
Abstract
Species-rich calcareous grasslands in Europe strongly declined during the twentieth century due to drastic land use changes. Many grasslands were converted into more productive pastures or are covered by shrubs or forests today, since they were overgrown after abandonment or afforested. Restoration of calcareous grasslands by shrub or forest clearing and subsequent recolonization of grassland species from adjacent grasslands is, therefore, an important conservation approach. Restored populations of calcareous grassland species may, however, differ from their source populations in genetic diversity and differentiation due to potential founder and bottleneck effects. In our study we analyzed, therefore, the impact of restoration by forest clearing and natural recolonization on the genetic variation of three common calcareous grassland species (Agrimonia eupatoria, Campanula rotundifolia, and Knautia arvensis) without a contribution of persistent seed bank, in South Western Germany. We used molecular markers AFLPs (Amplified fragment length polymorphisms) to compare genetic diversity within and differentiation between spontaneously recovered subpopulations with adjacent historically old, natural subpopulations at eight study sites. Restored parts of the grasslands have been re-established during the 1990s. Molecular markers revealed broadly similar levels of genetic diversity in source and restored subpopulations of the study species. Only A. eupatoria exhibited slightly higher diversity in restored subpopulations, which may be explained by higher dispersal potential due to the hooky fruits of the species. Genetic differentiation between source and restored subpopulations was not significant, indicating strong gene flow between the subpopulations. Our study underlines, therefore, that restoration of calcareous grasslands by natural recolonization after forest clearing is an efficient method to re-establish genetically variable subpopulations comparable to their sources.
Similar content being viewed by others
Introduction
Semi-natural calcareous grasslands rank among the most species-rich, but also highly vulnerable and endangered ecosystems across Europe. Due to the tremendous changes of land-use practices during the nineteenth and twentieth century, this habitat has rapidly declined in area and quality (Poschlod and WallisDeVries 2002; Pullin et al. 2009; Huber et al. 2017). Calcareous grasslands were abandoned, afforested or converted into intensively managed agricultural land (WallisDeVries et al. 2002). Due to the extraordinary biodiversity of calcareous grasslands, they are an important target habitat for the biodiversity maintenance at the European scale (European Community 1992). Since the 1990s, in some cases even earlier, numerous restoration schemes have been launched to re-establish species rich calcareous grassland communities.
Several methods of grassland restoration as well as techniques to enhance species richness have been frequently applied to recover near-natural grasslands following cropland, quarry and mining areas abandonment (Tȍrȍk et al. 2011) or forest felling (Poschlod et al. 1998; Pärtel et al. 1998; Bisteau and Mahy 2005). First, the common and widely used restoration practice is sowing regional seed mixtures (Durka et al. 2019; Höfner et al. 2021; Kaulfuß et al. 2022), either in high-diversity or low-diversity option (Kirmer et al. 2012). This method helps to restore grassland communities in a comparatively short time horizon (Kövendi‐Jakó et al. 2019). It is especially vital in cases when the local species pool does not enable spontaneous grassland regeneration via natural diaspore input from the seed rain and seedbank (Willems and Bik 1998) or, if effective dispersal vectors (DiLeo et al. 2017) are lacking, respectively. Second, translocation of plant material, e.g. green hay, raked litter, threshed seeds or barn chaff (Kaulfuß and Reisch 2021) have been applied to introduce target species and to enhance species richness in degraded grasslands, or to initiate new grasslands (Poschlod et al. 1997; Kiehl et al. 2010; Albert et al. 2019). Third, planting seedlings, mature plants or belowground parts, respectively, is a technique often applied to additionally enhance species richness and propagule availability in sites restored using other methods (Guerrant Jr and Kaye 2007). This approach usually yields faster maturity and population establishment than seed sowing (Dalrymple et al. 2012) but requires more time and financial costs. Use of plants also implies multiple steps in a production process and may unintentionally cause erosion of genetic diversity (Basey et al. 2015).
Finally, spontaneous colonization on fallow lying old fields (review Rejmanek and van Katwyk 2005), abandoned quarry areas (Ilves et al. 2015) or following scrub or woodland removal (Kiefer and Poschlod 1996; Kiefer 1998; Blanckenhagen and Poschlod 2005) imposes an important grassland restoration tool. This method is a natural and low cost way of grassland restoration (Prach and Hobbs 2008), as it relies on spontaneous seed dispersal processes from locally available propagule sources (Bakker et al. 1996; Kirmer et al. 2008; Redhead et al. 2014). Natural colonization is usually combined with other techniques, e.g. sowing seed mixtures (Johanidesová et al. 2015; Kaulfuß et al. 2022), creation of artificial biodiversity hotspots (Kiss et al. 2021), as well as topsoil removal (Řehounková et al. 2021). However, the occurrence of source grasslands in the nearby surrounding landscape plays a crucial role in colonization of restored grasslands by target species (Prach et al. 2015; Aavik and Helm 2018; Kiss et al. 2021).
In comparison with other restoration methods, natural (re)colonization intrinsically involves the advantage of matching the gene pool of nearby populations, due to the seed rain from surrounding grasslands (McKay et al. 2005). For restoration purposes, the importance of regional seed material has been generally recommended (Mijnsbrugge et al. 2010) and thoroughly documented (Bucharova et al. 2022; Höfner et al. 2021). The local origin of diaspores is desirable to preserve patterns of genetic variation, because mixing strongly differing genotypes may lead to the outbreeding depression (Hufford and Mazer 2003; Frankham et al. 2011). This may bring about decreased fitness and vitality in restored populations, since coadapted gene complexes could break down and local adaptations get lost (Montalvo and Ellstrand 2001; Hufford and Mazer 2003).
Regeneration process of restored sites always imposes several risks to genetic makeup of newly established populations. At the present time, especially genetic issues caused by bringing in propagules, particularly if those were bred specifically for this purpose have been studied rigorously (Durka et al. 2017; Bucharova et al. 2022; Höfner et al. 2021).
However, restoration by colonization from adjacent habitats may also impose the risk of genetic diversity loss, because founder groups may consist of just a limited number of individuals and comprise only a part of the source populations genetic diversity (Franklin and Frankham 1998; Vandepitte et al. 2012). Further, the loss of alleles and changes in their frequencies may additionally occur due to random genetic drift in small populations (Franklin 1980). Finally, founder events may not only lead to the locally decreased genetic diversity, but also to considerably enhanced genetic divergence between populations (Vandepitte et al. 2012). The magnitude of a founder effect and subsequent genetic differentiation as a consequence of an extinction and recolonization process, has been studied explicitly in the context of metapopulation theory (Slatkin 1977a; Wade and McCauley 1988; Whitlock and McCauley 1990).
