
Abstract
In genetics study, the genotypes or phenotypes can be missing due to various reasons. In this paper, the impact of missing genotypes is investigated for high resolution combined linkage and association mapping of quantitative trait loci (QTL). We assume that the genotype data are missing completely at random (MCAR). Two regression models, “genotype effect model” and “additive effect model”, are proposed to model the association between the markers and the trait locus. If the marker genotype is not missing, the model is exactly the same as those of our previous study, i.e., the number of genotype or allele is used as weight to model the effect of the genotype or allele in single marker case. If the marker genotype is missing, the expected number of genotype or allele is used as weight to model the effect of the genotype or allele. By analytical formulae, we show that the “genotype effect model” can be used to model the additive and dominance effects simultaneously, and the “additive effect model” can only be used to model the additive effect. Based on the two models, F-test statistics are proposed to test association between the QTL and markers. The non-centrality parameter approximations of F-test statistics are derived to calculate power and to compare power, which show that the power of the F-tests is reduced due to the missingness. By simulation study, we show that the two models have reasonable type I error rates for a dataset of moderate sample size. However, the type I error rates can be very slightly inflated if all individuals with missing genotypes are removed from analysis. Hence, the proposed method can help to get correct type I error rates although it does not improve power. As a practical example, the method is applied to analyze the angiotensin-1 converting enzyme (ACE) data.






Similar content being viewed by others

References
Abecasis GR, Cardon LR, Cookson WOC (2000a). A general test of association for quantitative traits in nuclear families. Am J Hum Genet 66:279–292
Abecasis GR, Cherny SS, Cookson WOC, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101
Abecasis GR, Cookson WOC, Cardon LR (2000b). Pedigree tests of linkage disequilibrium. Eur J Hum Genet 8:545–551
Allison DB (2001) Joint tests of linkage and association for quantitative traits. Theor Popul Biol 60:239–251
Almasy L, Blangero J (1998) Multipoint quantitative trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211
Almasy L, Williams JT, Dyer TD, Blangero J (1999) Quantitative trait locus detection using combined linkage/disequilibrium analysis. Genet Epidemiol 17(Suppl 1):S31–S36
Amos CI (1994) Robust variance-components approach for assessing linkage in pedigrees. Am J Hum Genet 54:534–543
Amos CI, Elston RC (1989) Robust methods for the detection of genetic linkage for quantitative data from pedigrees. Genet Epidemiol 6:349–360
Boerwinkle E, Chakraborty E, Sing CF (1986) The use of measured genotype information in the analysis of quantitative phenotype in man. I. models and analytical methods. Ann Hum Genet 50:181–194
Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics, 4th edn. Longman, London
Fan RZ, Jung JS (2003) High resolution joint linkage disequilibrium and linkage mapping of quantitative trait loci based on sibship data. Hum Hered 56:166–187
Fan RZ, Jung JS, Jin J (2006) High resolution association mapping of quantitative trait loci, a population based approach. Genetics 172:663–686
Fan RZ, Spinka C, Jin L, Jung JS (2005) Pedigree linkage disequilibrium mapping of quantitative trait loci. Eur J Hum Genet 13:216–231
Fan RZ, Xiong MM (2003) Combined high resolution linkage and association mapping of quantitative trait loci. Eur J Hum Genet 11:125–137
Farrall M, Keavney B, MckKenzie CA, Delèpine M, Matsuda F, Lathrop GM (1999) Fine mapping of an ancestral recombination break-point in DCP1. Nat Genet 23:270–271
Feingold E (2002) Invited editorial: regression-based quantitative-trait-locus mapping in the 21st century. Am J Hum Genet 71:217–222
