
Abstract
In recent years, medical data analysis has become paramount in delivering accurate diagnoses for various diseases. The plethora of medical data sources, encompassing disease types, disease-related proteins, ligands for proteins, and molecular drug components, necessitates adopting effective disease analysis and diagnosis methods. Soft computing techniques, including swarm algorithms and machine learning (ML) methods, have emerged as superior approaches. While ML techniques such as classification and clustering have gained prominence, feature selection methods are crucial in extracting optimal features and reducing data dimensions. This review paper presents a comprehensive overview of soft computing techniques for tackling medical data problems through classifying and analyzing medical data. The focus lies mainly on the classification of medical data resources. A detailed examination of various techniques developed for classifying numerous diseases is provided. The review encompasses an in-depth exploration of multiple ML methods designed explicitly for disease detection and classification. Additionally, the review paper offers insights into the underlying biological disease mechanisms and highlights several medical and chemical databases that facilitate research in this field. Furthermore, the review paper outlines emerging trends and identifies the key challenges in biomedical data analysis. It sheds light on this research domain’s exciting possibilities and future directions. The enhanced understanding of soft computing techniques and their practical applications and limitations will contribute to advancing biomedical data analysis and support healthcare professionals in making accurate diagnoses.
Similar content being viewed by others

1 Introduction
Biomedical data analysis is crucial in healthcare and disease management, enabling healthcare professionals to make informed decisions for diagnosis, treatment, and patient care. The increasing reliance on data-driven approaches stems from recognizing that medical data holds valuable insights that can enhance our understanding of diseases and improve patient outcomes. As a result, researchers and clinicians increasingly turn to advanced analytical techniques to extract meaningful information from the vast amount of available medical data. Recent advancements in medical data analysis have paved the way for more accurate and efficient disease diagnosis and treatment. Machine learning (ML) techniques, such as deep learning algorithms, have remarkably succeeded in various healthcare applications. These algorithms can extract intricate patterns and relationships from large-scale medical datasets, enabling predictive modeling, risk stratification, and early disease detection.
Soft computing techniques have emerged as powerful tools for addressing the challenges posed by medical data analysis. Medical data’s inherent complexity and heterogeneity require flexible and adaptive approaches that effectively handle uncertainties, noise, and incomplete information. Soft computing techniques, such as swarm algorithms and ML methods, can deal with imprecise, uncertain, and incomplete data, making them well-suited for medical data analysis tasks. These techniques offer robustness, flexibility, and the ability to handle non-linear relationships, enabling accurate modeling and analysis of medical data.
1.1 Biomedical domain
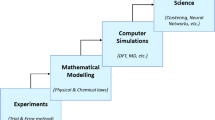
Biomedical informatics is a branch of computational biology that applies computer science concepts to analyze medical data. It aims to improve human health and enhance healthcare through computational biology methods. Several successful medical applications have been developed to aid in disease diagnosis and prediction (Yap et al. 2016; Houssein and Sayed 2023). Biomedical research focuses on studying how drugs and medical procedures impact the biological systems of living organisms (Tarle et al. 2016). It is an interdisciplinary field that combines biology, computer science, chemistry, and information technology to perform comprehensive analyses (as shown in Fig. 1). Basic tasks in biomedical informatics include acquiring, cleaning, storing, organizing, analyzing, and visualizing medical data. With the increasing prevalence and rapid spread of diseases, collecting diverse data is crucial for accurate diagnosis (Hedayati et al. 2021).

Biomedical research fields
Medical data analysis employs various techniques to improve case diagnosis and treatment. Sentiment analysis, for instance, utilizes user reviews to anticipate opinions on the side effects and effectiveness of pharmaceutical products. Classification-based sentiment analysis, employing transfer learning methodologies, allows for the suggestion of user reviews by illustrating commonalities across domains. Transferability of trained classification models between domains has been explored to overcome challenges arising from the absence of annotated training data (Satapathy et al. 2017).
Another analysis approach involves mining protein-protein interactions from biological literature to discover patterns (Ma and Liao 2020). The use of DNA sequences in investigating gene functions, particularly transcription factors (TFs) that control genetic information transcription rates, is also prominent. Optimization methods such as the artificial bee colony algorithm can be applied to identify new transcription factor-binding sites in DNA sequences, yielding excellent results (Avik et al. 2020; Karaboga and Aslan 2016).
In the field of medical data mining, various techniques can be employed to extract valuable information. Classification machine learning methods like K-nearest neighbors (k-NN), support vector machines (SVM), and decision trees (DT) can be integrated with optimization algorithms to extract suitable modular descriptors and provide optimal results (Houssein et al. 2020; Houssein et al. 2021).
Biomedical computational methods play a crucial role in analyzing disease relationships across different levels of abstraction and utilizing various forms of biomedical data. These methods provide a comprehensive understanding of diseases by examining both the observed characteristics of organisms (phenotype) and the genetic variations in specific genes or genetic locations (genotype) (Ashima and Kumar 2021).
Computer-aided diagnosis (CAD) is a vital computational method in the medical field, enhancing the efficiency and performance of radiologists, particularly in terms of sensitivity rate (Halalli and Makandar 2018). Current research focuses on developing medical imaging and analysis systems that employ artificial intelligence (AI) techniques and digital image processing tools. These systems aim to identify and classify abnormal features in medical images and provide visual confirmations to radiologists (Winkel et al. 2019).
Computational biomedical methods are not only used for disease diagnosis but also for understanding disease mechanisms and analyzing protein effects. Proteins are crucial in medicine as they control a cell’s structure and overall shape (Cheng et al. 2019). Predicting protein structures, cellular activities, and interactions between targets provides researchers with insights into the fundamental basis of molecular biological activity (Theodora et al. 2016; Barabási et al. 2011).
Gene expression refers to the process in which the information of a gene is used to synthesize a functional gene product, such as proteins or non-coding RNA, which contribute to phenotype modification (Han et al. 2019). Gene selection is also essential in classifying complex multidimensional samples and genes found in microarray medical data as shown in Fig. 2.