This theory predicts that the severity of a founder effect depends on two major parameters. First, the number of colonists arriving to a restored site, and their proportion in comparison with the number of migrants exchanged among extant populations. If the number of founders exceeds the number of migrants exchanged between established populations more than twice, then the founder effect is expected to be weak (Wade and McCauley 1988). Second, the number (single or multiple) of source populations contributing to the formation of a colonist group is also determining regarding the founder effect magnitude (the “propagule pool” and the “migrant pool” model; Slatkin 1977b; Whitlock and McCauley 1990). The likelihood of founder effects will be reduced if more than one source populations appeared under seed sources (Slatkin 1977a), and, further, if high migration rates into restored populations took place, enabled through spatial vicinity (Helsen et al. 2013), permeability and intrinsic species dispersal capacity, or availability of a suitable dispersal vector, respectively (DiLeo et al. 2017). Moreover, fast population growth following founding events also helps to rapidly recover population genetic diversity, owing to new mutations (Nei et al. 1975). The broad population genetic diversity is a necessary raw material for the population’s adaptability to changing environments. If levels of genetic diversity are reduced, then populations would be at risk in the long term due to the loss of their evolutionary potential (Franklin and Frankham 1998; Bucharova et al. 2022).
Spontaneous colonization processes and their impact on genetic properties of founder populations were investigated in numerous studies, often in natural landscapes, e.g. new volcanic deposits (Bishop 1996; Yang et al. 2008), glacier forelands (Raffl et al. 2006) and floodplains (Van Looy et al. 2009; Honnay et al. 2009). Vandepitte et al. (2007) and Jacquemyn et al. (2009) investigated the impact of gene flow, isolation and genetic drift on genetic variation in forest herbs with contrasting mating systems. Colonization of abandoned quarries and its consequences for genetic variation and fitness of a rare herb was studied by Ilves et al. (2015). However, only a few studies have highlighted the genetic consequences of colonization processes in temperate grasslands after scrub and tree removal. In addition, results of these studies were inconsistent, most probably due to different configuration and initial number of source and restored populations, as well as due to differences in species traits. Thus, Vandepitte et al. (2012) found that a limited number of available remnant source populations led to the reduced genetic diversity as well as inflated genetic differentiation in founder populations. On the other hand, Helsen et al. (2013) stated that sufficient number of source populations occurring in the surroundings of colonized spots may result in neither decreased genetic diversity, nor increased genetic differentiation between founder populations. However, this outcome was most probably also due to the gene flow and recruitment from the long-term seedbank.
A broader knowledge is thus still lacking in terms of genetic patterns in recolonized calcareous grasslands, without any contribution of viable seeds in soil. To address this gap, we highlight the impact of the recolonization on common grassland species Agrimonia eupatoria, Campanula rotundifolia and Knautia arvensis. Specifically, we asked following questions: (i) what are possible inter-specific differences in genetic diversity between the study species relating to differences in their mating systems? (ii) Are levels of genetic diversity comparable in source and restored subpopulations? (iii) Is there a significant genetic differentiation between source and restored subpopulations? And, finally, (iv) is recolonization after clear-cutting an appropriate method to maintain local genetic variation patterns of common grassland species without persistent soil seed bank?
Materials and methods
Study sites, study species and sampling design
The study is based on a restoration project situated in South-Western Germany, in the region of the Neckar basin and the Swabian Alb (Kiefer and Poschlod 1996; Poschlod et al. 1998). Here, former calcareous grasslands overgrown for at least one decade (Kiefer 1998) with trees and shrubs such as pine (Pinus sylvestris) and blackthorn (Prunus spinosa), or deliberately afforested with Pinus sylvestris and Picea abies were restored in the 1990s by clear-cutting. Trees, scrub, as well as the entire herbaceous vegetation layer were removed during the restoration. The sites were then left open for grassland recovery through spontaneous recolonization by species immigrating from adjacent well-preserved remnant grasslands and, to some extent, also from the soil seedbank (Blanckenhagen and Poschlod 2005).
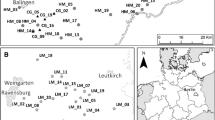
For our study, we selected eight study sites located in South Western Germany near Stuttgart city, on the hills of the Swabian Alb and its northern surroundings in the Neckar River basin (Fig. 1). The altitude of the study sites ranges from 250 to 700 m above sea level. The climate in the study area is temperate; the mean precipitation varies between 650 mm in the Neckar basin and 900 mm in the Swabian Alb, respectively. The bedrock is formed by Malm series, which belongs to the upper Jurassic formation and consists mostly of the reef limestone. The prevailing soil type is Rendzina, sometimes developed as brown soil.

Geographic location of the study sites in South Western Germany. Source and restored subpopulations of the study species A. eupatoria, C. rotundifolia and K. arvensis were investigated at six study sites each. A. eupatoria was sampled in EH, FB, HA, HE, RR and WS; C. rotundifolia was sampled in BK, EH, FB, HA, HE and RR; K. arvensis was sampled in ER, FB, HA, HE, RR and WS. BK, Botenklinge; EH, Eichhalde; ER, Eselrain; FB, Furtberg; HA, Haarberg; HE, Heulerberg; RR, Roter Rain; WS, Weiler-Schlätterle
In our study, we consequently applied a paired study design in all study sites (source vs. restored subpopulations present at each study site). This means that we have strictly chosen sites where restored and source grasslands were simultaneously present. All subpopulation pairs in our study were adjacent, without any barriers between them. Zoochory was the most probable dispersal modus, though dispersal by gravity was also possible. Grazing occurred sporadically during regeneration and contributed to the seed dispersal. We consequently use the term subpopulation with regard to the ancient and restored parts of the grasslands. Recently, both the source and restored subpopulations build one single population occupying particular study locations.
In our study, we investigated not only one but three study species. Moreover, we selected taxa with different pollination strategies, thus enabling broader conclusions regarding impact on genetic variation. Suitable study species were selected according to the following criteria: first, widespread species which occur in each part (source and restored) of the grasslands. This enabled us to consequently apply the paired design of source/restored grassland parts in all study sites. Second, taxa which do not regenerate from the soil seed bank, and, third, species which are likely to represent a gradient of genetic diversity, due to the different mating systems (mainly selfing, mixed-mating and outcrossing species). Agrimonia eupatoria and Knautia arvensis are known to only possess a transient soil seed bank. Campanula rotundifolia maintains a short-term persistent seed bank (Poschlod et al. 2003; Kleyer et al. 2008), i.e. the seeds should remain viable until at least the sixth germination season after dispersal (Walck et al. 2005). However, the restored sites were overgrown or afforested for at least ten years. Moreover, remaining individuals under the tree or scrub canopy did not contributed to the soil seed bank, because C. rotundifolia is a light demanding species (Ellenberg et al. 1991). This feature made it possible that exclusively genetic properties of subpopulations arisen from the recolonization process were assessed, without a contribution of propagules from the soil seedbank (compare Helsen et al. 2013). A. eupatoria is a perennial, mainly selfing species (Chrtek JR 2018) occurring commonly in dry grasslands, mesic pastures and meadows, but also in the forest fringe communities, heathlands and scrub. Seed dispersal takes places predominantly through zoochory (Fischer et al. 1996; Römermann et al. 2005). C. rotundifolia is a perennial, insect-pollinated, predominantly outcrossing species. Self-incompatibility was also reported (Chrtek JR 2018). Seeds are dispersed mainly due to gravity, but zoochory is also possible especially by grazing sheep (Fischer et al. 1996). C. rotundifolia occurs in similar habitats as A. eupatoria, but also in the vegetation of screes and walls. K. arvensis is a perennial, mainly outcrossing species (Chrtek JR 2018), occurring in meadows and mesic pastures, as well as in acidophilous grasslands and pine forests. Seeds are dispersed by gravity, but zoochory, especially transport by sheep flocks plays an important role (Fischer et al. 1996; Poschlod et al. 1998; Chytrý et al. 2021).
