boetius and Wenzhöfer 2013; Lamaitre et al. 2014).
The main objective of this study was to investigate how the biogeochemical processes in pockmarks, i.e. methanogenesis, anaerobic methane oxidation, and groundwater seepage, affect the transformation of iron and the mineral composition of the sediment. For this purpose, we investigated Fe speciation and mineral phases of sediments and pore water composition in two pockmarks and two reference stations in the Baltic Sea. Similar structures were found in multiple estuaries globally, e.g. in NW Spain (García-Gil et al. 2002), NE USA (Kelley et al. 1994), Kongo (Gay et al. 2003), Taiwan (Chuang et al. 2010) and Black Sea (Knab et al. 2009). These coastal methane reservoirs may be formed e.g. due to organic matter accumulation caused by strong riverine input, so we hypothesize that the results presented in this paper could be applied to such regions. We focused on two pockmarks with different morphology and type of activity. One of the investigated pockmarks is small but has very high activity manifested by intensive gas and groundwater seepage. The second of the studied pockmarks is more extensive, but at the same time, it is characterized by lower activity, mainly manifested by gas outflow.
Materials and methods
Study area
The study area is the Gulf of Gdansk—a shallow (average depth—59 m) coastal body of water in the southern part of the semi-closed Baltic Sea (Majewski 1990). The area is characterized by low surface water salinity (7–8 PSU), halocline at the depth of 60–80 m and high concentration of dissolved and particulate organic matter (OM) due to freshwater inflow from the land and eutrophication (Majewski and Klusek 2014; Chubarenko et al. 2017). High productivity (the primary production in the gulf is estimated to be ca. 156 g cm−2 y−1; Kruk-Dowgiałło and Szaniawska 2008) leads to hypoxia or anoxia, especially in deep parts of the Gulf of Gdansk (Łukawska-Matuszewska 2016). The anoxic conditions can be improved during inflows of more saline and oxygenated waters from the North Sea through the Danish Straits. The Gulf of Gdansk is under strong anthropogenic pressure due to intensive agriculture in the drainage basin and strongly urbanized coastline.
The present study was conducted in the deep (79–95 m) central part of the Gulf of Gdańsk (southern Baltic Sea). Previous hydroacoustic surveys revealed several pockmarks present on the seabed of this area (Fig. 1)—of which MET1-BH and MET1-MP are the most representative and significant. The MET1-BH is a relatively small circular depression with a diameter of ca. 50 m and a relative depth of 9 m below the sea bottom. The MET1-MP is a much bigger depression of irregular shape with ca. 1500 m of length, ca. 500 m of width, and only 2 m of relative depth. Gas seeps (flares) have been observed above these pockmarks (Majewski and Klusek 2014; Idczak et al. 2020). They are considerably more intensive in the case of MET1-BH (Fig. 1S in Supplementary material). In addition, groundwater infiltrates surface sediments of both pockmarks: MET1-BH and MET1-MP. However, previous observations and research revealed that groundwater outflow to the near-bottom water occurs occasionally only at MET1-BH. In this study, for comparative purposes, two reference stations (MET1-BH-REF and MET1-MP-REF) were designated based on previous detailed geophysical and ROV studies (e.g. Idczak et al. 2020; Brodecka-Goluch et al. 2022), close (ca. 100 m) to the pockmarks, at similar depths, but in the areas without gas seepage from the bottom (confirmed by hydroacoustic water column studies). Reference stations were located clearly outside the pockmarks, at 9 m (MET1-BH-REF) and 2 m (MET1-MP-REF) shallower depths as compared to pockmark stations MET1-BH and MET1-MP, respectively. The precision of ship positioning and, consequently, sediment sampling was possible thanks to the DP1 system available on RV Oceanograf used in the present study.

Map of the study area with location of the sampling stations—pockmarks MET1-BH and MET1-MP and corresponding reference stations MET1-BH-REF and MET1-MP-REF
Sampling
During two cruises on RV Oceanograf (vessel of the University of Gdańsk) in November 2020 and February 2021, samples of near-bottom water, sediment, and pore water were collected from two pockmarks (MET1-MP and MET1-BH) and two reference stations (MET1-MP-REF and MET1-BH-REF). Sediment cores were collected using a Rumohr Lot core probe with plexiglass tubes (length 150 cm, diameter 7.5 cm). Four sediment cores were taken from each station. Pore and near-bottom waters were collected from intact and sealed sediment cores (without any contact with the air) using Rhizon® samplers and syringes. The samplers were inserted through holes drilled in the walls of the liners at 5 or 10-cm intervals. The first one was placed a few centimeters above the sediment–water interface to collect sample of near-bottom water. Samples for methane analyses were taken from a separate sediment core, within 0.5 h after core retrieval, through the holes previously made on the sides of core liners, using 3 ml syringes with the luer-tip removed and transferred into 20 ml vials, according to the standard procedure described by Jørgensen et al. (2001). The core for solid phase analyses was cut into 5 cm-long segments, of which every second was taken for the analyses and placed in PE zip-locked bags. Sediment samples were kept in a freezer (-21 °C) until the analyses.
Near-bottom and pore-water parameters
Oxygen concentration (dO2), temperature (T), pH, redox potential (Eh), and salinity (S) in near-bottom water were measured immediately after taking the cores. Measurements were made using a multimeter with an optical oxygen probe and pH and Eh glass electrodes. Salinity was measured with a conductivity meter. Analysis of hydrogen sulfide (ΣH2S), phosphates (ΣPO43−) and ammonia (ΣNH4+) was performed immediately after sampling using the standard spectrophotometric methods (Grasshof et al. 1999). The detection limit was 0.1 µmol/l for ΣH2S, 0.02 µmol/l for ΣPO43− and 0.1 µmol/l for ΣNH4+. The precision (RSD) of ΣH2S, ΣPO43− and ΣNH4+ analyses was 2, 2, and 3%, respectively. Samples for the analysis of chlorides (Cl−), sulfates (SO42−), dissolved iron (dFe) and dissolved inorganic carbon (DIC) were stored in sealed syringes in a refrigerator until analysis. Samples for dFe analysis were acidified with HNO3 to pH 2. Cl−, SO42−, and dFe were measured in a commercial laboratory according to PN-EN ISO 10304-1:200 and PN-EN ISO 17294-2:2016-1, on an ion chromatograph (DIONEX ICS 1100/RFIC1600/20) in the case of chlorides and sulfates, and on an inductively excited plasma optical emission (ICP-OES) spectrometer (Perkin-Elmer—OPTIMA 8300) in case of dFe. Concentrations of DIC were measured on the TOC analyzer VarioTOC Cube (Elementar GmbH) equipped with a non-dispersive infrared (NDIR) detector. Samples were acidified (1% H3PO4) inside the apparatus, and DIC was purged in the sparger and measured on the detector. The calibration of the equipment was performed using a certified reference solution of TIC (ICC-033) from Ultra Scientific, Inc. The accuracy levels for DIC (percentage of recovery) was 98%. The precision of DIC assay in near-bottom and pore water samples was ≤ 1%.
Sediment parameters
In the sediment, the following parameters were measured: concentration of methane, loss on ignition (LOI), total iron (FeTOT), iron speciation and mineral composition.