Protein structure
The prediction of protein structures, cellular activities, and interactions between targets has been instrumental in understanding the underlying mechanisms of molecular biological activity (Theodora et al. 2016; Barabási et al. 2011). Fig. 3 illustrates the life cycle of diseases, highlighting the journey from disease identification to determining suitable treatments. Each disease is associated with a specific gene, and proteins are derived from these genes. Proteins become the target, and specific ligands or drugs can interact with them to combat the disease (Lynch and Dawson 2020).

Diseases life cycle
The main objective of drug design is to propose new treatments. Drug development aims to identify and validate potential medications that interact with specific therapeutic targets (Miguel et al. 2016). Traditional drug development processes are expensive, prompting the need for target prioritization based on molecular functions. In this regard, computational approaches, particularly in silico methods, offer faster and more efficient drug design (Maurer Tristan et al. 2020; Brogi et al. 2020). In drug design, computational methods are employed to identify disease processes and drug targets, facilitating the development of novel treatments. The high costs associated with the drug development process necessary methods that prioritize targets based on their molecular functions. The study of chemical interactions can be done in various environments, including in vivo and in vitro, but computational methods in silico environments have gained prominence. In silico approaches provide a faster and more efficient means of drug development compared to other methods. The drug design process involves identifying disease processes, understanding physiologic mechanisms, and proposing novel treatments that target specific disease-related factors. Improving methods for target prioritization is crucial for the long and expensive drug development process in the pharmaceutical industry. Knowledge about how drugs function at the molecular level informs the development of candidate drugs. Factors such as cost, screening facilities, drug development facilities, medicinal chemistry requirements, accessibility, safety, and efficacy of target distribution influence drug design (Maurer Tristan et al. 2020; Jean-Paul et al. 2016; Gasteiger 2016; Zhang et al. 2020).
Computational methods play a crucial role in drug design, aiding in identifying disease processes and drug targets for developing novel treatments. CADD involves several procedures, such as identifying disease mechanisms, validating screening tools, and building molecule databases, to guide the selection of suitable treatments (Yu and MacKerell 2017). Ligand-based and structure-based virtual screening, multiple docking programs, and QSAR analysis are employed in CADD to assess the interactions between drugs and targets (Masand and Rastija 2017). CADD plays a vital role in selecting appropriate treatments by facilitating the diagnosis system for various diseases and aiding in the selection of optimal drugs. In this context, drugs act as ligands that interact with biological functions, particularly proteins. Fig. 4 illustrates the interrelation between bioinformatics and cheminformatics in the drug design process.

Cheminformatics is a consequence of bioinformatics in drug design
Key techniques utilized in computer-aided drug design (CADD) include Quantitative Structure-Activity Relationship (QSAR) analysis and docking, which enable the prediction and evaluation of drug efficacy (Yu and MacKerell 2017; Torres Pedro et al. 2019; Khan Asad 2016).
Docking is an important type of CADD techniques is (Torres Pedro et al. 2019; Khan Asad 2016). Firstly, a disease target is identified, followed by the development of a small chemical library to test against a molecular target, the use of docking to evaluate interactions, and finally the submission of selected medications to pharmacokinetic studies (Eastgate et al. 2017). Optimization algorithms enhance docking operations, and molecular databases aid in identifying potential drugs (Di Muzio et al. 2017; Shuguang et al. 2016; Stefano et al. 2016).
Chemical substances, protein structures, and ligands that are used in pharmacophore modelling or docking techniques are determined from protein bank databases (Burley Stephen et al. 2019). To determine the most effective drugs, first, the ligand and protein are separated using pymol software (Shuguang et al. 2016), and then the energy is calculated using outodock software (Stefano et al. 2016). Docking is done by binding between molecular proteins (receptors or inhibitors) and ligands, so finding the actual chemical conformations for the active site is the main task of docking (Di Muzio et al. 2017; Shuguang et al. 2016; Stefano et al. 2016). Many optimization algorithms are used to enhance docking operations as psovina (Ng et al. 2015). A particle swarm algorithm is used to build a new model that is used for enhancing docking.
The QSAR model, on the other hand, which allows for different descriptions of correlation between structural elements in a molecule’s collection and target reaction, is considered a CADD development (Toropova et al. 2015). The QSAR generates the appropriate descriptors by comparing the data gathered from a group of active and passive compounds to the targets (Masand and Rastija 2017). A mathematical model (QSAR or SAR) are utilised to show the relationship between biological activity and chemical structure (Ma et al. 2018; Khan Asad 2016). Searching through molecular databases, molecules have been identified, and chemical libraries have proven helpful in building a model as a two-stage component. A molecular graph, which is eventually transformed into a vector of features, is the fundamental form of a molecular structure. Meta-heuristics are utilized to choose the best chemical descriptors and compound activities during preprocessing. The second part involves building an ML model for cheminformatics to identify a critical function. It helps with the property mapping of feature vectors. The best medicine can be predicted using several ML techniques (Hussien et al. 2017). In cheminformatics, chemical descriptors and ML models are utilized to predict the effectiveness of different drugs (Hussien et al. 2017).
1.2 Soft computing overview
Soft computing techniques have gained significant popularity in healthcare due to their ability to efficiently diagnose diseases, propose suitable treatments, and deliver superior results compared to conventional approaches. These techniques exhibit adaptable behavior, which allows them to adapt their strategies based on the specific problem at hand (Shalini et al. 2016). Soft computing techniques, including machine learning, have become increasingly popular in healthcare due to their effectiveness in diagnosing diseases, proposing treatments, and delivering superior outcomes compared to traditional approaches (Shalini et al. 2016). These adaptable techniques allow them to tailor their strategies to the specific problem. ML, a subfield of artificial intelligence, plays a crucial role in soft computing. It enables systems to learn and improve from experience without explicit programming (Andino et al. 2018). ML algorithms make informed decisions by leveraging data, observations, or previous experiences.
Deep learning, a subfield of machine learning, is considered a part of soft computing techniques. It involves training artificial neural networks with multiple layers to learn hierarchical representations of data, automatically extracting intricate features from raw input data without manual feature engineering (Houssein et al. 2022). As a broader framework, soft computing encompasses various computational techniques inspired by human-like intelligence to handle complex and uncertain problems. These techniques include fuzzy logic, neural networks, evolutionary computing, and probabilistic reasoning. Deep learning, with its neural network architecture and ability to learn from large-scale datasets, aligns with the principles of soft computing.