Each species was sampled at six study sites. At each site, we sampled subpopulations of study species in the historically old parts of grasslands which served as seed sources. Similarly, we evenly sampled restored subpopulations of the same species in grassland parts which developed after clearcutting. Geographic position was measured by means of global positioning system (GPS). In summer 2018, we collected fresh young leaves from eighteen individuals per subpopulation per species and dried them over silica gel. We collected the samples randomly within the whole subpopulation area, applying, if necessary, a threshold limit of three meters between the samples. We then compared the genetic variation of plants from subpopulations occurring in the ancient (source) and in the restored parts.
DNA ploidy level of the study species
Flow cytometrical analyses give information about the amount of DNA contained in nuclei of the study species. For all study species, polyploid complexes were described (Slavik 2000). Since possible mating barrier between different ploidy levels may affect genetic variation and thus bias our results, we assessed DNA ploidy levels at all sites for all study species. Flow cytometrical analyses (Suda et al. 2006) were carried out according to the two-step protocol after Dolezel et al. (2007). We analyzed individuals in all six study sites per species, for each species separately. Nuclei from the leaf tissue were isolated from three individuals per subpopulation (both source and restored) in each study location. From each subpopulation, samples were randomly chosen for analyses. In total, we analyzed 132 individuals of three study species. In one site (Eichhalde), subpopulations of A. eupatoria contained individuals with a double amount of DNA, presumably caused by occurrence of a congener octoploid species Agrimonia procera at the same grassland. We therefore analyzed further twelve individuals from both subpopulations and then decided to exclude the Eichhalde study location from further analyses. In the remaining five study locations, A. eupatoria occurred as a tetraploid. In C. rotundifolia, we revealed the presence of diploids. For K. arvensis, we detected occurrence of tetraploids. Thus, we excluded co-occurrence of several ploidy levels in our study system, for all three study species. For detailed description of the flow cytometric analyses see Supplementary.
AFLP analyses
The AFLPs yielded 149 analyzed individuals of A. eupatoria, 184 individuals of C. rotundifolia, and 175 individuals of K. arvensis, a total of 508 plants (Vos et al. 1995). This number is lower than that of sampled individuals due to the losses during the lab procedure. Genomic DNA for AFLPs was isolated from silica gel dried plant material following the CTAB protocol (Rogers and Bendich 1994), using adaptations as described in previous studies (Reisch et al. 2005). Concentration of DNA stock solutions was detected using a nano-spectrophotometer and these were subsequently diluted with water to a concentration of 7.8 ng/μl. AFLP analyses were performed following the standardized protocol of Beckmann Coulter as described formerly (Bylebyl et al. 2008; Reisch 2008). Selective amplifications were carried out using three primer combinations. For A. eupatoria we used following primer combinations: MseI-CTG/EcoRI-AAC (D2), MseI-CTG/EcoRI-AAG (D3), MseI-CTC/EcoRI-ACT (D4), for C. rotundifolia: MseI-CAC/EcoRI-ACC (D2), MseI-CAT/EcoRI-AGG (D3), MseI-CAT/EcoRI-ACT (D4), and for K. arvensis MseI-ACC/EcoRI-CAG (D2), MseI-AGG/EcoRI-CTT (D3), MseI-ACT/EcoRI-CTT (D4), respectively. The fluorescence-labeled samples were separated by capillary gel electrophoresis on an automated sequencer (GenomeLab GeXP, Beckman Coulter). Raw data were examined applying the GeXP software (Beckman Coulter), exported as synthetic gel files (.crv files) and analyzed using the software Bionumerics 4.6 (Applied Maths). Across all samples, each band was scored as either present or absent. For quality control of the AFLP procedure, a genotyping error rate was calculated (Bonin et al. 2004), which was 0.18% for A. eupatoria, 4.94% for C. rotundifolia and 1.84% for K. arvensis.
Statistical analyses
Based on the AFLP data, a binary matrix (0/1) was generated using the software Bionumerics 4.6 (Applied Maths). If present, fragments of a particular length were classified as 1 and in case of absence as 0. Using the 0/1 matrix, the percentage of polymorphic loci (PPL) was calculated across the whole dataset as a ratio ni/N, where ni is number of fragments containing polymorphism, N represents the full number of fragments (loci). Percentage of polymorphic loci (PPL), a frequency-based estimator of genetic diversity within subpopulations was computed using PopGene1.32 (Yeh et al. 1997). Genetic diversity was further estimated using the sample size independent formula based on the AMOVA measurements, SSWP/n-1 (sum of squares within subpopulations divided by the subpopulation sample size reduced by one). Thanks to the sample size-sensitivity of this genetic diversity measure, we were able to additionally compare subpopulations irrespective of whether or not the sample sizes were equal. We further calculated the level of rarity using the frequency-down-weighted marker values (DW), an index usually used as a measure of the rare fragment accumulation within a subpopulation (Schönswetter and Tribsch 2005), which is an equivalent to range-down-weighted species values (Crisp et al. 2001). The number of occurrences of each AFLP marker in a subpopulation was divided by the number of occurrences of that particular marker in the entire dataset. These values were eventually summed.
Bayesian cluster analysis was performed using the program STRUCTURE 2.3.4. (Pritchard et al. 2000, 2009). The population structure of the whole data set was inferred based on the clustering of individuals. An admixture model with correlated allele frequencies was assumed. A number of Markov Chain Monte Carlo simulations was set to 100000 and the burn-in period to 10000 iterations. Number of clusters K was set to 13 for C. rotundifolia and K. arvensis, and 11 for A. eupatoria. This corresponds to the number of subpopulations plus one. Analyses were run independently 20 times for each K to assess the amount of variation of the likelihood for each value of K. The best estimate of K for the given data set was specified according to the model, which gave a consistent results for all 20 runs (Kopelman et al. 2015). The program Harvester (Earl and vonHoldt 2012) was used to summarize results. An estimate of the posterior probability of the data Pr (X|K) for a particular K was calculated (Pritchard et al. 2000). To identify the real number of clusters K, ad hoc statistic ΔK was used which was calculated as a second order rate of change of probability of the data Pr (X|K) with respect to K (Evanno et al. 2005).