Fulker DW, Cherny SS, Cardon LR (1995) Multiple interval mapping of quantitative trait loci, using sib-pairs. Am J Hum Genet 56:1224–1233
Fulker DW, Cherny SS, Sham PC, Hewitt JK (1999) Combined linkage and association sib-pair analysis for quantitative traits. Am J Hum Genet 64:259–267
George V, Tiwari HK, Zhu XF, Elston RC (1999) A test of transmission/disequilibrium for quantitative traits in pedigree data, by multiple regression. Am J Hum Genet 65:236–245
Goldgar DE (1990) Multipoint analysis of human quantitative genetic variation. Am J Hum Genet 47:957–967
Graybill FA (1976) Theory and application of the linear model. Pacific Grove, California
Harville DA (1997) Matrix algebra from a statistician’s perspective. Springer
Haseman JK, Elston RC (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19
Hedrick PW (1987) Gametic disequilibrium measures: proceed with caution. Genetics 117:331–341
Jung JS, Fan RZ, Jin L (2005) Combined linkage and association mapping of quantitative trait loci by multiple markers. Genetics 170:881–898
Keavney B, MckKenzie CA, Connell JM, Julier C, Ratcliffe PJ, Sobel E, Lathrop M, Farrall M (1998) Measured haplotype analysis of the angiotension-1 converting enzyme gene. Hum Mol Genet 7:1745–1751
Lange K (2002) Mathematical and Statistical methods for genetic analysis, 2nd edn. Springer
Li M, Boehnke M, Abecasis GR (2005) Joint modeling of linkage and association: identifying SNPs responsible for a linkage signal. Am J Hum Genet 76:934–49
Little RJA, Rubin DB (2002) Statistical analysis with missing data, 2nd edn. Wiley Inter-Science, Wiley, Inc., Publication
Pinheiro JC, Bates DM (2000) Mixed-effects models in S and S-plus. Springer
Pratt SC, Daly M, Kruglyak L (2000) Exact multipoint quantitative-trait linkage analysis in pedigrees by variance components. Am J Hum Genet 66:1153–1157
Sham PC, Cherny SS, Purcell S, Hewitt JK (2000) Power of linkage versus association analysis of quantitative traits, by use of variance-components models, for sibship data. Am J Hum Genet 66:1616–1630
Wang T, Elston RC (2005) The bias introduced by population stratification in IBD based linkage analysis. Hum Hered 60:134–142
Xiong MM, Jin L (2000) Combined linkage and linkage disequilibrium mapping for genome screens. Genet Epidemiol 19:211–234
Acknowledgments
The research was supported by the National Science Foundation Grant DMS-0505025. We thank two anonymous reviewers for very detailed and thoughtful critiques, which make the paper better.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Pak Sham.
Appendices
Appendix A
Multiplying both sides of the “genotype effect model” (1) by \(1_{(G_{Aij}=A_gA_h)}\) and taking expectation lead to
Let G Qij be genotype of the j-th individual of the i-th family at the trait locus Q. A true random effect model describing the trait value is y ij = w ij γ + g ij + H ij + e ij , where
Since the missing mechanism is missing completely at random, we have
Utilizing relations \(P(Q_1A_g)=-D_{A_gQ}+P_{A_g}q_1\) and \(P(Q_2A_g)=-D_{A_gQ}+P_{A_g}q_2,\) we have
Equating Eqs. 17 and 18, we show the Eq. 5 when g = h. Now assume that g ≠ h. Since the missing mechanism is missing completely at random, we have
Utilizing relations \(P(Q_1A_g)=D_{A_gQ}+P_{A_g}q_1,\,\, P(Q_2A_g)=-D_{A_gQ}+P_{A_g}q_2,\,\, P(Q_1A_h)=D_{A_hQ}+P_{A_h}q_1,\,\, P(Q_2A_h)=-D_{A_hQ}+P_{A_h}q_2,\) we have
Equating Eqs. 17 and 18, we show the Eq. 5 when g ≠ h.
Appendix B
In relations (17), replacing β gh by α g + α h and taking summation lead to
Since the missing mechanism is missing completely at random, one has E(y ij 1(G_Aij ≠ ?)) = E(y ij |G Aij ≠ ?)(1 − ɛ A ) = (1−ɛ A )Ey ij = (1 − ɛ A )(w ij γ + μ). Thus, \(\sum_{g=1}^m \alpha_gP_{A_g} = \mu/2.\)
Again, replacing β gh by α g + α h in relations (17) and taking summation with respect to h lead to
Notice \(\sum_{g=1}^m D_{A_g Q} =0.\) Taking summation of relations (18) and (19) leads to
Equating the right-hand terms of relations (20) and (21) leads to (6).