Deep learning has achieved remarkable success in diverse fields, such as computer vision, natural language processing, and speech recognition. Its capability to process vast amounts of data and extract high-level representations has revolutionized domains such as healthcare, finance, and autonomous systems. By leveraging deep neural networks, soft computing techniques, including deep learning, provide advanced solutions for complex data analysis, pattern recognition, and decision-making tasks. Their ability to handle unstructured data and learn from experience has significantly improved the accuracy and performance of numerous applications.
1.3 Motivation
The motivation behind exploring soft computing techniques for biomedical data classification arises from the increasing need for effective and accurate analysis of complex medical datasets. Biomedical data encompasses a wide range of information, including clinical records, genomic data, imaging data, and molecular data. Such data’s sheer volume and complexity pose significant challenges for traditional analytical methods. Soft computing techniques, which encompass various computational intelligence approaches such as machine learning, fuzzy logic, genetic algorithms, and swarm intelligence, offer promising solutions for tackling the complexities of biomedical data analysis. These techniques can effectively handle the data’s non-linearity, uncertainty, and noise, enabling researchers and practitioners to extract meaningful patterns, make accurate predictions, and gain valuable insights. The potential applications of soft computing techniques in the biomedical field are extensive. They can aid disease diagnosis, prediction, and prognosis, drug discovery and development, personalized medicine, and understanding complex biological processes. By harnessing the power of soft computing, researchers can uncover hidden patterns, identify biomarkers, and optimize treatment strategies, ultimately leading to improved healthcare outcomes. Furthermore, the growing availability of large-scale biomedical datasets and advancements in computing power have opened up new avenues for applying soft computing techniques. These techniques can leverage the abundance of data to train robust and accurate models, enabling researchers to develop reliable decision-support systems and clinical decision-making tools. Despite the promising nature of soft computing techniques, research gaps, and challenges still need to be addressed. These include the need for more comprehensive and diverse datasets, standardized evaluation metrics, interpretability of models, and the integration of multiple soft computing approaches for improved performance. By addressing these gaps, researchers can further enhance soft computing techniques’ practical applicability and effectiveness in biomedical data analysis. In conclusion, the motivation behind exploring soft computing techniques in the biomedical domain stems from the need for robust and accurate analysis of complex medical datasets. By leveraging the power of computational intelligence, researchers can unlock valuable insights, improve disease diagnosis and treatment, and contribute to advancements in healthcare. Addressing the challenges and research gaps in this field will enable the development of innovative solutions that have the potential to revolutionize biomedical research and clinical practice.
1.4 Contribution
Previous papers on soft computing techniques for biomedical data classification have identified several research gaps and limitations. One crucial area that requires attention is the availability of comprehensive and diverse datasets. Many studies rely on small and specific datasets, which may not adequately represent the complexity and variability of real-world biomedical data. To address this, researchers need access to more extensive and varied datasets to ensure the generalizability of classification models.
Additionally, the lack of standardized evaluation metrics and benchmark datasets poses a challenge for comparing the performance of different soft computing techniques across studies. Without consistent metrics and benchmark datasets, it becomes difficult to assess the effectiveness and reliability of these techniques accurately. Standardization in evaluation methods would facilitate better comparison and understanding of the strengths and weaknesses of various soft computing approaches.
Another significant challenge lies in selecting appropriate feature selection and extraction methods. Different biomedical problems may require different techniques for identifying relevant features, and finding the most effective approach can be challenging. Further research is needed to determine which feature selection and extraction methods work best for specific biomedical datasets and classification tasks.
Moreover, interpretability is a concern when using soft computing models. Understanding the decision-making process and the important features driving classification becomes difficult due to the complex nature of these models. Developing techniques for enhancing the interpretability of soft computing models would provide valuable insights and increase trust in their application in biomedical data classification.
Existing surveys in the literature have touched upon some aspects of soft computing techniques for biomedical data analysis, but they often lack comprehensiveness and fail to address the identified research gaps. For example, a previous survey (Garg and Mago 2021) focused only on ML without discussing other soft computing methods and did not discuss the real limitation. In contrast, another survey (Zhijun et al. 2019) study only user-generated content (UGC) information that social media users data. Furthermore, a previous review (Suganyadevi et al. 2022) focused only on deep learning with medical images, not comparison with other datasets and methods. In contrast, another survey (Haleem et al. 2022) focused only on self-organizing map (SOM) artificial neural network for COVID-19, not other methods.
Addressing these research gaps and limitations will contribute to soft computing techniques’ advancement and practical applicability in biomedical data classification. This review aims to bridge these gaps by examining recent studies and proposing a novel taxonomy that classifies soft computing applications into two groups: machine learning techniques for biomedical data analysis and swarm intelligence algorithms and their applications. By summarizing the findings and identifying new trends in disease diagnosis and prognosis techniques based on microarray gene expression data, this review aims to provide valuable insights for researchers in the field.
So, the main Contributions of this review are listed as follows:
-
Comprehensive Assessment: This review comprehensively assesses various soft computing methods and their application in the medical data domain. It defines and evaluates these methods, offering a valuable resource for researchers and practitioners in the field.
-
Data Collection and Analysis: The review collects and discusses a wide range of popular medical datasets from diverse resources. It emphasizes the importance of understanding the nature of medical data to extract relevant and valuable information.
-
The review also explores preprocessing methods and techniques for mapping medical data to features, facilitating effective data preparation.
-
Optimization Algorithms: The review highlights the significance of applying optimization algorithms to enhance the performance of classification models in medical data analysis. It delves into the use of these algorithms to optimize the accuracy and efficiency of classification tasks, contributing to improved diagnostic capabilities and decision-making.
-
Swarm Algorithms and ML Techniques: This review explores the recent advancements in swarm algorithms and machine learning techniques and their applicability to medical data analysis. It discusses how these innovative approaches can be effectively utilized in solving various medical problems, offering insights into their potential benefits and limitations.
-
Challenges and Limitations: The review also addresses the challenges and limitations of using different medical datasets for disease diagnosis or drug proposal. By acknowledging these challenges, researchers can better understand the complexities and constraints of applying soft computing methods to medical data analysis.