Genetic differentiation between subpopulations and regions (groups of subpopulations) was detected using analysis of molecular variance AMOVA (Excoffier et al. 1992) implemented in the program GenAlex 6.41 (Peakall and Smouse 2006). Computations based on pairwise Euclidian distances between samples. Significance values related to variance components rest upon 999 permutations of individuals supposing no genetic structure. We tested partitioning of genetic variation within subpopulations, between subpopulations and between regions, applying two-level and three-level hierarchical AMOVAs.
A multivariate analysis (principal coordinate analysis, PCoA) was calculated separately for each study species and plotted using GenAlEx 6.41 (Peakall and Smouse 2006). Computations based on Jaccard similarities between individuals, CJ = a / [a + b + c], where a is the number of fragments shared between two individuals and b and c are the numbers of fragments present in only one individual.
Results
Genetic diversity of source and restored subpopulations
For A. eupatoria, AFLP genotyping of 149 plants resulted in 146 fragments, of which 58% were polymorphic. The percentage of polymorphic loci (PPL) in source subpopulations ranged from 26.03 to 31.51% (mean 27.63%), in restored subpopulations between 27.40 and 32.88 (mean 30.03). The frequency-down-weighted marker value (DW) ranged between 7.93 and 8.86 (mean 8.44) in the source subpopulations, in restored subpopulations from 8.16 to 9.24 (mean 8.56). In the source subpopulations, SSWP/n−1 (sum of squares within a subpopulation divided by n−1, where n is a subpopulation size) ranged from 5.17 to 7.41 (mean 6.42). In the restored subpopulations, values of SSWP/n−1 were slightly but significantly higher (mean 7.14; p = 0.03) (Table 1a). We observed, however, no significant differences in PPL and DW.
In C. rotundifolia, AFLP analysis of 184 plants yielded 179 fragments, of which 90.5% were polymorphic. The percentage of polymorphic loci in source subpopulations varied between 62.27 and 74.30% (mean 68.24%), in restored subpopulations between 68.16 and 73.74% (mean 70.67%). The frequency-down-weighted marker value (DW) in source subpopulations ranged between 12.61 and 14.16 (mean 13.39), in restored subpopulations between 13.40 and 14.04 (mean 13.61). SSWP/n−1 in source subpopulations ranged from 20.08 to 23.79 (mean 21.86), in restored subpopulations from 20.70 to 22.99 (mean 22.10). Genetic diversity in restored subpopulations of C. rotundifolia was slightly but not significantly higher than in source subpopulations (Table 1b).
For K. arvensis, AFLP analysis of 175 plants yielded 146 fragments, 82% of them were polymorphic. The percentage of polymorphic loci in source subpopulations ranged between 47.95% and 57.53% (mean 52.62%), in restored subpopulations between 42.47% and 62.33% (mean 50.34%). The frequency-down-weighted marker value (DW) in source subpopulations varied between 9.75 and 10.85 (mean 10.09), in restored subpopulations between 9.66 and 10.28 (mean 9.91). SSWP/n−1 in source subpopulations ranged from 12.03 to 14.13 (mean 13.20) in restored subpopulations from 10.55 to 14.98 (mean 12.41). The difference between source and restored subpopulations was not significant (Table 1c).
Genetic differentiation between subpopulations
In the Bayesian cluster analysis, individuals of all three species were assigned to two groups. However, we did not detect any population grouping according to the study location or according to the feature source/restored (Fig. 2). For each species, outputs for K = 2 of all twenty iterations were equal. For A. eupatoria, ∆K was 170.68; for C. rotundifolia, ∆K was 563.89; for K. arvensis, ∆K was 110.71.

Bayesian cluster analysis for a A. eupatoria based on 146 AFLP fragments. b C. rotundifolia based on 179 AFLP fragments. c K. arvensis based on 146 AFLP fragments. Populations of all three species were assigned to two groups
The PCoA did not reveal any separation of source and restored subpopulations for all study species A. eupatoria, C. rotundifolia and K. arvensis. Source and restored subpopulations were admixed and no groups could be identified (Fig. 3a, b, c). Similarly, no grouping according to the study locations was detected.

Results of the PCoA (principal coordinates analysis) based on Jaccard similarities. Black circles represent individuals of source subpopulations. Hollow circles show restored subpopulations. Individuals of source and restored subpopulations were mixed in all three species. a A. eupatoria: axis 1 explained 25.52% of variance in the data set. axis 2 explained 19.98%, respectively. b C. rotundifolia: axis 1 explained 37.12% of variance and axis 2 explained 16.58% of variance in the dataset. c K. arvensis: axis 1 explained 28.22% of variance, axis 2 explained 21.12%
Applying AMOVA, we detected low overall genetic differentiation between subpopulations of the study species. Genetic differentiation (ΦPT) between all subpopulations of A. eupatoria was 0.08 (Table 2a). Differentiation between all source subpopulations was 0.10, and between all restored subpopulations 0.06. Hierarchical partitioning of molecular variance revealed that its largest amount was comprised within subpopulations (92%). We detected only a weak variation between subpopulations within the groups of the source and the group of restored subpopulations (8%), and zero genetic variation between the two groups of the source and restored subpopulations.
For C. rotundifolia, ΦPT between all subpopulations was 0.04 (Table 2b). Differentiation between all source subpopulations was 0.04 and among all restored subpopulations 0.05. We detected no genetic differentiation between the groups of the source and restored subpopulations.
For K. arvensis, ΦPT between all subpopulations was 0.03 (Table 2c). Genetic variation between all source subpopulations was 0.02 and between all restored subpopulations 0.04. Comparably to A. eupatoria and C. rotundifolia, no genetic variation between the two groups of the source and the restored subpopulations was be detected.
Discussion
In our study, we detected apparent differences in genetic variation between the study species A. eupatoria, C. rotundifolia and K. arvensis. The species followed genetic variation patterns observed previously for outcrossing, mixed-mating and selfing taxa. The first, A. eupatoria, is a highly selfing species and showed the lowest levels of genetic diversity and the highest levels of genetic differentiations. The second, C. rotundifolia, which is predominantly outcrossing displayed the highest genetic diversity and low differentiation levels. The last, K. arvensis, is a mixed-mating but predominantly outcrossing species, which showed intermediate genetic diversity levels as well as low genetic differentiation (Table 1, Table 2). A significant, profound impact of the mating system on genetic variation within and among populations has been reported previously (Reisch and Bernhardt-Römermann 2014). Outcrossing and mixed-mating species display considerably higher levels of variation within, and lower variation among populations. And vice versa, selfing species show conspicuously lower levels of genetic variation within populations, and stronger population differentiation than outcrossing or mixed-mating taxa. Thus, our results support the previous observation that mating system has a significant impact on the patterns of genetic variation in plant species.