Methane concentration analysis was performed on sediment samples (collected during the cruise into 20 ml vials pre-filled with 6 ml of 2.5% NaOH, closed with butyl rubber stoppers and aluminum seals), using standard headspace techniques on a gas chromatograph (Perkin Elmer) equipped with a flame ionization detector (FID) and an HP-INNOWAX column (30 m, 0.32 mm, 0.5 µm). High-purity helium at a flow rate of 2 ml min−1 was used as a carrier gas. For the calibration curve, six standard solutions with CH4 in a headspace were prepared in sealed 20 ml vials with 6 ml of 2.5% NaOH as serum. The detection limit was 0.25 μM. Methane results were corrected for sediment porosity. The porosity of sediments at each station was calculated based on organic matter content (LOI) and water content (a detailed description can be found in Brodecka et al. 2013).
LOI was obtained by igniting sediment samples at 550 °C to the constant mass. Freeze-dried, homogenized samples (250–300 mg) for FeTOT analysis were put into mineralization bombs and acidified with conc. HNO3, conc. HF and conc. HCl (v:v:v ratio = 1:2:1 ml). The vessels were placed in a microwave mineralizer (ETHOS One, Milestone Srl) for 1.5 h at 180 °C. Afterward, samples were placed into HDPE tubes and diluted 1:100 with 0.1 M HNO3. Measurements were performed on an atomic absorption spectrometer (ASA) with flame atomization (Perkin Elmer). The analysis was carried out in triplicate. The precision of determination was ≤ 5%. The analysis of total Fe content in certified reference material (BCR-320R) from the Institute for Reference Materials and Measurements, according to the same procedure, gave recovery of 98%.
For Fe speciation, the sequential extraction scheme by Poulton et al. (2005) was applied. The sediment samples used for Fe speciation analysis were lyophilized and homogenised in a ball mill. Approximately 100 mg of sediment was weighed out and placed into plastic tubes and a four stage sequential extraction was performed: 1 M Na-acetate (pH 4.5) for Fe carbonates (FeCARB), 1 M hydroxylamine-HCl (pH < 2) for labile Fe oxyhydroxides i.e. ferrihydrite and lepidocrocite (FeOX1), 0.3 M Na-dithionite (pH 4.8) for crystalline Fe hydroxides like goethite, hematite and akagenéite (FeOX2) and 0.2 M ammonium oxalate (pH 3.2) for magnetite (FeMAG). Extracts were diluted (1:10) with 0.1 M HNO3 and analysed on an ASA with flame atomization. The difference between total iron (FeTOT) and four forms of Fe extracted is the sum of iron bound to poorly reactive silicate sheets and iron in form of sulfides (FeS+PRS).
Several sediment sample mineral compositions were determined using a powder X-ray diffractometer (PXRD) and scanning electron microscopy equipped with energy dispersive X-ray spectroscopy (SEM/EDS). Powder X-ray diffraction (PXRD) measurements were performed using a 3rd generation Malvern Panalytical Empyrean diffractometer. The X-ray source is a tube with the Cu anode, and the generator was set on the 45 kV and 40 mA as the detector is used PIXcel3D Detector with Medipix3. Diffraction patterns of soil samples were collected using a gonio scan with a step size of 0.013° and scanning angle (o2Theta) from 2 to 70. For data treatment, the compiled pattern was corrected for systematic errors (external Si standard). The qualitative interpretation of the XRD pattern was compared to standard patterns contained in the database PDF2 (ICDD, PDF2 Released 2020), using HighScore Plus. SEM/EDS analysis was conducted under a low vacuum using the Jeol JEM-7610F Plus microscope equipped with the Oxford Ultim Max 65 SDD X-ray analyzer for the EDS analysis. Acceleration voltage and pressure were up to 1 kV and 4.4 × 10–4 Pa, respectively, for collecting secondary electron images (SEI), and voltage was in the range from 15 up to 20 kV for elemental analysis.
Quantitative analysis of sediments was calculated by using the Rietveld method. To apply the Rietveld method of quantitative calculation of individual phases, CIF files from the Inorganic Crystal Structure Database (ICSD) were used for the following phases Chamosite, Illite, Kaolinite, Muscovite 2M1, Feldspar, Albite, Quartz, Tridymite, Ankerite, Siderite, Pyrite, Lepidocrocite, Goethite, Hematite, Gwihabaite and for Riebeckite CIF file was used from Crystallography Open Database (COD). The Rietveld refinement was carried out by using the HighScore Plus program. The value of the parameter weighted R profile (Rwp) after the refinement of the multiphase system was in the range from 5.1 to 9.3, while the goodness of fit (GOF) was in the range from 1.03 up to 1.48 and an example of the fitting is shown in Fig. 2S in Supplementary information.

Concentration of methane (CH4), chloride (Cl−), sulfate (SO42−) and hydrogen sulfide (ΣH2S) in pore water of MET1-BH and MET1-MP pockmarks and corresponding reference stations MET1-BH-REF and MET1-MP-REF (values at y = 0 are near-bottom water concentrations)
Results
Parameters in near-bottom and pore water
Table 1 presents data on the parameters in near-bottom water, and Figs. 1 and 2 show the concentrations of the analyzed parameters in pore water. While the salinity, temperature, sulfate, and chloride in near-bottom water of pockmarks and their surroundings were similar, the concentrations of the remaining parameters in pockmarks differed from the values recorded at the reference stations (Table 1). The near-bottom water nutrients, i.e. ammonia, phosphate and iron, displayed higher values in pockmarks than in their surroundings. A similar situation was observed for hydrogen sulfide in the case of MET1-BH and MET1-BH-REF stations (near-bottom water at MET1-MP and MET1-MP-REF was free of hydrogen sulfide). On the other hand, the concentration of dO2 and DIC in pockmarks was lower than at the reference stations. Notably, the highest concentration of nutrients was found in the near-bottom water of MET1-BH, which is a small but very active pockmark.
Methane concentrations in sediments of the investigated stations ranged from < LOD to 9.323 mmol/l, and they increased with sediment depth (Fig. 2). Generally, the highest values were recorded in both pockmark sediments (mean values for ca. 80 cm long sediment profiles were 4.567 and 5.651 mmol/l, respectively for MET1-MP and MET1-BH). However, the highest peak value of 9.323 mmol/l occurred at 69 cm bsf of MET1-BH-REF. The pore water concentration of DIC was 2.44–30.15 mmol/l. Pore water chloride (Cl−) concentration was in the range of 85.97–201.83 mmol/l. In general, concentration of Cl− in pore water in pockmarks was lower than at the corresponding reference stations (Fig. 2). Sulfate (SO42−) concentrations in pore water of all the stations varied from 0.03 to 10.46 mmol/l. At all stations an exponential, very similar to each other decrease of SO42−concentration with depth was observed (Fig. 2). The concentration of H2S in pore water varied from 0.0 to 1849.5 µmol/l. At all four stations, there was an increase of hydrogen sulfide in a shallow sediment layer, about 5–25 cm bsf (Fig. 2). Moreover, concentrations at the reference stations were significantly higher than at the pockmark stations. The H2S was present at all core depths only in the case of MET1-MP-REF.
Porewater ammonia (ΣNH4+) concentration at all stations varied in the range of 130 to 10 377 µmol/l, with the highest values observed at MET1-BH (Fig. 3). The concentrations increased with depth at all the stations, especially in the layer 0–20 bsf (below sediment surface), but at different rates. Concentration in pockmarks were several times higher than in reference stations. The phosphate concentration (ΣPO43−) in pore water ranged from 18.1 to 803.0 µmol/l, with values several times higher in pockmarks than in their surroundings (Fig. 3). At all stations, the concentration increased with sediment depth, more rapidly in pockmarks than in reference stations. The maximum values in pockmarks were reached at a depth ca. 20 cm below the surface.