-
Future Research Directions: The review presents potential future research directions in the field, identifying areas where further investigations and advancements are needed. It serves as a valuable reference for researchers looking to contribute to advancing soft computing techniques in the medical domain.
1.5 Review structure
The remainder of this review is organized as follows. Section 2 provides a detailed overview of the methodology and strategies employed to gather relevant study details, including research questions, keywords, and study selection criteria. Section 3 of the paper provides a comprehensive overview of the fundamental concepts and background information related to soft computing methods, various biomedical databases, and evaluation metrics. It serves as a foundation for understanding the subsequent sections. In Sect. 4, the focus shifts towards discussing the application of soft computing techniques, particularly ML, in analyzing different diseases and treatment approaches within the biomedical field. Additionally, the section explores the utilization of meta-heuristic methodologies in this context. Section 5 presents the findings and insights derived from the previous discussions, aiming to provide a comparative analysis of the different soft computing techniques and their applications in the biomedical domain. This section offers a comprehensive overview of these methods’ strengths, weaknesses, and comparative performance. Moving forward, in Sect. 6, future trends and various challenges within the field of soft computing in biomedicine are explored. The section sheds light on the limitations and obstacles researchers and practitioners may encounter when implementing these techniques, highlighting areas requiring further investigation and improvement. Finally, Sect. 7 summarizes the entire review, encapsulating the key insights and contributions discussed throughout the paper. It provides a cohesive conclusion to the review, emphasizing the significance of the reviewed soft computing techniques in the biomedical field.
For a visual representation of the paper’s structure and organization, please refer to Fig. 5, which outlines the guidelines and flow of the paper.

Review structure
2 Review methodology
The methodology employed in this review paper involved a systematic search and selection process based on specific criteria. The search was conducted to ensure the inclusion of relevant and comprehensive studies for analysis and evaluation. The following criteria were considered during the search process to identify suitable literature for inclusion in the review:
-
Relevance: Studies focusing on soft computing techniques for biomedical data classification were considered for inclusion. The aim was to gather a comprehensive collection of research papers that address the application of these techniques in the medical domain.
-
Quality: High-quality studies published in reputable journals or conference proceedings were given priority. Peer-reviewed articles were considered to ensure the reliability and validity of the included studies.
-
Diversity: Studies covering various soft computing methods, such as ML techniques, neural networks, and swarm intelligence, were included to provide a comprehensive overview of the field.
2.1 Literature search and article selection
To conduct a comprehensive review, we conducted a thorough literature search using the Scopus and Web of Science databases. Our search strategy employed relevant keywords, including ”Soft Computing techniques,” ”medical data analysis,” ”feature selection,” ”feature extraction,” ”machine learning algorithms,” and ”biomedical data classification based on soft computing techniques.” We applied specific eligibility criteria to ensure the selected studies’ relevance and currency. First, we limited our search results to articles published between 2010 and 2023. This time frame allowed us to focus on recent advancements in the field and capture the most up-to-date research. Additionally, we filtered the results based on article type, prioritizing full-text articles over other formats. Most eligible articles that met our criteria were journal articles, which provide in-depth analysis and rigorous peer review. Focusing on this publication type, we aimed to include high-quality studies that contribute significantly to biomedical data analysis. Combining systematic keyword selection, database exploration, and eligibility criteria allowed us to gather a comprehensive range of relevant studies for this review. The selected articles provide valuable insights into applying soft computing techniques for biomedical data analysis. Fig. 6 explains different research steps in our field.

Paper selection mechanism
2.2 Analysis of search results
Researchers have recently focused on applying soft computing techniques such as ML and swam optimization algorithms to biomedical data analysis. Moreover, several metaheuristic algorithms and ML techniques have been presented in the last decade for biomedical data analysis. This research discusses the most recent deep architectures, significant metaheuristic algorithms, and ML techniques that can be used in biomedical applications. According to data from the Web of Science (WoS), fig. 9 presents statistics of ML techniques research over the past ten years (2012 to 2022). Fig. 7 shows the various study areas for ML across all databases in this field and how diverse fields can be combined in biomedical research depending on data from the Web of Science databases. Additionally, based on data from Scopus, Fig.8 displays several trends for all databases ML in drug design from 2012 to 2022.

Several research areas for ML in biomedical

Histogram of ML for classification biomedical field

Several graphical analyses for ML in the biomedical field
2.3 Research questions
The primary objective of this study is to address the following research questions and gain insights into the field of soft computing techniques for biomedical data classification:
-
1.
What are the commonly utilized soft computing techniques in bioinformatics?
-
2.
What types of data are typically utilized to classify medical data?
-
3.
Which databases are commonly employed in classification models for biomedical data analysis?
-
4.
What ML approaches are currently applied to categorize biomedical data using meta-heuristic and feature selection techniques?
-
5.
What ML architectures are employed for medical data classification?
-
6.
What are the recent hybridization methods that combine ML with other approaches for improved results in medical data classification?
-
7.
What metrics are commonly used to evaluate the effectiveness of classification models in biomedical data analysis?
By addressing these research questions, we aim to enhance our understanding of the current state-of-the-art soft computing techniques for biomedical data classification and provide valuable insights for future research and application.
3 Basics and background
This section identifies the main topic of our research. Many concepts that will be used in biomedical field analysis will be discussed.
3.1 Soft computing techniques
Soft computing consists of many methods, including swarm optimization algorithms, artificial intelligence (AI), ML, deep learning, evolutionary computation, artificial neural networks (ANNs), and fuzzy computing. They are considered the most widely applied methods in several domains. These methods attempt to find the best optimal solution (Ibrahim 2016). Specifically, this review will detail the ML and swarm optimization algorithms. The soft computing techniques categories are discussed, as indicated in Fig. 10.

Soft computing categories
3.2 Machine learning techniques
Machine learning (ML) is a fundamental technique in soft computing. ML is divided into supervised, unsupervised, and Semi-supervised Learning. The classification and regression methods are ML-supervised, but clustering and association are ML-unsupervised. Semi-supervised has many models as both self-training and co-training Models (Houssein et al. 2021).
Figure 11 indicates ML techniques architectures.

ML category
All ML methods are widely used in various fields, especially in the biomedical field, because of their ability to predict or classify several diseases and treatments. This part explains the main machine learning methods in the biomedical domain (Cheng-Sheng et al. 2020).