Apart from the mating system, genetic diversity and differentiation of plant populations can also strongly be affected by the restoration process, specifically by founder events during the colonization process (Vandepitte et al. 2012), because the number of propagules arriving in a restored site may be limited due to the restricted availability of source populations in the close vicinity (Willems and Bik 1998; Poschlod et al. 2005; Prach et al. 2015) or, by the lacking effective dispersal vector such as grazing sheep flocks (Fischer et al. 1996; Fischer et al. 1996; Römermann et al. 2005).
Moreover, only a limited number of propagules succeeds to establish as a mature plant for several reasons (lack of suitable microsites, biotic and abiotic conditions and their stochasticity predation etc.) (Roberts and Feast 1973; Matos and Watkinson, 1998; Goldberg and Werner 1983). Consequently, genetic diversity of restored populations may only partially represent the entire diversity of extant source stands.
However, we detected no significant decrease of genetic diversity in restored subpopulations of A. eupatoria, C. rotundofolia and K. arvensis, in comparison with the source subpopulations. All measures of genetic diversity, i.e. SSWP, PPL as well as DW were marginal and not significant, or showed only negligible differences between the source and restored subpopulations, respectively (Table 1).
Previous studies reported decreased genetic diversity in populations founded through natural colonization in comparison with long-standing, established populations (Bishop 1996; Jacquemyn et al. 2009; Vandepitte et al. 2012), but this was not always the case. In accordance with our results, Vandepitte et al. (2007), Van Looy et al. (2009) as well as Ilves et al. (2015) and Helsen et al. (2013) revealed that genetic diversity in newly founded populations was not lowered. Two main factors may have played the major role in the process of enhancing genetic diversity and unification of the restored and source subpopulations. First, the spatial proximity of ancient grassland parts containing source subpopulations, which have served as a propagule and pollen donors. Occasionally grazing sheep flocks served additionally as mobile dispersal vectors accelerating the natural dispersal process via epizoochory (Rico et al. 2014; Lehmair et al. 2020). All three study species are insect pollinated, and, therefore, the physical closeness was a crucial factor for insects to reach newly established subpopulations in the restored parts (Zurbuchen et al. 2010). Second, restored fragments not only directly adjoined ancient source grassland parts. Beyond that, several grassland fragments were detected in the close surroundings within a radius of 700 m from the restored grassland. These grasslands could have served as additional propagule sources which might have contibuted to the unreduced genetic diversity in the restored subpopulations, due to gene flow (compare Slatkin 1987; Wade and McCauley 1988). Here, not only distance, but also possible dispersal barriers and dispersal vectors could have played a role; however, this would require a separate study.
Thus, the gene flow played a key role in the unification process between the source and restored subpopulations and contributed to the enhancement of genetic diversity in the restored stands (Dlugosch and Parker 2008). This underpins our assessment that colonization from nearby sources can be a reasonable approach to restore populations without loss of local populations´ genetic diversity properties.
Interestingly, we detected slightly but significantly higher genetic diversity levels in restored subpopulations in A. eupatoria, in comparison with its source subpopulations. This result was not massively surprising because of the seed morphology, i.e. hooks on the surface of A. eupatoria fruits, which can effectively be dispersed by zoochory. One of the most common species found in the sheep fur were fruits of A. eupatoria (Fischer et al. 1996). Comparable results were reported for newly founded populations of Geum urbanum, which has a similar fruit morphology (Vandepitte et al. 2007). However, this fact alone cannot explain why K. arvensis, similarly to A. eupatoria, did not show the same pattern of higher genetic diversity in restored stands. According to Hintze et al. (2013), K. arvensis possess even higher dispersal abilities than Agrimonia.
Most probably, numerous calcareous grassland fragments in the close surroundings could have served as supplementary propagule sources [compare the “migrant pool” colonization model described by Wade and McCauley (1988)]. Presumably both factors, i.e. good dispersal abilities and multiple seed sources in the close surroundings might have contributed to the increased genetic diversity levels in the restored subpopulations of A. eupatoria.
Restoration through colonization may not merely decrease genetic diversity within, but also enhance genetic divergence between populations. Low numbers of arriving propagules, and, generally, low levels of gene flow as well as limited establishment rates on restored sites may jointly increase genetic differentiation between source and restored grassland parts (Vandepitte et al. 2012). In this study, genetic differentiation between A. eupatoria, C. rotundifolia and K. arvensis subpopulations was, however, lower than previously reported for common species, and also lower than that detected for particular groups according to the mating system types (Reisch and Bernhardt-Römermann 2014). Moreover, and this was of even greater importance, genetic differentiation between the group of the source and the restored subpopulations was lower than differentiation between the source subpopulations. Lower levels of differentiation between restored subpopulations and their genetic similarity to the source subpopulations can be attributed, first, to high levels and long lasting gene flow between the source and restored grassland parts. Second, the entire vegetation layer removal in the course of restoration apparently reduced the density-dependent mortality of colonists in the restored sites and, thus, enabled population founding and growth, whereby the space in ancient (source) grasslands was already densely occupied (Helsen et al. 2013). Our study showed that any apparent and long-lasting founder effect as a consequence of the spontaneous recolonization process of the three common grassland species was avoided.
Conclusions
The results of our study provide evidence that, at least under the settings of the underlying restoration project, grassland restoration through recolonization neither necessarily induces erosion of genetic diversity, nor enhanced levels of genetic differentiation between the restored and source subpopulations. Hence we can deduce that any apparent founder or bottleneck effect due to colonization events after clear-cutting were avoided. Restored subpopulations may further act as valuable diaspore sources in landscapes containing endangered calcareous grassland fragments.
Potentially, this approach may even induce an increase in genetic diversity relative to the nearby source stands. This supports our perception that scrub and woodland removal and subsequent recolonization are helpful tools to create genetically variable, non-differentiated restored subpopulations of common species, maintaining their local genetic patterns. These characteristics are necessary prerequisites for a long-term, sustainable persistence of the restored subpopulations in landscapes with calcareous grasslands.