Concentration of ammonia (ΣNH4+), phosphate (ΣPO43−), total dissolved iron (dFe) and inorganic dissolved carbon (DIC) in pore water of MET1-BH and MET1-MP pockmarks and corresponding MET1-BH-REF and MET1-MP-REF reference stations (values at y = 0 are near-bottom water concentrations)
The dissolved iron (dFe) concentration in pore water varied in a wide range—from 0.02 to 47.72 µmol/l. At each station, a different pattern of changes with depth was observed (Fig. 3). At MET1-BH, two maxima were reached, at 30 and 60 cm bsf, and the concentration was much higher than at the corresponding reference site (Fig. 3). The concentration range in pore water of MET1-MP and MET1-MP-REF was similar. However, the maximum values were found at different depths in sediment—20 cm bsf in the pockmark and 5 cm bsf in its surrounding (Fig. 3). DIC concentrations in pockmarks increased exponentially with depth and generally were higher than at reference stations (Fig. 3). The highest values were recorded in pore water of MET1-BH, where the concentration reached ca. 30 mmol/l in the top 10 cm of sediment.
Mineral composition of sediment
The average LOI was higher at MET1-BH and MET1-MP (19.1 and 14.3%, respectively) than at their reference stations (12.3 and 11.8%, respectively). The changes in LOI were similar to each other at all the stations.
The mineral composition of the samples from MET1-MP-REF is shown in Fig. 4, and from pockmarks, MET-MP and MET1-BH, are demonstrated in Fig. 5 and as 2D patterns in Fig. 3S (Supplementary Information). It is evident from PXRD patterns that in all cases, at all depths, the presence of phyllosilicates Muscovite, Chlorite (Chamosite), Illite, and Kaolinite was observed. The iron present in the examined samples appeared in three structurally arranged crystal forms as Pyrite, Ferrian (Muscovite with the Fe into the structure of Muscovite) and Riebeeckite (Sodium Iron Silicate Hydroxide, which belongs to the Amphibole family). Other minerals found in the samples were quartz, halite, dolomite, and Gwihabaite (potassium ammonium nitrate).

Diffraction patterns of the sediment samples: a MET1-MP-REF reference station, 30–35 cm layer, and b all samples from MET1-MP-REF reference station, all sediment layers

Diffraction patterns of the samples from a MET1-MP and b MET1-BH pockmarks
The results of the quantitative analysis of minerals present in sediments of pockmarks and reference stations obtained by using the Rietveld method were summarized in Fig. 6 as well as in Table 1S in Supplementary material. At Fig. 6a, c, e and g, the results of quantitative analysis related to the proportion of the following phases: Chamosite, Illite, Kaolinite, Muscovite 2M1, Feldspar, Albite, Riebeckite, Quartz, and Tridymite, which represent clay and silicate minerals, are shown. The total mass fraction of those, mentioned above, structurally ordered phases in the examined samples represents an amount in the range from 92.1 up to 98.2 wt%. Other structurally ordered phases, which are shown in Fig. 6b, d, f and h are related to the content of the following minerals: Ankerit (Ca(Fe, Mg,Mn)(CO3)2), Siderite (Fe(CO)3), Pyrite (FeS2), Lepidocrocite (γ-FeO(OH)), Goethite (α-FeO(OH)), Hematite (α-Fe2O3), Gwihabaite ((NH4,K)NO3), Calcite (CaCO3) and Halite (NaCl). The total share of these phases ranged from 1.8 up to 8.0 wt%. In this group of compounds, the following minerals: Ankerite (isostructural with Dolomite) and Pyrite, present in the ranges of 0.3–3.8 wt% and 0.5 to 2.9 wt%, respectively, accounted for the largest share. Other compounds were present in a tiny proportion ranging from 0.1 to a maximum of 0.5 wt%.

Mineral composition of samples from different sediment depths of a, b MET1-BH and c, d MET1-MP pockmarks as well as e, f MET1-BH-REF and g, h MET1-MP-REF reference stations
Sediments of MET1-BH pockmark, from the depths down to 85 cm bsf contain mostly diatoms and clay minerals (Fig. 7a–f). Iron was present as pyrite (FeS2), in a spheroidal form of the framboids (Fig. 7a, b).

SEM images of MET1-BH sediments (0–55 cm) a, b—framboids, c, d, e, f—most abundant soil diatom species)
The iron sulfide occurred as framboids with diameters from 9.5 up to 15 μm, composed of smaller microcrystals with dimensions from 0.86 up to 1 μm. In the framboids, even though they are mainly composed of Fe and S, the presence of Si, Al, K, Ti, Ca, Mg, Na, Cl, and Zn was determined by EDS mapping of the characteristic area. At Fig. 4S, in the EDS spectra of S and Fe elements, it was clearly visible that pyrite (not recognized in the SEM images) was present as microcrystals in the area of highest intensity of the Fe and S spots. In the same way, the presence of dolomite was confirmed where the concentration of Ca and Mg was high.
Iron speciation in sediments
The FeTOT content in the samples ranged from 22.34 to 50.41 mg/g d.w., with average values (36.27–38.17 mg/g d.w.) and irregular changes with sediment depth similar at all stations (Fig. 5S). The FeCARB concentration ranged from 0.21 to 5.41 mg/g d.w., with the lowest mean value at MET1-MP-REF (0.79 mg/g d.w.) and the highest at MET1-BH (2.58 mg/g d.w.). In general, pockmark sediment was enriched in FeCARB compared to reference stations (Fig. 8). At the pockmark stations, changes in the content of FeCARB with depth were irregular. At the same time, there was a gradual decrease in this Fe form at the reference stations (Fig. 8).

Changes in the concentration of iron bound in carbonates (FeCARB), labile Fe oxyhydroxides (FeOX1), crystalline Fe hydroxides (FeOX2), and magnetite (FeMAG) with sediment depth in MET1-BH and MET1-MP pockmarks and corresponding reference stations MET1-BH-REF and MET1-MP-REF
FeOX1 content ranged from 1.50 mg/g d.w. to 5.89 mg/g d.w., with the lowest average value again occurring at MET-MP-REF (2.16 mg/g d.w.) and the highest at MET1-BH (3.97 mg/g d.w.). The concentration of FeOX1 in pockmarks was generally higher than at reference stations (except for the surface layer of the sediment at MET1-BH-REF). At all stations except for MET1-BH, there was an irregular decrease of FeOX1 content with depth, and at MET1-BH the concentration increased (Fig. 8).
FeOX2 concentrations ranged from 0.96 to 2.15 mg/g d.w. with the lowest average value occurring at MET1-BH (1.52 mg/g d.w.) and the highest at MET1-BH-REF (1.71 mg/g d.w.). A slight increase in the concentration of FeOX2 with the depth of the sediment was noted in the pockmarks (Fig. 8). At reference stations, the FeOX2 content in the sediment profile changed more irregularly. It is worth noting that the content of FeOX2 in the sediment of MET1-BH was generally lower than at the corresponding reference station (Fig. 8). In the case of MET1-MP and MET1-MP-REF, a higher concentration was recorded in the pockmark than in its vicinity, except for the upper 20 cm of sediment (Fig. 8).
FeMAG reached values in the range of 0.23–3.59 mg/g d.w. Average concentrations were very similar, with a minimum (2.14 mg/g d.w.) occurring at MET1-BH and a maximum (2.63 mg/g d.w.) at MET1-MP-REF.