Supervised learning: Supervised learning is using labeled datasets to train algorithms, enabling them to classify data or accurately predict outcomes. A common application of supervised learning is the classification of spam emails into separate folders from regular emails. This approach uses a training set containing inputs and corresponding labeled outputs to teach the models and guide them toward producing the desired results. The correctness of the algorithm is measured using a loss function, and iterations are performed to reduce the error until it reaches an acceptable level. Supervised learning can be further divided into two categories: classification and regression (Sen et al. 2020).
In the medical field, supervised learning approaches are on the rise, as they have been shown to deliver excellent results (Nadia and Huber Marco 2021). These techniques enable the development of models that can effectively analyze medical data, make accurate predictions, and support decision-making processes. By leveraging labeled datasets and the iterative learning process, supervised learning plays a crucial role in various medical applications, contributing to improved diagnosis, treatment selection, and patient care.
Classification is the most important method that is used in several fields. Data classification is analyzing and categorizing data based on file type, contents, and other metadata. The classification method aims to identify duplicate data, optimize search capability, discover trends inside data, and secure critical data (Aggarwal and Aggarwal 2015). SVM, k-NN, logistic regression, and naive Bayes are algorithms for classification methods. These techniques are commonly used in the medical field to provide the most accurate diagnosis and treatment (Ali et al. 2018).
K-nearest neighbors (K-NN): For applications that require prediction, the k-NN algorithm can be applied to both classification and regression (Han et al. 2015). It is often applied to classification tasks. It offers many benefits, including short calculation times, straightforward output interpretation, and predictive ability. The k-NN method is one of the machine learning approaches for comprehending and explaining and one of the supervised learning techniques for classifying various k-NN-based cases from an unknown sample. It offers a classification for the appropriate significance of the features that have been selected. Depending on the two main components, the k value, which also denotes the neighbors’ number as a factor, determines the metric applied to estimate the distance between two points. Choosing an appropriate value for k prevents overfitting and underfitting problems. The classification process depends on the nearest neighbors instead of learning a reasonable boundary separating the classes (Osama et al. 2022).
Today’s classification method affects how we understand a variety of biomedical phenomena. Single-cell RNA-seq (scRNA-seq), a new DNA sequencing technology with promising potential but considerable problems because of the large scale of the data created, serves as an illustrative example. The k-NN classifier is a suitable method for classifying scRNA-seq data. It is typically used for large prediction tasks because of its low parameterization and model-free nature (Baran et al. 2019).
In Anagnostou et al. (2020), a specific methodology designed for high dimensional data is focused on and advocated for using approximate closest neighbor search methods for k-NN classification tasks in scRNA-seq data. The experiment results provide the possibility of broader applicability, which supports the main assumption.
Chemical applications are important because they use molecules as position vectors in feature spaces (Houssein et al. 2020). If the parameter is provided, neighbors can be used to calculate the inclusion and feature space’s distance. The data points may even be multidimensional vectors or scalars, representing everything in a metric space. This approach is built in Euclidean distance, whereas other metrics may be established based on the Jaccard distance.
Support Vector Machines (SVMs): Support Vector Machines (SVMs) are a supervised learning technique commonly used for classification tasks (Rodríguez-Perez 2017). SVMs employ a kernel function and nonlinear concepts to map data into a high-dimensional space. By determining the optimal method for separating two classes, SVMs can effectively solve linear and nonlinear problems, making them suitable for real-world applications. The primary task of SVMs is to find a dividing line or hyperplane that can separate data points into their respective classes. The algorithm outputs a line representing the class division, maximizing the margin between support vectors and the hyperplane. In a two-dimensional space, the hyperplane divides the plane into two parts, with each part corresponding to a different class. The SVM’s output is controlled by a few parameters, such as C and Gamma, defined by the designer during the classifier’s construction. These tuning parameters influence the SVM’s performance and are chosen carefully to avoid overfitting or underfitting.
In biomedical machine learning, SVMs are widely utilized (Deepak et al. 2022). One notable variant is the Fuzzy-based Lagrangian Twin Parametric-Margin SVM (FLTPMSVM), which aims to mitigate the effects of outliers in biomedical data. FLTPMSVM assigns weights to each data sample based on fuzzy membership values, reducing the impact of outliers on the model. SVMs also find application in predicting toxicity-related features, such as HERF blockage, mutagenic toxicity, and phospholipidosis toxicity (Beibei et al. 2020). The versatility and effectiveness of SVMs make them a valuable tool in various biomedical applications, providing insights and assisting in decision-making processes. SVM graphical explanation is given in Fig. 12.

Applying SVM for drug design
Naive Bayes Classifier: The naive Bayes classifier is based on the Bayes theorem. It offers significant independence between the properties, which have probabilistic classifier backgrounds. These classifiers are commonly used in ML classification because they are straightforward to implement. This classifier is built using this equation (1).
One of the Naive Bayes classification models is the Gaussian model. Continuous data are normally distributed (Gaussian). The Gaussian model makes it simpler to get statistical results from the training database (Kamble and Dale 2022).
Naive Bayes classification models are widely used in biomedical data because of their ability to predict the target class for many problems accurately. Many classification algorithms are used to analyze medical data, for example, the probability of a person developing a chronic disease (Jena et al. 2020).
Some medications can cause the cochlear and/or vestibular systems to degenerate cellularly, resulting in temporary or permanent hearing loss, dizziness, ear infections, hyperacusis, vertigo, nystagmus, and other ear problems (Li et al. 2020). Therefore, it is crucial to accurately estimate the toxicity of chemicals in drugs. The silico toxicity prediction model is created using the naive Bayes classifier technique and 2612 compounds. A collection of seven molecular descriptors crucial for toxicity was chosen using a genetic algorithm. Specific structural alerts for toxicity were discovered. The constructed prediction model reached an overall training set accuracy of 90.2% and an external test set prediction accuracy of 88.7%. The created model should be implemented accurately and computationally efficient to detect and screen chemical-induced toxicity in drug development. These essential details about toxic drugs’ chemical structures may provide theoretical guidance for leading optimization in drug design (Zhang et al. 2020).