We assume that for the restoration success, the direct spatial proximity was particularly important. This enabled the propagule availability from the sources and subsequent gene flow uniting the source and restored stands via further seed dispersal and pollen transfer. However, species with low dispersal abilities, as well as rare taxa should be analyzed separately due to different levels of gene flow. Moreover, in future studies, it would definitely be interesting to look at the population genetics of natural regeneration sites that are not directly adjacent to source populations.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Aavik T, Helm A (2018) Restoration of plant species and genetic diversity depends on landscape-scale dispersal. Restor Ecol. https://doi.org/10.1111/rec.12634
Albert Á-J, Mudrák O, Jongepierová I, Fajmon K, Frei I, Ševčíková M, Klimešová J, Doležal J (2019) Grassland restoration on ex-arable land by transfer of brush-harvested propagules and green hay. Agric Ecosyst & Environ 272:74–82
Bakker JP, Poschlod P, Strykstra RJ, Bekker RM, Thompson K (1996) Seed banks and seed dispersal: important topics in restoration ecology. Acta Botanica Neerl 45(4):461–490
Basey A, Fant J, Kramer A (2015) Producing native plant materials for restoration: 10 rules to collect and maintain genetic diversity. Native Plants J 16:37–53
Bishop JG (1996) Demographic and population genetic variation during colonization by the herb Lupinus lepidus on Mount St. Helens. https://www.proquest.com/openview/c3a4d3e7c59cd3c507920d3255e1cab8/1?pq-origsite=gscholar&cbl=18750&diss=y Accessed 12 May 2021
Bisteau E, Mahy G (2005) Vegetation and seed bank in a calcareous grassland restored from a pinus forest. Appl Veg Sci. https://doi.org/10.1111/j.1654-109X.2005.tb00642.x
von Blanckenhagen B, Poschlod P (2005) Restoration of calcareous grasslands: the role of the soil seed bank and seed dispersal for recolonisation processes. Biotechnol Agron Soc Environ 9:143–149
Bonin A, Bellemain E, Eidesen P, Pompanon F, Brochmann C, Taberlet P (2004) How to track and assess genotyping errors in population genetics studies. Mol Ecol 13:3261–3273
Bucharova A, Bossdorf O, Hoelzel N, Kollman J, Prasse R, Durka W (2019) Mix and match: regional admixture provenancing strikes a balance among different seed-sourcing strategies for ecological restoration. Conserv Genet 20(1):7–17
Bylebyl K, Poschlod P, Reisch C (2008) Molecular Ecology 17(14):3379-3388. https://doi.org/10.1111/j.1365-294X.2008.03836.x
Chrtek J. JR (2018) Generative reproduction type. www.pladias.cz. Accessed 22 December 2021
Chytrý M, Danihelka J, Kaplan Z, Wild J, Holubová D, Novotný P, Řezníčková M, Rohn M, Dřevojan P, Grulich V, Klimešová J, Lepš J, Lososová Z, Pergl J, Sádlo J, Šmarda P, Štěpánková P, Tichý L, Axmanová I, Bartušková A, Blažek P, Chrtek J, Fischer FM, Guo WY, Herben T, Janovský Z, Konečná M, Kühn I, Moravcová L, Petřík P, Pierce S, Prach K, Prokešová H, Štech M, Těšitel J, Těšitelová T, Večeřa M, Zelený D, Pyšek P (2021) Pladias Database of the Czech flora and vegetation. Preslia 93(1):1–87. https://doi.org/10.23855/preslia.2021.001
Crisp MD, Laffan S, Linder HP, Monro A (2001) Endemism in the Australian Flora. J Biogeogr. https://doi.org/10.1046/j.1365-2699.2001.00524.x
Dalrymple SE, Banks E, Stewart GB, Pullin AS (2012) A meta-analysis ofthreatened plant reintroductions from across the globe. In: Maschinski J, Haskins KE, Raven PH (eds) Plant reintroduction in a changing climate. Island Press/Center for Resource Economics, Washington, pp 31–50
DiLeo MF, Rico Y, Boehmer HJ, Wagner HH (2017) An ecological connectivity network maintains genetic diversity of a flagship wildflower, Pulsatilla vulgaris. Biol Conserv 212:12–21
Dlugosch KM, Parker IM (2008) Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol Ecol 17(1):431–449
Dolezel J, Binorava P, Lucretti S (1989) Analysis of nuclear DNA content in plant cells by flow cytometry. Biol Plant 31(2):113–120
Dolezel J, Greilhuber J, Suda J (2007) Estimation of nuclear DNA content in plants using flow cytometry. Nat publ Group 2(9):2233–2244
Durka W, Bossdorf O, Bucharova A, Frenzel M, Hermann J-M, Hoelzel N, Kollmann J, Michalski S (2019) Regionales saatgut von wiesenpflanzen: genetische unterschiede, regionale anpassung und interaktion mit insekten. Natur und Landschaft 94:146–153
Durka W, Michalski SG, Berendzen KW, Bossdorf O, Bucharova A, Hermann JM, Hölzel N, Kollmann J, Wan S (2017) Genetic differentiation within multiple common grassland plants supports seed transfer zones for ecological restoration. J Appl Ecol 54(1):116–126. https://doi.org/10.1111/1365-2664.12636
Earl D, vonHoldt B (2012) Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ellenberg H, Weber HE, Düll R, Wirth V, Werner W, Paulißen D (1991) Zeigerwerte von pflanzen in mitteleuropa. Scr Geol 18:1–248
European Community (1992) The Habitats Directive. 92/43/92/43/EEC of the European Community. https://ec.europa.eu/environment/nature/legislation/habitatsdirective/index_en.htm. Accessd 16 April 2021
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial dna restriction data. Genetics 131(2):479–491
Fischer SF, Poschlod P, Beinlich B (1996) Experimental studies on the dispersal of plants and animals on sheep in calcareous grasslands. J Appl Ecol 33:1206–1222
Frankham R, Ballou J, Eldridge M, Lacy R, Ralls K, Dudash M, Fenster C (2011) Predicting the probability of outbreeding depression. Conserv Biol. https://doi.org/10.1111/j.1523-1739.2011.01662.x
Franklin IR (1980) Evolutionary change in small populations. In: Soulé ME, Wilcox BA (eds) Conservation biology, an evoluitonary and ecological perspective. Sinauer Associates Inc., Sunderland, pp 135–150
Franklin IR, Frankham R (1998) How large must populations be to retain evolutionary potential? Animal Conserv 1:69–70
Guerrant E Jr, Kaye T (2007) Reintroduction of rare and endangered plants: common factors, questions and approaches. Aust J Bot 55:362
Helsen K, Jacquemyn H, Hermy M, Vandepitte K, Honnay O (2013) Rapid buildup of genetic diversity in founder populations of the gynodioecious plant species origanum vulgare after semi-natural grassland restoration. PLoS One. https://doi.org/10.1371/journal.pone.0067255
Honnay O, Jacquemyn H, Van Looy K, Vandepitte K, Breyne P (2009) Temporal and spatial genetic variation in a metapopulation of the annual Erysimum cheiranthoides on stony river banks. J Ecol 97:131–141
Höfner J, Klein-Raufhake T, Lampei C, Mudrak O, Bucharova A, Durka W (2021) Populations restored using regional seed are genetically diverse and similar to natural populations in the region. J Appl Ecol. https://doi.org/10.1111/1365-2664.14067