Discussion
Effects of groundwater outflow on pore water chemistry of pockmarks
A decrease in chloride concentration with sediment depth in pockmarks is a result of upward groundwater infiltration of sediments, and this phenomenon can be found, e.g. in the south-eastern part of the Baltic Sea, in the areas where sediments from the Late Cretaceous period are situated relatively shallow below the bottom (outcrops) and groundwater from that aquifer may easily infiltrate surface marine sediments (Piekarek-Jankowska 1996; Idczak et al. 2020). An approx. 20 and 50% decrease in chloride concentration with sediment depth was measured respectively at MET1-MP and MET1-BH in the present study, which, taking into account data from Idczak et al. (2020) for MET1-BH, points to the regular presence of groundwater infiltration in this area of the seabed. Interestingly, only at MET1-BH a decrease in Cl− and salinity occurred also in near-bottom water which was probably the effect of intense and more pronounced activity of this pockmark, with gas flares in the water column above the pockmark and clear outflow of groundwater from sediments registered during ROV inspections of the bottom in this pockmark area.
Sulfate accounts for about 7.7% of dissolved salts in seawater, but its concentration is reduced by groundwater infiltration. Sulfate in sediments is used by sulfate-reducing bacteria (SRB) and archaea, which finally reduce this ion to sulfide (Jørgensen et al. 2001; Rozan et al. 2002). Dilution of pore waters with groundwater decreases the intensity of sulfate reduction, leading to lower H2S concentrations at pockmark stations as compared to reference stations (Fig. 2). Sulfate depletion and high OM accumulation promote shallow methanogenesis in these sediments, which is confirmed by a relatively sharp increase in methane concentrations (Fig. 2) just below the sediment surface (2–4 mmol/l of methane already at 5 cm bsf). Higher CH4 concentrations at MET1-BH compared to MET1-MP may also be a result of higher organic matter content of the sediment (the average LOI at MET1-BH is 19.1%, while at MET1-MP is 14.4%) combined with the reduced availability of sulfate for its decomposition as a result of groundwater outflow. Similar correlations between sulfate depletion, high organic matter content, and methanogenesis were found in the Black Sea (Knab et al. 2009) and NW Spain (García-Gil et al. 2002). In addition, in active pockmarks, as in the case of MET1-BH and MET1-MP, as a result of ebullition, methane from deeper layers is transported upward, and the gas concentration at the surface tends to be high (Idczak et al. 2020). Moreover, a lower supply of sulfates also affects the pool of oxidants for anaerobic methane oxidation. Therefore, the sediment layer where this process occurs may migrate up from deeper sediments to be closer to the sediment surface. Consequently, the AOM coupled to sulfate reduction (SO42−-AOM) may be less efficient, which was previously observed by Brodecka-Goluch et al. (2022) for MET1-MP. In the case of active pockmarks, high demand for electron acceptors causes oxidants other than sulfate to get involved in the process of AOM. In the areas where iron oxy(hydro)oxides are common in surface sediments (e.g. in most regions of the Gulf of Gdańsk), they may become additional significant oxidants for the AOM. Their involvement in this process may generate substantial changes to the iron cycle in marine sediments (Beal et al. 2009; Yang et al. 2021) and subsequently produce different minerals in sediments (Teichert et al. 2005; Akam et al. 2020).
Impact of intensive anaerobic decomposition of organic matter in pockmarks on the chemical composition of pore waters
Over the past few years, several pockmarks have been discovered in the Gulf of Gdansk (Idczak et al. 2020), but their geochemical characteristics have not been sufficiently studied. Due to their characteristic shape and morphology (shallow depressions), pockmarks are generally characterized by a higher accumulation of organic matter that needs to decompose. In the present study, it was found that organic matter content was 1/3 higher at MET1-BH and 1/5 higher at MET1-MP than at their reference stations. Mineralization of organic detritus of both autochthonous and allochthonous origin is the primary source of nutrients in pore waters. Thus, higher accumulation of organic matter, among others, increases ammonia and phosphate concentrations in pore waters of pockmark sediments.
In reductive, anaerobic conditions, almost all inorganic nitrogen is assumed to be NH4+/NH3 (Canfield 1993). The concentration of ΣNH4+ increased just below the sediment surface (5–20 cm) at MET1-BH and also at MET1-MP, while at the reference stations, there was no zone of intensive ammonia production, but only a prolonged quite regular growth with sediment depth (Fig. 3). Ammonia concentrations, being on average 2–4 times higher in pockmark pore waters than at reference stations, point to a significant internal source of this nutrient in the areas of pockmarks. Similarly, phosphate concentrations at the pockmark stations are much higher than at the reference stations (Fig. 3) and, like in the case of ΣNH4+ concentrations, reach their maximum around 20 cm bsf and then stabilize. Phosphate release into pore waters can occur through both mineralization of organic matter and reductive dissolution of Fe (oxy)hydroxides (Canfield et al. 1993; Jensen et al. 1995) that strongly bind phosphate under aerobic conditions (Mortimer 1941). For this nutrient, again, there is a large pool in pore waters of pockmark sediments which after being released upward to the water column may boost primary production. Based on phosphate concentrations at MET1-MP and to some extent at MET1-BH, it can be concluded that there was a significant enrichment of the near-bottom water with that nutrient. The reductive dissolution of Fe (oxy)hydroxides also increases the concentration of ferrous iron in pore waters (Sundby et al. 1986; Rozan et al. 2002; Raiswell and Canfield 2012). This was particularly evident in the case of MET1-BH, where below 10 cm bsf, the Fe2+ concentration increased to about 50 µmol/l (Fig. 3).
Another manifestation of the intensive decomposition of organic matter in pockmarks was the high concentration of DIC (Fig. 3). In layers 5–70 cm bsf, concentration of DIC was 1.5–6 times higher than at the reference stations where no dynamic changes occurred. Such high values were partly due to the anaerobic oxidation of methane (Łukawska-Matuszewska et al. 2022), which leads to the formation of significant amounts of DIC.
Apart from diffusion, the release of nutrients from pockmark sediments can be further promoted by gas bubbles (Moore 2010), which occasionally or regularly (as in the case of gas flares) escape from sediments (e.g. in the process of gas ebullition). Moreover, significantly higher concentrations of dissolved substances in pore waters of pockmark sediments as compared to the reference sites may also have some implications for iron cycling and minerals formation.
Processes in active pockmarks and their effect on iron speciation and authigenic mineral phases in sediments
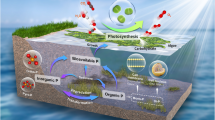
Upward methane seepage creates a characteristic zonation of microbial assemblages (Boetius et al. 2000) where carbon cycling is coupled with iron and sulfur cycles (Judd and Hovland 2009; Suess 2018). The processes occurring in active pockmarks and their impact on the chemical composition of sediment and pore water as well as on the formation of authigenic minerals are schematically shown in Fig. 9.

A simplified representation of microbiological processes in methane-charged sediments and their impact on pore water chemistry and formation of authigenic mineral phases in pockmarks. DSR dissimilatory sulfate reduction, AOM anaerobic methane oxidation, OM organic matter, DIC dissolved inorganic carbon, FeOX iron (oxydr)oxides, FeCARB iron carbonates, FeFeS iron monosulfides, FeFeS2 pyrite iron
In pockmarks MET1-BH and MET1-MP sulfates are present in the 0–25 cm layer of sediment (Fig. 2), and below this layer, their importance as an electron acceptor decreases. Based on pore water profiles of CH4 and SO42−, we can conclude that anaerobic oxidation of methane coupled to sulfate reduction (SO42−-AOM) occured in both pockmarks in the sulfate-methane transition zone (SMTZ) extending over a sediment interval of ca. 5 to 25 cm bsf (Fig. 2). In the SMTZ we observed a rapid depletion of sulfate and a peak in hydrogen sulfide concentration. Within the SMTZ as well as below this layer there was an accumulation of DIC, with concentrations reaching approx. 30 mmol/l (Fig. 3). Such high concentrations in pore waters were produced by conversion of the methane carbon to dissolved inorganic carbon during AOM (Reeburgh 2007; Akam et al. 2020).