Appendix C
Assume that there are no covariates, and the dataset is a population sample. Then the model matrix of “genotype effect model” (1) is \(X_i=X_{Ai1}^{\tau}= (x_{Ai1}^{(11)},\ldots, x_{Ai1}^{(mm)}, x_{Ai1}^{(12)},\ldots, x_{Ai1}^{(1m)},\ldots,x_{Ai1}^{(m-1,m)}), i=1,\ldots, N.\) To show non-centrality parameter approximation (7), we first notice the following relation
where v is a column vector given by \( v^{\tau} =\left(P_{A_2}^2,\ldots, P_{A_m}^2, 2P_{A_1}P_{A_2},\ldots, 2P_{A_1}P_{A_m},\ldots,2P_{A_{m-1}}P_{A_m} \right).\) In addition, \(\hbox{diag} (P_{A_1}^2,v^{\tau})\) is a diagonal matrix, whose elements on the diagonal are given by the elements of \((P_{A_1}^2,v^{\tau}).\) We may verify (22) by \(E [(x_{A11}^{(gh)})^2] = \hbox{E} 1_{(G_{{A11}=A_gA_h)}}+P(A_gA_h)^2 \hbox{E} 1_{(G_{A11}=?)}=P(A_gA_h)(1-\varepsilon_A)+P(A_gA_h)^2\varepsilon_A,\) and for \((g,h) \neq (k,l), \ E [x_{A11}^{(gh)} x_{A11}^{(kl)} ] = P(A_gA_h)P(A_kA_l) \hbox{E} 1_{(G_{A11}=?)} =P(A_gA_h)P(A_kA_l) \varepsilon_A.\)
Let us denote \(u=\left(P_{A_2}^{-2},\ldots, P_{A_m}^{-2}, [2P_{A_1}P_{A_2}]^{-2},\ldots, [2P_{A_1}P_{A_m}]^{-2}, \cdots, [2P_{A_{m-1}}P_{A_m}]^{-2} \right).\) Applying the large number law and a fact of inverse matrix \((M+a b^{\tau})^{-1} = M^{-1}-(M^{-1} a) (b^{\tau} M^{-1})/ (1+ b^{\tau} M^{-1} a),\) we can calculate the following approximation
Utilizing above relation, we may show non-centrality parameter approximation (7) in the same way as Appendix III, Fan et al. (2006).
Appendix D
Assume that there are no covariates, and the dataset is a population sample. Then the model matrix of “additive effect model” (3) is \(X_i=Z_{Ai1}^{\tau}= (x_{Ai1}^{(1)},\ldots, x_{Ai1}^{(m)}), i=1,\ldots, N.\) To show non-centrality parameter approximation (8), we first notice the following relation
which can be verified by \(E [(x_{A11}^{(g)})^2] = 4\hbox{E} 1_{(G_{A11}=A_gA_g)}+\sum_{h \neq g} \hbox{E} 1_{(G_{A11} =A_gA_h)} + 4P_{A_g}^2 \hbox{E} 1_{(G_{A11}=?)} =2(1-\varepsilon_A)P_{A_g} [1+ P_{A_g}] +4P_{A_g}^2\varepsilon_A,\) and for \(h \neq g, \ E [x_{A11}^{(g)} x_{A11}^{(h)} ]=(1- \varepsilon_A)\,\cdot\,2P_{A_g}P_{A_h} + 4P_{A_g}P_{A_h} \varepsilon_A.\) Let \(X=(Z_{A11},\ldots, Z_{AN1})^{\tau}.\) Applying the large number law and a fact of inverse matrix \((M+a b^{\tau})^{-1} = M^{-1}-(M^{-1} a) (b^{\tau} M^{-1})/ (1+ b^{\tau} M^{-1} a),\) we can calculate the following approximation
Utilizing above relation, we may show non-centrality parameter approximation (8) in the same way as Appendix IV, Fan et al. (2006).