Regression methods: The regression method is based on a statistical concept that determines the strength between variables. Regression is a useful analytical inference tool that has also been used to predict future outcomes based on past evidence. This method is widely used in several fields. Linear regression, ridge regression, and ordinary least squares are methods for the regression technique (Judd et al. 2017).
Linear regression is a core and widely used type of predictive analysis. Determining a linear relationship between one or more predictors is done using linear regression. This method is applied to predict the value of one variable based on the value of another variable. Simple regression and multiple regression (MLR) are the two types of linear regression (Maulud and Abdulazeez 2020).
This method is becoming more important in the medical field. In Kandel et al. (2013), indicates the role of regression method in the medical field. A novel method is provided for extrapolating cognitive or other continuous-variable information from medical imaging.
Unsupervised learning: Unsupervised learning analyses and groups unlabeled datasets using machine learning algorithms. These algorithms identify hidden patterns or data clusters without the assistance of a human. It is the best option for exploratory data analysis, cross-selling, consumer segmentation, and picture identification because of its capacity to find similarities and differences in information (Meng et al. 2020). Unsupervised learning is divided into clustering and association roles. One of the important unsupervised learning applications is Medical imaging. Unsupervised machine learning gives medical imaging equipment crucial features including image identification, classification, and segmentation that are utilized in radiology and pathology to promptly and effectively identify patients (Sughasiny and Rajeshwari 2018).
Clustering: Clustering is dividing the population or set of data points into a number of groups so that the groups’ members are more similar to one another and different from those in other groups. On the basis of its similarities and differences, it is essentially a collection of objects (Gan et al. 2020). K-mean and K-median are examples of clustering methods. These methods are used in the medical field. In Clayman et al. (2020), the K-mean method is applied to gene microarray data to predict gene expression.
Association Rules: A rule-based approach for identifying connections between variables in a particular dataset is called an association rule. Market basket analysis usually employs these techniques, which help businesses comprehend the connections between various items. Association rule-based algorithms include AIS, SETM, Apriori, and variants of the latter (Akbar et al. 2020). In this research (Sa and Vadivu 2017), a new method for identifying the optimum association rule mining algorithm utilizing multiple-criteria decision analysis is proposed for extracting association rules from medical records. The proposal’s goal is to find relationships between diseases, diseases and symptoms, and medications.
Semi-supervised Learning: ML includes the field of semi-supervised learning. Semi-supervised learning, as the name implies, is a hybrid technique between supervised and unsupervised learning. It is a broad category of machine learning techniques that makes use of both labeled and unlabeled data. The fundamental principle of semi-supervision is to treat data points differently depending on whether they are labeled or not. For labeled points, the algorithm will update the model weights using traditional supervision; for unlabeled points, the algorithm minimizes the difference in predictions between other similar training examples. Semi-supervised learning has many approaches, such as co-training and self-training. Speech recognition, web content classification, and text document classification are examples of semi-supervised learning (Van Engelen and Hoos 2020).
Semi-supervised learning is used increasingly in the medical field. This method is used to propose automated decisions for Dynamic treatment regimes, medical diagnosis, Healthcare resource scheduling and allocation, drug discovery, design, and development (Huynh et al. 2022).
Artificial neural network (ANN): Artificial Neural Networks are the basic building blocks of deep learning. It is a sub-class of machine learning. ANNs are computational algorithms (Rem et al. 2019; Emam et al. 2023). They aim to mimic the actions of neuron-based biological systems. They are computer models that are inspired by the central nervous system mechanism of pattern recognition. ANNs were described as networks of neurons capable of computing values from inputs as oriented graphs. Using a biological analogy, they consist of nodes connected by arcs representing neurons, and they are related to dendrites and synapses. Each arc has a weight assigned to it at each node. The received node data must be applied as input, the activation function for the incoming arcs must be specified, and the arc weights must be considered. A neural network is an ML technique based on the design of the human neuron. The human brain consists of millions of neurons, which send and process electrical and chemical impulses. These neurons are interconnected by synapses, which have a unique structure. Neurons can transmit messages through synapses. An enormous number of simulated neurons are used to create neural networks. ANN is an information processing technique. It functions similarly to how the brain processes information. It is a collection of several connected processing units that work together to process data and produce useful outcomes. Its use is not limited to classification. Regression of continuous target attributes is also applicable (Kotsovsky et al. 2020).
A basic neural network consists of the following three types of layers :
Input layer : this layer contains the raw data that will be fed into the network, which is indicated by the activity of the input units.
Hidden layer: this layer is used to establish the activity of each hidden unit. The operations of the input units and the weights on the ties connecting the input and hidden units. The network might have one or more hidden layers.
Output layer this layer is always concerned with the classification of the data. The activity of the hidden units and the weights between the hidden and output units affect the behavior of the output units (Barredo et al. 2020).
Deep examination and disease classification are necessary for effective disease diagnosis. The exponential growth of biological data during the past two decades has presented several researchers with many opportunities.
ANNs are the most effective models for automatic pattern identification of almost all AI techniques. A model acquires its properties from inputs and estimates the outcome when a data set is introduced into it Jhalia and Swarnkar (2021).
3.3 Swarm optimization algorithms
An optimization process includes identifying the best values for particular system parameters to fulfill the system design as efficiently as possible (Baykasoğlu and Ozsoydan 2015). The traditional optimization techniques have drawbacks, such as convergent local optima and unexplored search space. In addition, they only have a single-based solution. Swarm intelligence (SI) algorithms are one of the soft computing techniques that mimic the nature of algorithms applied to solve several problems (Laith et al. 2021).
Swarm Optimization algorithms can be applied to tackle many problem types (Sörensen et al. 2018; Emam et al. 2023). Optimization can be proposed for minimization or maximization problems, and the best solution is obtained by applying several optimization techniques. In real life, optimization is used to produce ideal paths. Several optimization methods are applied to get the optimal solution (Emam et al. 2023).
Swarm optimization algorithms are optimization tools that employ several approaches to improve the efficacy of search procedures (Houssein et al. 2022; Singh et al. 2022; Houssein et al. 2021a, b). Although it can be difficult to determine the exact solution, algorithms can provide the greatest overall solution (Algorithm and applications 2019). Continuous, discrete, and binary optimization descriptions are possibly described depending on the search space (Crawford et al. 2017; Majdi et al. 2023). The solutions can be divided into three groups: 1. continuous variables, which constantly change and have a continuous range of values; 2. discrete variables, which can take variables with either an integer or a binary value. 3. Mixed variables can have many reals or integers, making it a mixed problem. By conforming to a few essential criteria, swarm intelligence algorithms simulate collective behavior for more interconnected agents.