The supersaturation of pore waters with DIC creates favorable conditions for the precipitation of authigenic carbonates (Akam et al. 2020). As a result, the FeCARB content of pockmark sediments in the present study was higher than at reference stations (Fig. 8). At MET1-BH, the FeCARB content was the highest around 25 cm bsf, just below the sulfate reduction zone defined by the hydrogen sulfide concentration peak. At MET1-MP, on the other hand, the layer with highest FeCARB content was slightly deeper, where the DIC maximum occurred.
The main carbonate mineral in the sediments of MET1-BH and MET1-MP pockmarks located on the bottom of the Gulf of Gdańsk was ankerite with the chemical formula Ca(Fe, Mg, Mn)(CO3)2. The sediment content of this carbonate mineral reached 3.8 wt%, while calcite was at most 0.8 wt% and siderite—0.3 wt% (Tab. 1S). Ankerite is isostructural with dolomite and is formed in suboxic to anoxic pore water, where magnesium is replaced by varying amounts of reduced iron and manganese. When comparing the mineralogical composition of sediments from the reference MET1-BH-REF station with MET1-BH pockmark (Fig. 6), it is evident that the content of ankerite down to the 40 cm of sediment depth was lower in the reference station. In the MET1-BH sediments, the percentage of this mineral was 1.8–2.6 wt% with the maximum at a depth of 15–25 cm bsf, which roughly agrees with the FeCARB maximum determined by the chemical method. The MET1-MP pockmark sediments contained 0.3–3.8 wt% of ankerite, with the maximum in the subsurface layer. The ankerite content of the MET1-MP sediment was more variable than at MET1-BH. However, even so, the content of this mineral in the MET1-MP pockmark was higher than at the MET1-MP reference station (Fig. 6).
The source of iron for the formation of this mineral in the sediments of the Gulf of Gdańsk is most likely AOM mediated by Fe oxy(hydroxides) (Fe-AOM) which, in addition to sulfate, can act as an electron acceptor in this process (Beal et al. 2009). In estuarine settings with high rate of organic matter deposition, sulfates rarely occur below one meter in sediments. Thus, they cannot oxidize all the methane formed, which then diffuses toward shallower layers (Martens and Klump 1980). In such case, also other oxidants are involved in AOM, including iron and manganese oxides and hydroxides (Beal et al. 2009; Cai et al. 2018). It can be expected that in pockmarks, where there is high accumulation of methane and the availability of sulfate is limited due to groundwater drainage (see Sect. “Effects of groundwater outflow on pore water chemistry of pockmarks”), the involvement of an alternative oxidant will be even greater than in typical anoxic marine sediments.
The concentration of Fe oxy(hydroxides) in the present study was determined by sequential extraction as FeOX1, FeOX2, and FeMAG, and their contribution to the total iron pool in the studied sediments was 13–30%. In both pockmarks, below the SMT zone, there were low or undetectable sulfate concentrations and, at the same time, reactive Fe oxyhydroxides were present, which creates favourable conditions for the Fe-mediated AOM (Riedinger et al. 2014; Rooze et al. 2016).
The MET1-BH sediment showed depletion in reactive iron (FeOX1, FeOX2, and FeMAG) in the upper 25 cm (Fig. 8), into which methane diffused from deeper layers. Based on the substance profiles in pore water (Figs. 2 and 3) and sediment (Fig. 8), it can be concluded that Fe-AOM also occurred deeper in the sediment, below 30 cm, leading to a sharp increase in dissolved Fe and a decrease in methane concentration as well as a slight loss of reactive Fe. In the MET1-MP, the depletion in the most reactive forms of Fe (FeOX1) was present deeper in the sediment, below 20 cm (Fig. 8). In the surface layers of the sediment, FeOX1 concentrations were even higher than at the corresponding reference station (MET1-MP-REF). This was probably a manifestation of the intense redox recycling of Fe at this station—reduced iron diffused from deeper layers toward the sediment–water interface, it was oxidized and precipitates as amorphous iron hydroxides. Similar enrichment was not seen at MET1-BH, which on the one hand may be due to the use of freshly precipitated Fe compounds to oxidize methane (found in more significant quantities at MET1-BH), and on the other hand may be related to different morphology of the pockmarks. The MET1-MP bottom is located at a shallower depth, and the pockmark itself is also shallower and more extensive than MET1-BH, so the oxygenation at the water–sediment interface of MET1-MP was probably higher than at MET1-BH, which favors the accumulation of Fe hydroxides in the sediments.
Fe-AOM leads to the release of significant amounts of Fe2+ into the pore water (Beal et al. 2009; Yang et al. 2021). This was particularly evident at MET1-BH, where ca. 30 cm bsf the concentration of dissolved iron in the pore waters reached 50 µmol/l (Fig. 3). Similar ferrous iron enrichment of pore waters has been observed in other marine regions with an active Fe-dependent oxidation of methane diffusing from deeper sediment layers toward the surface (D’Hondt et al. 2004; Slomp et al. 2013; Riedinger et al. 2014).
Ferrous iron originating from Fe-AOM can precipitate with DIC to form authigenic carbonates (as described above) or diffuse upward and react with hydrogen sulfide produced during SO42− AOM, leading to the formation of Fe-sulfides (Fig. 9). In addition, hydrogen sulfide released in sulfate reduction can react directly with iron oxides to form iron sulfides such as greigite and/or pyrite (Kasten et al. 1998; Neretin et al. 2004). In the studied sediments, the most abundant form of sulfide was pyrite (Fig. 6). The content of this mineral in MET1-BH sediment was generally lower than at the reference station (Fig. 6), except for the subsurface layer. The pyrite content in MET1-BH sediment was about 1%, while at MET1-BH-REF it reached 3.9%. This indicates the limiting role of hydrogen sulfide in the formation of pyrite, as its formation occurs mainly in the layer of hydrogen sulfide occurrence. Insufficient availability of hydrogen sulfide combined with intensive Fe reduction led to the accumulation of Fe2+ in pore waters of MET1-BH (Fig. 3). The situation was different in the MET1-MP pockmark, where Fe2+ was completely removed from the pore waters (Fig. 3) and the pyrite content was similar to that at the reference station (Fig. 6). Since the concentration of FeOX1 constituting the most reactive forms of Fe at MET1-MP was similar or even higher than at MET1-MP-REF (Fig. 8), it can be assumed that iron reduction at MET1-MP was not as intense as in sediments of the second pockmark MET1-BH. As a result, less ferrous iron is produced there and the amount of hydrogen sulfide and DIC generated in sediments is sufficient to almost completely remove Fe from the pore waters in the form of pyrite and carbonates (ankerite and siderite), the concentrations of which increase below 40 cm (Fig. 7).