Appendix E
For g = 1,2,…,m, k = 1,…,n, let us denote \(D_{A_gB_k}=P(A_gB_k)-P_{A_g}P_{B_k},\)which are measures of LD between markers A and B. Here, P(A g B k ) is frequency of haplotype A g B k . It can be shown that for g ≠ h, k ≠ l, h ≠ h′, l ≠ l′, (g,h) ≠ (g′,h′), (k,l) ≠ (k′,l′)
The quantities in (23) imply that the elements of V A are given by
Since \(\hbox{E}\,Z_{A \cup B}^{(ij)}\) is a vector of 0s by the quantities in (23), it can be shown that \(V_D =\hbox{Cov}\left(Z_{A \cup B}^{(ij)}, Z_{A \cup B}^{(ij)}\right) = \hbox{E} \left(Z_{A \cup B}^{(ij)} (Z_{A \cup B}^{(ij)})^\tau\right).\) Moreover, the quantities in (23) imply that the covariance matrix \(\hbox{Cov}\left(X_{A \cup B}^{(ij)}, Z_{A \cup B}^{(ij)}\right)\) is a 0 matrix. In addition, the covariances between the trait value y ij and variables \(x_{Aij}^{(g)}, x_{Bij}^{(k)}, z_{Aij}^{(gh)}\) and \(z_{Bij}^{(kl)}\) are
Taking variance–covariance between y ij and \(x_{Aij}^{(g)}, x_{Bij}^{(k)}, z_{Aij}^{(gh)}, z_{Bij}^{(kl)}\) based on relation (12), we may get the regression coefficients (13) of models (10) and (12).
Appendix F
Notice \(\Upsigma_i^{-1}= \frac 1 {\sigma^2} (\gamma_{hj})_{(s+2) \times (s+2)}.\) Let X i be the model matrix of family i = 1, 2, …, I. Then
Denote \(\gamma= \sum_{k=1}^{s+2} \sum_{l=1}^{s+2} \gamma_{kl}.\) Applying large number law leads to an approximation as
where O i , i = 1,2,3,4 are zero vectors or matrices, and \(\hbox{E}\left( X_{A \cup B}^{(11)}\right) = (2 P_{A_1},\ldots, 2 P_{A_{m-1}}, 2 P_{B_1},\ldots, 2 P_{B_{n-1}})^{\tau}.\)
Let
be the test matrix corresponding to hypothesis H ABad0, and \({\phi=}({\alpha,}\,\alpha_{A1},\ldots, \alpha_{A(m-1)}, \alpha_{B1},\ldots, \alpha_{B(m-1)},\delta_{A12},\ldots, \delta_{A(m-1)m},\delta_{B12},\ldots, \delta_{B(n-1)n})^{\tau}\) be the column vector of regression coefficient of “genotype effect model” (12). Utilizing regression coefficients (13), we may show (15) by plugging approximation (24) into \(\lambda_{ABad} =(S \phi)^{\tau}[S(\sum_{i=1}^I X_i^{\tau}\Upsigma_i^{-1} X_i)^{-1} S^{\tau}]^{-1}(S \phi).\) One may want to notice that we may use Theorem 8.5.11, Harville (1997), to calculate the inverse of the right-hand matrix of (24).
Appendix G
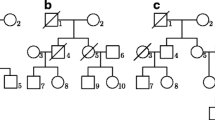
For pedigrees in graph A of Fig. 1, the constants b 1 and b 2 of λ AB,ad in (16) are given by
For pedigrees in graph B of Fig. 1, constants b 1 and b 2 are given by
Rights and permissions
About this article
Cite this article
Fan, R., Liu, L., Jung, J. et al. Combined Linkage and Association Mapping of Quantitative Trait Loci with Missing Completely at Random Genotype Data. Behav Genet 38, 316–336 (2008). https://doi.org/10.1007/s10519-008-9194-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-008-9194-3