Huber S, Huber B, Stahl S, Schmid C, Reisch C (2017) Species diversity of remnant calcareous grasslands in south eastern Germany depends on litter cover and landscape structure. Acta Oecol 83:48–55
Hufford KM, Mazer SJ (2003) Plant ecotypes: genetic differentiation in the age of ecological restoration. Trends Ecol Evol 18:147–155
Ilves A, Metsare M, Tali K, Kull T (2015) The impact of recent colonization on the genetic diversity and fine-scale genetic structure in Orchis militaris (L.). Plant Syst Evol 301(7):1875–1886
Jacquemyn H, Vandepitte K, Roldán-Ruiz I, Honnay O (2009) Rapid loss of genetic variation in a founding population of Primula elatior (Primulaceae) after colonization. Ann Bot 103:777–783
Johanidesová E, Fajmon K, Jongepierová I, Prach K (2015) Spontaneous colonization of restored dry grasslands by target species: restoration proceeds beyond sowing regional seed mixtures. Grass Forage Sci 70(4):631–638
Kaulfuß F, Reisch C (2021) Restoration of species-rich grasslands by transfer of local plant material and its impact on species diversity and genetic variation-Findings of a practical restoration project in southeastern Germany. Ecol Evol 11(18):12816–33
Kaulfuß F, Rosbakh S, Reisch C (2022) Grassland restoration with local seed mixture. new evidence from a practical 15 years old study (subm.). App Veg Sci. 25(2):e12652
Kiefer S (1998) Untersuchungen zur Wiederherstellung brachgefallener oder aufgeforsteter Kalkmagerrasen. PhD. Thesis. Heimbach, Ostfildern
Kiefer S, Poschlod P (1996) Restoration of fallow or afforested calcareous grasslands by clear-cutting. In: Settele J, Margules C, Poschlod P, Henle K (eds) Species survival in fragmented landscapes. Springer, Dordrecht, pp 209–218
Kiehl K, Kirmer A, Donath TW, Rasran L, Hölzel N (2010) Species introduction in restoration projects - Evaluation of different techniques for the establishment of semi-natural grasslands in Central and Northwestern Europe. Basic Appl Ecol 11(4):285–299
Kirmer A, Baasch A, Tischew S (2012) Sowing of low and high diversity seed mixtures in ecological restoration of surface mined-land. Appl Veg Sci 15:198–207
Kirmer A, Tischew S, Ozinga WA, von Lampe M, Baasch A, van Groenendael JM (2008) Importance of regional species pools and functional traits in colonization processes: predicting re-colonization after large-scale destruction of ecosystems. J Appl Ecol 45(5):1523–1530
Kiss R, Deák B, Tóthmérész B, Miglécz T, Tóth K, Török P, Lukács K, Godó L, Körmöczi Z, Radócz S, Kelemen A, Sonkoly J, Kirmer A, Tischew S, Švamberková E, Valkó O (2021) Establishment gaps in species-poor grasslands: artificial biodiversity hotspots to support the colonization of target species. Restor Ecol. https://doi.org/10.1101/2020.01.23.916155
Kleyer M, Bekker R, Knevel IC, Bakker J, Thompson K, Sonnenschein M, Poschlod P, Van Groenendael JM, Klimes L, Klimesova J, Klotz S, Rusch GM, Hermy M, Adriaens D, Boedeltje G, Bossyut B, Dannemann A, Endels P, Götzenberger L, Hodgson JG, Jackel A-K, Kühn I, Kunzmann D, Ozinga WA, Römermann C, Stadler M, Schlegelmilch J, Steendam HJ, Tackenberg O, Wilmann B, Cornelissen J, Eriksson O, Garnier E, Peco B (2008) The LEDA Traitbase. A database of life-history traits of Northwest European flora. J Ecol 96(6):1266–1274
Kopelman N, Mayzel J, Jakobsson M, Rosenberg N, Mayrose I (2015) CLUMPAK: a program for identifying clustering modes and packaging population structure inference s across K. Mol Ecol Resour. https://doi.org/10.1111/1755-0998.12387
Kövendi-Jakó A, Halassy M, Csecserits A, Hülber K, Szitár K, Wrbka T, Török K (2019) Three years of vegetation development worth 30 years of secondary succession in urban-industrial grassland restoration. Appl Veg Sci 22(1):138–149
Lehmair TA, Pagel E, Poschlod P, Reisch C (2020) Surrounding landscape structures, rather than habitat age, drive genetic variation of typical calcareous grassland plant species. Landsc Ecol. https://doi.org/10.1007/s10980-020-01120-7
McKay J, Christian C, Harrison S, Rice K (2005) ‘“How local is local?”’. A review of practical and conceptual Issues in the genetics of restoration. Restor Ecol 13(3):432–440
Mijnsbrugge KV, Bischoff A, Smith B (2010) A question of origin: where and how to collect seed for ecological restoration. Basic Appl Ecol 11(4):300–311
Montalvo A, Ellstrand N (2001) Nonlocal transplantation and outbreeding depression in the subshrub Lotus scoparius (Fabaceae). Am J Bot. https://doi.org/10.2307/2657017
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution. https://doi.org/10.1111/j.1558-5646.1975.tb00807.x
Pärtel M, Kalamees R, Zobel M, Rosén E (1998) Restoration of species-rich limestone grassland communities from overgrown land: the importance of propagule availability. Ecolo Eng 10(3):275–286
Peakall R, Smouse P (2006) genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295
Poschlod P, Kiefer S, Traenkle U, Bonn S (1998) Plant species richness in calcareous grasslandsas affected by dispersability in space and time. Appl Veg Sci. https://doi.org/10.2307/1479087
Poschlod P, Kleyer M, Jackel A-K, Dannemann A, Tackenberg O (2003) BIOPOP—a database of plant traits and internet application for nature conservation. Folia Geobot Phytotax 38(3):263–271
Poschlod P, Tackenberg O, Bonn S (2005) Plant dispersal potential and its relation to species frequency and coexistence. In: van der Maarel E (ed) Vegeation Ecology. John Wiley & Sons, New York, pp 147–171
Poschlod P Tränkle U Böhmer J Rahmann H (1997) Steinbrüche und Naturschutz. Sukzession und Renaturierung. ecomed, Landsberg
Poschlod P, WallisDeVries MF (2002) The historical and socioeconomic perspective of calcareous grasslands—lessons from the distant and recent past. Biol Conserv 104(3):361–376
Prach K, Hobbs RJ (2008) Spontaneous succession versus technical reclamation in the restoration of disturbed sites. Restor Ecol. https://doi.org/10.1111/j.1526-100X.2008.00412.x
Prach K, Karešová P, Jírová A, Dvořáková H, Konvalinková P, Řehounková K (2015) Do not neglect surroundings in restoration of disturbed sites. Restor Ecol 23(3):310–314
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Pritchard J Wen X Falush D (2009) Documentation for structure software. Version 2.3 http://web.stanford.edu/group/pritchardlab/software/structure_v.2.3.1/documentation.pdf
Pullin AS, Báldi A, Can OE, Dieterich M, Kati V, Livoreil B, Lövei G, Mihók B, Nevin O, Selva N, Sousa-Pinto I (2009) Conservation focus on Europe: major conservation policy issues that need to be informed by conservation science. Conserv Biol 23(4):818–824
Raffl C, Schönswetter P, Erschbarmer B (2006) ‘Sax-sess’- genetics of primary succession in a pioneer species on two parallel glacier forelands. Mol Ecol 15(9):2433–2440. https://doi.org/10.1111/j.1365-294X.2006.02964.x