Carbonate minerals are a frequent autogenous component of sediments where oxidation of methane (both thermogenic and biogenic origin) occurs (Hovland and Judd 1988; Vogt et al. 1997; Stakes et al. 1999; Judd and Hovland 2009; Feng et al. 2010). Extensive areas of authigenic carbonate crust as well as authigenic carbonate nodules are common, for example, within mud volcanoes in the eastern Mediterranean (Aloisi et al. 2000). The calcite, aragonite and dolomite found in this area showed large 13C depletions, which indicates that AOM was a major carbon source for the carbonate. O’Reilly et al. (2014) described authigenic methane-derived carbonates (MDACs) in the shallow waters of the western Irish Sea, with aragonite as the main component. High-Mg-calcite or aragonite were also a major component of the layered carbonate crusts in gas bearing sediments on the shelf and slope of the northwestern Black Sea (Peckmann et al. 2001). Also in that case, the 13C depletion indicated that the carbonates were mainly from microbial oxidation of methane. Authigenic carbonates, composed mainly of aragonite and high-Mg calcite, were also collected from methane seeps on the northern Congo deep-sea fan (Feng et al. 2010).
Formation of authigenic carbonate minerals in methanic sediments is a significant pathway of marine carbon burial. On the global scale, carbonate autogenesis is responsible for the sequestration of about 3.93 (from 2.34 to 5.80) Tmol C yr−1 in marine sediments (Akam et al. 2020). Thus, it can be considered as one of the main mechanisms of C burial in marine sediments (the third largest according to calculations of Akam et al. 2020). This component of the carbon budget in the Baltic Sea has not been defined yet. Further research is required, especially since the Baltic is a brackish sea with high productivity, which in turn creates conditions favorable for methane production in sediments. Baltic water is unsaturated with respect to CaCO3, which negatively affects calcifying organisms (Thomsen et al. 2018). Biogenic carbonates occur mainly as shell debris, easily mechanically degradable and dissolved under unsaturated CaCO3 conditions. Therefore, the content of calcium carbonate in the sediment usually does not exceed 1% (Uścinowicz 2011). This implies that carbonate autogenesis is probably an important component of the C cycle in the Baltic Sea, which has so far been overlooked.
Conclusions
Geochemical conditions in pockmarked sediments are significantly different from those in the typical muddy sea bottom of the Gulf of Gdańsk. In the areas of deepwater (> 70 m) pockmarks, sediment can be characterized by higher organic matter content than in the surrounding bottom outside pockmarks. Intensive decomposition processes in these shallow depressions lead to increased ammonia and phosphate concentrations in the pore and near-bottom waters. Moreover, the infiltration of groundwater which occurs in pockmark sites affects the availability (or concentrations) of oxidants for organic matter mineralization or for AOM, and in consequence may lead to the migration of SMT zone into shallower sediment layers and/or cause the involvement of different oxidants (e.g., Fe oxyhydroxides instead of diluted and not available sulfate) in those processes. The dilution of seawater within the sediment, causing lower sulfate concentrations in pore waters, also leads to a decrease in hydrogen sulfide production. In the 0–15 cm layer, sulfate was the primary oxidant of organic matter and methane, but below, in the 15–25 cm layer, oxidation of OM and methane was driven mainly by Fe (III) minerals like ferrihydrite, goethite, and lepidocrocite. This process leads to the reduction of FeOX1 and FeOX2 content, and finally, produced Fe2+ may diffuse upward, where it can be reoxidized or precipitated to sulfides (like pyrite and pyrrhotine) within euxinic sediment. No significant changes in FeMAG concentrations were observed, as it is poorly reactive with hydrogen sulfide. Dissolved inorganic carbon formed as a result of AOM was precipitated with Fe2+ to form authigenic carbonate (FeCARB) which increased concentrations were observed in the pockmarks examined in this study. The main authigenic carbonate minerals identified were ankerite and siderite.
Data availability
The dataset generated and analysed during the current study is available in the Mendeley Data repository (https://data.mendeley.com/datasets/jnp8yn7pmw/1).
References
Akam SA, Coffin RB, Abdulla HAN, Lyons TW (2020) Dissolved inorganic carbon pump in methane-charged shallow marine sediments: state of the art and new model perspectives. Front Mar Sci 7:206
Aloisi G, Pierre C, Rouchy JM, Foucher JP, Woodside J (2000) Methane-related authigenic carbonates of eastern Mediterranean sea mud volcanoes and their possible relation to gas hydrate destabilisation earth planet. Sci Lett 184:321–338
Archer D, Johnson EK (2000) A model of the iron cycle in the ocean. Glob Biogeochem Cycl 14:269–279
Beal EJ, House CH, Orphan VJ (2009) Manganese- and iron-dependent marine methane oxidation. Science 325:184–187. https://doi.org/10.1126/science.1169984
Boetius A, Wenzhöfer F (2013) Seafloor oxygen consumption fuelled by methane from cold seeps. Nature Geosci 6:725–734
Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A, Amann R, Jørgensen BB, Witte U, Pfannkuche O (2000) A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407:623–626
Brocławik O, Łukawska-Matuszewska K, Brodecka-Goluch A, Bolałek J (2020) Impact of methane occurrence on iron speciation in the sediments of the Gdansk Basin (southern Baltic Sea). STOTEN 721:137718. https://doi.org/10.1016/j.scitotenv.2020.137718
Brodecka A, Majewski P, Bolałek J, Klusek Z (2013) Geochemical and acoustic evidence for the occurrence of methane in sediments of the polish sector of the southern Baltic Sea. Oceanologia 55:951–978. https://doi.org/10.5697/oc.55-4.951
Brodecka-Goluch A, Łukawska-Matuszewska K, Kotarba MJ, Borkowski A, Idczak J, Bolałek J (2022) Biogeochemistry of three different shallow gas systems in continental shelf sediments of the South-Eastern Baltic Sea (Gulf of Gdańsk): carbon cycling, origin of methane and microbial community composition. Chem Geol 597:120799
Bussmann I, Suess E (1998) Groundwater seepage in Eckernförde Bay (western Baltic Sea): effect on methane and salinity distribution of the water column. Cont Shelf Res 18:1795–1806
Cai C, Leu AO, Xie G-J, Guo J, Feng Y, Zhao J-X et al (2018) A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. ISME J 12:1929–1939. https://doi.org/10.1038/s41396-018-0109-x
Canfield DE, Jørgensen BB, Fossing H, Glud R, Gundersen J et al (1993) Pathways of organic carbon oxidation in three continental marine sediments. Mar Geol 113:27–40
Carstensen J, Andersen JH, Gustafsson BG, Conley DJ (2014) Deoxygenation of the Baltic Sea during the last century. Proc Natl Acad Sci USA 15(111):5628–5633