3.4 Biomedical datasets analysis
In this part, some popular datasets in the medical field are discussed. A biomedical database is a collection of organized data that stores the properties of diseases or molecular structures (Oughtred et al. 2021). Several operations are done by retrieving or archiving this data. Examples of biomedical databases include drug bank databases, cheminformatic.org, ZINC databases, the World Health Organization (WHO), NCBI, PubMed, BLAST, and gene ontology. Chemical and biological data should be integrated, so this field focuses on diseases based on molecular analysis and creating molecules that interact with disease agents to decrease the disease’s effects. One of the critical problems is the expansion of this research area due to the variety of features that can be found in data sets. So the traditional database method is not applicable. Recent methods are proposed, such as big data and cloud-based methodologies (Lavecchia 2015).
Biomedical datasets can be collected from several database systems such as Machine learning repository (UCI) (Karabatak and Mustafa 2018) and Kaggle (Charanasomboon and Viyanon 2019). FDA-Approved Drugs (Ciociola et al. 2014), Drug bank database (Wishart and Feunang 2018), NCBI (Schoch et al. 2020), cheminformatic.org (Bender and Cortes-Ciriano 2021), BLAST (Mingzhang et al. 2020), PubMed (Michael and Haddaway 2020), and ZINC (Haider et al. 2020) database. Examples of the biomedical dataset are as follows:
Firstly, from UCI as:
-
Gene Expression Cancer RNA-Seq: Instances are arranged in rows. The properties of each instance are the RNA-Seq gene expression levels determined by the Illumina HiSeq platform. For 801 samples, there are 20531 attributes. It is available onFootnote 1.
-
QSAR Biodegradation: The classification of 1055 chemical compounds in this dataset is based on 41 characteristics (molecular descriptors). With 356 readily biodegradable samples and 699 not readily biodegradable patterns, this data is utilized to distinguish between two chemical classes. This information can also be used in QSARs to determine the relationship between chemical design and molecular biodegradation. It is available onFootnote 2.
-
Drug Review: Patient reviews in this dataset describe both diseases. They are related to a particular drug. A rating that represents overall patient satisfaction has been provided by 10 patients. The details were discovered while browsing online drug reviews. The goal was to learn something new. The best results are obtained when this data is divided into training (75%) and testing (25%) parts. It is available onFootnote 3.
-
Drug consumption: The database has 1885 respondents. Initial categorization of all input qualities is followed by quantification. The use of 18 legal and illegal drugs, including nicotine, alcohol, crack, ecstasy, heroin, ketamine, legal highs, LSD, methadone, mushrooms, and one fictitious drug to identify over-claimers, was also inquired about by participants. It is available onFootnote 4.
-
QSAR androgen receptor: This dataset was applied to generate classification QSAR models for the separation of binder/positive (199) and non-binder/negative (1488) molecules using a variety of ML techniques as part of the CoMPARA collaborative modeling project, which aims to establish QSAR models to detect binders to the androgen receptor. It is available onFootnote 5.
-
Immunotherapy: This dataset contains 8 attributes and 90 instances of wart treatment results. It is available onFootnote 6.
-
Anti-cancer Peptides: Due to their capacity to prevent cellular resistance and get over frequent obstacles like cytotoxicity and the side effects of chemotherapy, membrane-bound anti-cancer peptides (ACPs) are becoming more and more popular as prospective cancer treatments. This dataset describes the anticancer activity of peptides on breast and lung cancer cell lines (annotated for their one-letter amino acid code). It reaches high standards (active, moderately active, experimentally inactive, virtually inactive). It is available onFootnote 7.
-
Relative location of computed tomography (CT) slices on the axial axis Data Set: This data set has 53500 instances with 386 attribute CT images. It is available onFootnote 8.
From Kaggle website’s some datasets as:
-
Magnetic resonance imaging (MRI) is another technique to detect cancer cells early, Primary Tumor: One of the three domains offered by the Oncology Institute that has frequently been referenced in ML literature is this one. It has 17 properties, which apply to 339 instances. It is available onFootnote 9.
-
Alzheimer Features: The features of Alzheimer’s are described using this dataset. There are 347 instances and 10 features. It can be collected fromFootnote 10.
-
Eye Disorder: This dataset includes ocular conditions. For 16383 features, it is utilized to describe 101 instances. It is available onFootnote 11.
-
PET radionics: This data is for cancers of the head and neck, which are becoming a domestic pandemic with 20,000 cases. It is found onFootnote 12.
-
Brain MRI Images for Brain Tumour Detection: This data is based on MRI images from 98 files with several images. It is available onFootnote 13. from cheminformatics.org as:
-
Monoamine Oxidase (MAO): An enzyme that is widely present in the major tissues provides the dataset. The inactivation and oxidation of monoamine neurotransmitters can be facilitated by it. This data set is taken fromFootnote 14. By using the open Babel software (Andersen et al. 2016), MOA is converted to SMILES (Simplified Molecular-Input Linear EntrySystem). Using E-Dragon’s (Khan Asad 2016), the molecular descriptors (MD) are subsequently calculated. With 68 compounds split into two groups, it has 1665 characteristics (MD).
From FDA
-
The FDA data set: It includes 5909 FDA-approved drugs (Mohamed et al. 2020) with a range of therapeutic effects, including the management of hypertension, the treatment of cancer, and nutritional supplementation. Additionally, 31 molecular traits from drugs are extracted using the DataWarrior software (Shehabeldeen Taher et al. 2020). This data set is taken fromFootnote 15.
3.5 Biomedical tools
This section provides an overview of several popular tools in the field of biomedical research.
One important bioinformatics software is Bioclipse, which is based on the Eclipse-rich client platform. Bioclipse offers a visual platform for chemo- and bioinformatics and allows scripting in JavaScript, Python, and Groovy. A unique feature of Bioclipse is its plugin system, which provides domain-specific functionality to the scripting language (Willighagen 2021).