Redhead JW, Sheail J, Bullock JM, Ferreruela A, Walker KJ, Pywell RF (2014) The natural regeneration of calcareous grassland at a landscape scale: 150 years of plant community re-assembly on Salisbury Plain. UK. Appl Veg Sci 17(3):408–418
Řehounková K, Jongepierová I, Šebelíková L, Vítovcová K, Prach K (2021) Topsoil removal in degraded open sandy grasslands: can we restore threatened vegetation fast? Restor Ecol 29:e13188
Reisch C (2008) Glacial history of Saxifraga paniculata (Saxifragaceae): molecular biogeography of a disjunct arctic-alpine species from Europe and North America. Biol. J Linn Soc 93(2):385–398. https://doi.org/10.1111/j.1095-8312.2007.00933.x
Reisch C, Anke A, Röhl M (2005) Molecular variation within and between ten populations of Primula farinosa (Primulaceae) along an altitudinal gradient in the northern Alps. Basic Appl Ecol 6:35–45
Reisch C, Bernhardt-Römermann M (2014) The impact of study design and life history traits on genetic variation of plants determined with AFLPs. Plant Ecol 215(12):1493–1511
Rejmanek M van Katwyk KP (2005) Old field succession: a bibliographic review (1901–1991)
Rico Y, Holderegger R, Boehmer HJ, Wagner HH (2014) Directed dispersal by rotational shepherding supports landscape genetic connectivity in a calcareous grassland plant. Mol Ecol 23(4):832–842
Rogers S, Bendich A (1994) Extraction of total cellular DNA from plants, algae and fungi. In: Gelvin S, Schilperoort R (eds) Plant molecular biology manual, 2nd edn. Springer, Dordrecht, pp 183–190
Römermann C, Tackenberg O, Poschlod P (2005) How to predict attachment potential of seeds to sheep and cattle coat from simple morphological seed traits. Oikos. https://doi.org/10.1111/j.0030-1299.2005.13911.x
Schönswetter P, Tribsch A (2005) Vicariance and dispersal in the alpine perennial Bupleurum stellatum L (Apiaceae). Taxon. https://doi.org/10.2307/25065429
Slatkin M (1977) Gene flow and genetic drift in a species subject to frequent local extinctions. Theor Pop Biol 12(3):253–262
Slatkin M (1977) Gene flow and genetic frequent drift in a species subject to local extinctions. Theor Pop Biol 12(3):253–262
Slatkin M (1987) Gene flow and the geographic structure of natural populations. Science 236:787–792
Slavík B, Chrtek J jun, Štěpánková J [eds] (2000) Květena České republiky. 6. Academia, Praha
Suda J, Krahulcova A, Travnicek P, Krahulec F (2006) Ploidy level versus DNA ploidy level: an appeal for consistent terminology. Taxon. https://doi.org/10.2307/25065591
Török P, Vida E, Deak B, Lengyel S, Tothmeresz B (2011) Grassland restoration on former croplands in Europe: an assessment of applicability of techniques and costs. Biodivers Conserv 20:3211–2332
van Looy K, Jacquemyn H, Breyne P, Honnay O (2009) Effects of flood events on the genetic structure of riparian populations of the grassland plant Origanum vulgare. Biol Conserv 142(4):870–878
Vandepitte K, Gristina AS, de Hert K, Meekers T, Roldán-Ruiz I, Honnay O (2012) Recolonization after habitat restoration leads to decreased genetic variation in populations of a terrestrial orchid. Mol Ecol 21:4206–4215
Vandepitte K, Jacquemyn H, Roldán-Ruiz I, Honnay O (2007) Landscape genetics of the self-compatible forest herb Geum urbanum: effects of habitat age, fragmentation and local environment. Mol Ecol 16(19):4171–4179
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Friters A, Pot J, Paleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucl Acids Res 23:4407–4414
Wade MJ, McCauley D (1988) Extinction and recolonization: their effects on the genetic differentiation of local populations. Evolution. https://doi.org/10.1111/j.1558-5646.1988.tb02518.x
Walck JL, Baskin JM, Baskin CC, Hidayati SN (2005) Defining transient and persistent seed banks in species with pronounced seasonal dormancy and germination patterns. Seed Sci Res 15(3):189–196. https://doi.org/10.1079/SSR2005209
WallisDeVries MF, Poschlod P, Willems JH (2002) Challenges for the conservation of calcareous grasslands in northwestern Europe: integrating the requirements of flora and fauna. Biol Conserv 104(3):265–273
Whitlock MC, McCauley DE (1990) Some population genetic consequences of colony formation and extinction genetic correlations within founding groups. Evolution 44(7):1717–1724
Willems JH, Bik L (1998) Restoration of high species density in calcareous grassland: the role of seed rain and soil seed bank. Appl Veg Sci. https://doi.org/10.2307/1479088
Yang S, Bishop JG, Webster MS (2008) Colonization genetics of an animal-dispersed plant (Vaccinium membranaceum) at Mount St Helens Washington. Mol Ecol 17(3):731–740. https://doi.org/10.1111/j.1365-294X.2007.03625.x
Yeh F, Yang R-C, Boyle T, Ye ZH, Mao JX (1997) POPGENE, the user-friendly shareware for population genetic analysis. Biol Biotechnol Centre Univ Atlanta Edmonton Canada 10:295–301
Zurbuchen A, Landert L, Klaiber J, Müller A, Hein S, Dorn S (2010) Maximum foraging ranges in solitary bees: only few individuals have the capability to cover long foraging distances. Biol Conserv 143(3):669–676
Acknowledgements
The authors are obliged to the Regional council of Baden-Württemberg in Stuttgart for the permission to enter protected areas of Haarberg-Wasserberg, Eichhalde, Heulerberg and Roter Rain. We are acknowledged to Patricia Krickl, Helene Kleijn, Marie van Vught, Julia Eberl and Petr Kalík, who kindly helped to collect plant material. HK, MvV and JE many thanks for their work in Lab. We further thank Petra Schitko for her assistance in lab, as well as Sabine Fischer for her support with maps. Many thanks go out to Christoph Oberprieler for his generous support with flow cytometry. Two anonymous referees provided thoughtful comments on an earlier manuscript. We are also oblidged to František Krahulec for numerous discussions and valuable comments on methodological approach.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
CR and PP conceived the study. All authors contributed to the study design. Material preparation and data collection, as well as data analyses were performed by KI. The first draft of the manuscript was written by KI and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Authors declare that they have no competing interests.
Additional information
Communicated by Jens Dauber.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iberl, K., Poschlod, P. & Reisch, C. Restoration of calcareous grasslands by natural recolonization after forest clearing and its impact on the genetic variation of three common herb species. Biodivers Conserv 32, 671–690 (2023). https://doi.org/10.1007/s10531-022-02518-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-022-02518-2