Chuang P-C, Yang TF, Hong W-L, Lin S, Sun C-H, Lin AT-S, Chen J-C, Wang Y, Chung S-H (2010) Estimation of methane flux offshore SW Taiwan and the influence of tectonics on gas hydrate accumulation. Abs Geofluid 10:497–510
Chubarenko B, Domnin D, Navrotskaya S, Stont Z, Chechko V, Bobykina V, Pilipchuk V, Karmanov K, Domnina A, Bukanova T, Topchaya V, Kileso A (2017) Transboundary lagoons of the Baltic Sea. The diversity of Russian estuaries and lagoons exposed to human influence. Springer, Cham, pp 149–189
D’Hondt S, Jørgensen BB, Miller DJ, Batzke A, Blake R, Cragg BA, Cypionka H, Dickens GR, Ferdelman T, Hinrichs KU et al (2004) Distributions of microbial activities in deep subseafloor sediments. Science 306:2216–2221
Díaz RJ, Rosenberg R (2008) Spreading dead zones and consequences for marine ecosystems. Science 321:926–929. https://doi.org/10.1126/science.1156401
Egger M, Riedinger N, Mogollón JM et al (2018) Global diffusive fluxes of methane in marine sediments. Nature Geosci 11:421–425
Feng D, Chen D, Peckmann J, Bohrmann G (2010) Authigenic carbonates from methane seeps of the northern Congo fan: microbial formation mechanism. Mar Pet Geol 27:748–756
Froelich PN, Klinkhammer GP, Bender ML, Luedtke NA, Heath GR, Cullen D, Dauphin P, Hammond DE, Hartman B, Maynard V (1979) Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim Cosmochim Acta 43:1075–1090
García-Gil S, Vilas F, García-Garcia A (2002) Shallow gas features in incised-valley fills (Rı́a de Vigo NW Spain): a case study. Continent Shelf Res 22(16):2303–2315. https://doi.org/10.1016/S0278-4343(02)00057-2
Gay A, López M, Cochonat P, Sultan N, Cauquil E, Brigaud F (2003) Sinuous pockmark belt as indicator of a shallow buried turbiditic channel on the lower slope of the Congo basin, West African margin. Geol Soc London, Spec Publ 216:173–189
Giovannelli D, D’Errico G, Fiorentino F, Fattorini D, Regoli F, Angeletti L, Bakran-Petricioli T, Vetriani C, Yücel M, Taviani M, Manini E (2016) Diversity and distribution of prokaryotes within a shallow-water pockmark field. Front Microbiol 7:941
Graham LP (2004) Climate change effects on river flow to the Baltic Sea. Ambio 33:235–241
Grasshoff K, Ehrhardt M, Kremling K, Anderson LG (1999) Methods of seawater analysis. Wiley-VCH, Weinheim, p 600
Greinert J, Bohrmann G, Suess E (2001) Gas hydrate-associated carbonates and methane venting at hydrate ridge: classification, distribution and origin of authigenic lithologies. In: Paull C, Dillon W (eds) Natural gas hydrates: occurrence, distribution, and detection. American Geophysical Union, Washington, D. C., pp 99–113
Haverkamp THA, Hammer Ø, Jakobsen KS (2014) Linking geology and microbiology: inactive pockmarks affect sediment microbial community structure. PLoS ONE 9(1):e85990
Hinrichs K-U, Hayes JM, Sylva SP, Brewer PG, DeLong EF (1999) Methane-consuming archaebacteria in marine sediments. Nature 398:802–805. https://doi.org/10.1038/19751
Hovland M, Judd AG (1988) Seabed pockmarks and seepages: impact on geology, biology and the marine environment. Graham and Trotman, London
Hung C, Huang K, Shih Y et al (2016) Benthic fluxes of dissolved organic carbon from gas hydrate sediments in the northern South China Sea. Sci Rep 6:29597
Hyacinthe C, Van Cappellen P (2004) An authigenic iron phosphate phase in estuarine sediments: composition, formation and chemical reactivity. Mar Chem 91:227–251
Idczak J, Brodecka-Goluch A, Łukawska-Matuszewska K, Graca B, Gorska N, Klusek Z, Pezacki P, Bolałek J (2020) A geophysical, geochemical and microbiological study of a newly discovered pockmark with active gas seepage and submarine groundwater discharge (MET1-BH, central Gulf of Gdansk, southern Baltic Sea). Sci Tot Environ. https://doi.org/10.1016/j.scitotenv.2020.140306
Jensen HS, Mortensen PB, Andersen FO, Rasmussen E, Jensen A (1995) Phosphorus cycling in a coastal marine sediment, Aarhus Bay. Denmark Limnol Oceanogr 40:908–917
Jørgensen BB, Weber A, Zopfi J (2001) Sulfate reduction and anaerobic oxidation in Black Sea sediments. Deep-Sea Res Part I 48:2097–2120
Judd AG, Hovland M (2009) Seabed fluid flow: the impact on geology, biology and the marine environment. Cambridge University Press
Kabel K, Moros M, Porsche C, Neumann T, Adolphi F, Andersen TJ, Siegel H, Gerth M et al (2012) Impact of climate change on the Baltic Sea ecosystem over the past 1,000 years. Nat Clim Chang 2:871–874. https://doi.org/10.1038/nclimate1595
Kasten S, Freudenthal T, Gingele FX, Schulz HD (1998) Simultaneous formation of iron-rich layers at different redox boundaries in sediments of the Amazon deep-sea fan. Geochim Cosmochim Acta 62:2253–2264
Kelley J, Dickson S, Belknap D, Barnhardt W, Henderson M (1994) Giant sea-bed pockmarks: evidence for gas escape from Belfast Bay. Maine Geology 22:59–62
Knab NJ, Cragg BA, Hornibrook ERC, Holmkvist L, Pancost RD, Borowski C, Parkes RJ, Jørgensen BB (2009) Regulation of anaerobic methane oxidation in sediments of the Black Sea. Biogeosciences 6:1505–1518
Knittel K, Boetius A (2009) Anaerobic oxidation of methane: progress with an unknown process. Annu Rev Microbiol 63:311–334. https://doi.org/10.1146/annurev.micro.61.080706.093130
Kruglyakova R, Gubano Y, Kruglyakov V, Prokoptsev G (2002) Assessment of technogenic and natural hydrocarbon supply into the Black Sea and seabed sediments. Cont Shelf Res 22:2395–2408
Kruk-Dowgiałło L, Szaniawska A (2008) Gulf of Gdansk and Puck Bay. In: Schiewer U (ed) Ecology of Baltic coastal water, ecological studies. Springer-Verlag, Berlin Heidelberg, pp 139–166
Lemaitre N, Bayon G, Ondréas H, Caprais JC, Freslon N, Bollinger C, Rouget M, Prunelé AD, Ruffine L, Roy KO, Sarthou G (2014) Trace element behaviour at cold seeps and the potential export of dissolved iron to the ocean. Earth Planet Sci Lett 404:376–388
Levin LA (2005) Ecology of cold seep sediments: interactions offauna with flow, chemistry and microbes. Oceanogr Mar Biol Annu Rev 43:1–46
Łukawska-Matuszewska K (2016) Contribution of non-carbonate inorganic and organic alkalinity to total measured alkalinity in pore waters in marine sediments (Gulf of Gdansk, S-E Baltic Sea). Mar Chem 186:211–220
Łukawska-Matuszewska K, Brocławik O, Brodecka-Goluch A, Rzepa G, Manecki M, Bolałek J (2022) Biogeochemical and mineralogical effects of Fe-P-S dynamics in sediments of continental shelf sea: impact of salinity, oxygen conditions, and catchment area characteristics. Sci Total Environ 807:151035. https://doi.org/10.1016/j.scitotenv.2021.151035
Majewski A (ed) (1990) Ogólna charakterystyka morfometryczna Zatoki Gdańskiej. Zatoka Gdańska. Wyd. Geol., Warszawa, 10–15 (In Polish)
Majewski P, Klusek Z (2014) Parameters of echo signals originated from a gas seepage site in the southern Baltic Sea. Hydroacoustics 17:143–150
Martin J (1990) Glacial-interglacial CO2 change: the iron hypothesis. Palaeogeogr Palaeoclimatol 5:1–13. https://doi.org/10.1029/PA005i001p00001
Martens CS, Klump JV (1980) Biogeochemical cycling in an organic-rich coastal marine basin—I. Methane sediment-water exchange processes. Geochim. Cosmochim Acta 44:471–490
Mavromatis V, Botz R, Schmidt M, Liebetrau V, Hensen C (2012) Formation of carbonate concretions in surface sediments of two mud mounds offshore Costa Rica: a stable isotope study. Int J Earth Sci. https://doi.org/10.1007/s00531-012-0843-7
Meier HEM, Kniebusch M, Dieterich C, Gröger M, Zorita E, Elmgren R, Myrberg K, Ahola MP et al (2022) Climate change in the Baltic Sea region: a summary. Earth Syst Dynam 13:457–593. https://doi.org/10.5194/esd-13-457-2022
Moore WS (2010) The effect of submarine groundwater discharge on the ocean. Ann Rev Mar Sci 2:59–88
Mortimer CH (1941) The exchange of dissolved substances between mud and water in lakes. J Ecol 29(1):280–329. https://doi.org/10.2307/2256691
Neretin LN, Böttcher ME, Jørgensen BB, Volkov II, Luschen H, Hilgenfeldt K (2004) Pyritization processes and greigite formation in the advancing sulfidization front in the upper Pleistocene sediments of the Black Sea. Geochim Cosmochim Acta 68:2081–2093
O’Reilly SS, Hryniewicz K, Little CTS, Monteys X, Szpak MT, Murphy BT, Jordan SF, Christopher ACR, Kelleher BP (2014) Shallow water methane-derived authigenic carbonate mounds at the codling fault zone, western Irish sea. Mar Geol 357:139–150
O’Reilly SS, Jordan SF, Monteys X, Simpson AJ, Allen CCR, Szpak MT, Murphy BT, McCarron SG, Soong R, Wu B, Jenne A, Grey A, Kelleher BP (2021) Production of methane and gaseous compounds by surface microbial activity in a small pockmark field, Dunmanus Bay, Ireland. Estuar Coast Shelf Sci 255:107340
Peckmann J, Reimer A, Luth U, Luth C, Hansen BT, Heinicke C, Hoefs J, Reitner J (2001) Methane-derived carbonates and authigenic pyrite from the northwestern Black Sea. Mar Geol 177:129–150
Piekarek-Jankowska H (1996) Hydrochemical effects of submarine groundwater discharge to the Puck Bay (Southern et al.). Geogr Pol 67:103–119
Poulton S, Canfield D (2005) Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem Geol 214:209–221
Rabalais NN, Cai W-J, Carstensen J, Conley DJ, Fry B, Hu X, Quiñones-Rivera Z, Rosenberg R, Slomp CP, Turner RE, Voss M, Wissel B, Zhang J (2014) Eutrophication-driven deoxygenation in the coastal ocean. Oceanography 27:172–183
Raiswell R, Canfield DE (2012) The iron biogeochemical cycle past and present. Geochem Perspect 1:1–220. https://doi.org/10.7185/geochempersp.1.1
Reeburgh WS (2007) Oceanic methane biogeochemistry. Chem Rev 107:486–513. https://doi.org/10.1021/cr050362v
Riedinger N, Formolo MJ, Lyons TW, Henkel S, Beck A, Kasten S (2014) An inorganic geochemical argument for coupled anaerobic oxidation of methane and iron reduction in marine sediments. Geobiology 12:172–181
Ritger S, Carson B, Suess E (1987) Methane-derived authigenic carbonates formed by subduction-induced pore-water expulsion along the Oregon/Washington margin. Geol Soc Am Bull 98:147–156
Rooze J, Egger M, Tsandev I, Slomp CP (2016) Iron-dependent anaerobic oxidation of methane in coastal surface sediments: potential controls and impact. Limnol Oceanogr 61:S267–S282. https://doi.org/10.1002/lno.10275
Rozan TF, Taillefert M, Trouwborst RE, Glazer BT, Ma S, Herszage J, Valdes LM, Price KS, Luther GW III (2002) Iron-sulfur-phosphorus cycling in the sediments of a shallow coastal bay: implications for sediment nutrient release and benthic macroalgal blooms. Limnol Oceanogr. https://doi.org/10.4319/lo.2002.47.5.1346
Slomp CP, Mort HP, Jilbert T, Reed DC, Gustafsson BG, Wolthers M (2013) Coupled dynamics of iron and phosphorus in sediments of an oligotrophic coastal basin and the impact of anaerobic oxidation of methane. PLoS ONE 8(4):e62386
Söderberg P, Flodén T (1992) Gas seepages, gas eruptions and degassing structures in the seafloor along the Strömma tectonic lineament in the crystalline Stockholm Archipelago, east Sweden. Cont Shelf Res 12(10):1157–1171
Sørensen J (1982) Reduction of ferric iron in anaerobic, marine sediment and interaction with reduction of nitrate and sulfate. Appl Environ Microbiol 43:319–324
Stakes DS, Orange D, Paduan JB, Salamy KA, Maher N (1999) Cold-seeps and authigenic carbonate formation in Monterey Bay, California. Mar Geol 159:93–109
Suess E (2018) Marine cold seeps: background and recent advances. In: Wilkes H (ed) Hydrocarbons, Oils and lipids: diversity, origin, chemistry and fate. handbook of hydrocarbon and lipid microbiology. Springer, Cham
Sundby B, Anderson LG, Hall POJ, Iverfeldt A, Rutgers van der Loeff M et al (1986) The effect of oxygen on release and uptake of cobalt, manganese, iron and phosphate at the sediment-water interface. Geochim Cosmochim Acta 50:1281–1288
Teichert BMA, Bohrmann G, Suess E (2005) Chemoherms on hydrate ridge—unique microbially-mediated carbonate build-ups growing into the water column. Palaeogeogr Palaeoclimatol Palaeoecol 227:67–85
Thomsen J, Ramesh K, Sanders T, Bleich M, Melzner F (2018) Calcification in a marginal sea—influence of seawater [Ca2+] and carbonate chemistry on bivalve shell formation. Biogeosciences 15:1469–1482
Uścinowicz S (2011) Geochemistry of Baltic Sea surface sediments. Polish Geological Institute-National Research Institute, Warsaw
Vogt PR, Cherkashev G, Ginsburg G, Ivanov G, Milkov A, Crane K, Lein A, Sundvor E, Pimenov N, Egorov A (1997) Haakon Mosby Mud Volcano provides unusual example of venting. Eos 78:540
Weber T, Wiseman NA, Kock A (2019) Global ocean methane emissions dominated by shallow coastal waters. Nat Commun 10:4584
Whiticar MJ, Werner F (1981) Pockmarks: Submarine vents of natural gas or freshwater seeps? Geo-Mar Lett 1:193–199. https://doi.org/10.1007/BF02462433
Yang H, Yu S, Lu H (2021) Iron-coupled anaerobic oxidation of methane in marine sediments: a review. J Mar Sci Eng 9:875
Zeppilli D, Canals M, Danovaro R (2012) Pockmarks enhance deep-sea benthic biodiversity: a case study in the western Mediterranean Sea. Divers Distrib 18:832–846
Acknowledgements
The authors would like to thank the captain and the crew of R/V Oceanograf.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Material preparation, data collection, and analysis were performed by SK, KŁ-M, AČ, DJ and AB-G. The first draft of the manuscript was written by SK, KŁ-M, DJ, and AB-G and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Responsible Editor: Kate Lajtha.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kurowski, S., Łukawska-Matuszewska, K., Čović, A. et al. Effects of pockmark activity on iron cycling and mineral composition in continental shelf sediments (southern Baltic Sea). Biogeochemistry 167, 135–154 (2024). https://doi.org/10.1007/s10533-024-01127-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-024-01127-1