For PHP developers, BioPHP is a toolkit that includes classes for various bioinformatics tasks such as database processing, sequence alignment, and DNA/protein sequence analysis (Ye et al. 2014). The BioPHP toolkit consists of four projects, including genePHP, Functions, tools, and Minitools. These projects provide classes, functions, and scripts that facilitate bioinformatics operations and minimize code duplication.
In the field of virtual reality (VR), the Molecular Rift is a VR system that allows researchers to explore molecular structures using hand motion and the Oculus Rift head-mounted display. This technology provides an immersive experience and is particularly useful for studying ligand-protein complexes and drug discovery (Norrby et al. 2015).
BioVR is another interactive platform that combines DNA, RNA, and protein sequence and structure visualization. It utilizes Unity3D and the C# programming language, along with the Oculus Rift and leap motion hand movement, to enable intuitive navigation and analysis of biological data. BioVR includes a proof of concept software that integrates protein and nucleic acid data, allowing users to interactively explore molecular structures in VR (Zhang et al. 2019).
The Minitools project offers a set of PHP scripts designed to simplify minor and repetitive bioinformatics tasks (Stevens 2015).
When it comes to storing genomic data, the BED format is commonly used. The BED format employs text files to store genomic coordinates and annotations. It has become a de-facto standard in bioinformatics due to its widespread usage and compatibility with various software tools. A BED file consists of columns containing information such as chromosome names, start and end coordinates of sequences, and optional annotations (Diez-Fuertes et al. 2021).
During public health emergencies like the COVID-19 pandemic, the Food and Drug Administration (FDA) may grant Emergency Use Authorization (EUA) for the use of unapproved medical products or unapproved uses of approved medical products. For instance, the FDA authorized the use of ML-based COVID-19 non-diagnostic screening tools, such as the Tiger Tech COVID Plus monitor, which can detect biomarkers associated with various disorders including hypercoagulation. These tools aid in preventing the spread of SARS-CoV-2 and contribute to the early detection of infections and related conditions (Ison et al. 2020).
In the field of molecular docking, several software tools are widely used. AutoDock Vina is an open-source docking software that employs local search techniques to address conformation search problems (Di Muzio et al. 2017). The GOLD AutoDock program incorporates metaheuristic methods like genetic algorithms and particle swarm optimization to enhance the accuracy and optimization of ligand orientation and binding packet prediction (Wang et al. 2016).
Different optimization algorithms have been integrated into docking software to tackle complex problems. PSOVina combines the Particle Swarm Optimization (PSO) algorithm with the Broyden–Fletcher–Goldfarb–Shannon method, while FIPSDock utilizes a hybrid of Genetic Algorithms (GA) and PSO for ligand docking (Ng et al. 2015; Liu et al. 2013). Cuckoo Vina integrates cuckoo search and differential evolution algorithms to address docking problems, binding affinity, and root-mean-square deviation (RMSD) (Lin and Siu 2018). LightDock is another docking method that supports protein-protein docking with conformational flexibility and various scoring systems (Jiménez-García et al. 2018).
For calculating molecular descriptors, tools like E-Dragon and Mordred are commonly used. E-Dragon, a hybrid of KNIME and Dragon, enables the calculation of molecular descriptors numerically. It provides a wide range of molecular descriptors derived from different molecular representations, allowing researchers to select appropriate descriptors for their studies. Mordred, on the other hand, excels in calculating descriptors for large molecules and is favored for its performance, convenience, and extensive collection of descriptors (Mauri et al. 2006; Moriwaki et al. 2018).
Overall, these biomedical tools play a crucial role in various aspects of bioinformatics, molecular modeling, and drug discovery, enabling researchers to analyze and manipulate biological data effectively.
3.6 Evaluation metrics
Evaluation metrics play a crucial role in assessing the performance of algorithms and their ability to distinguish between different model results. These metrics provide valuable insights into the effectiveness and efficiency of the evaluated models.
Firstly, the method is validated and evaluated based on the best fitness value. The measurements below, denoted as fobj, are obtained at the i-th run.
-
1.
The mean value is calculated by averaging the fitness function values generated by the M technique over time. The following equation can be used to get the mean fitness function:
$$\begin{aligned} Mean = \frac{ \sum _{i=1}^{M} {fobj(i)} }{M} \end{aligned}$$(2) -
2.
The optimal fitness function is represented by the fitness function’s maximum value, which results from running the algorithm M times. The following formula can be used to determine the best fitness function’s value:
$$\begin{aligned} Best = \max _{i=1}^{M} {fobj(i)} \end{aligned}$$(3) -
3.
The fitness function that gives the lowest result after running the algorithm M times is the worst available fitness function so that this function can be calculated by (4).
$$\begin{aligned} Worst = \min _{i=1}^{M} {fobj(i)} \end{aligned}$$(4) -
4.
Standard deviation is applied to calculate the change in the fitness function value obtained from the running algorithm M times (STD). It provides a metric for the robustness and stability of an algorithm. A lower value implies that the method converges to the same value in most runs, whereas a higher value denotes wandering outcomes. The following formula can be used to calculate the standard deviation:
$$\begin{aligned} STD = \sqrt{\frac{1}{M-1}\Sigma _{{i=1}}^{M} (fobj(i)-mean) ^2} \end{aligned}$$(5) -
5.
CPU time: Each method’s computation time is as follows: n is the maximum number of iterations, and k is the number of iterations.
$$\begin{aligned} CP{U_{Time}} = {T_{best}^{(k)}} \; \;\; \; \;\; \; \;\; {k = 1,,2,3,......,n} \end{aligned}$$(6) -
6.
Average selection size of features (ASS): (ASS), which was used to measure how many of the chosen features were acquired each time.
$$\begin{aligned} A S S=\frac{1}{M} \sum _{i=1}^M \frac{ \text{ length } \left( Q_i\right) }{L} \end{aligned}$$(7)where L is the number of features in the initial dataset, Q is the highest score obtained thus far at each iteration, and M is the total number of iterations. Is required to do a statistical test on the significance of the findings acquired using various algorithms to evaluate the effectiveness of the proposed algorithm compared to existing SI algorithms. Based on accuracy metrics, testing algorithms show the p-values of the Wilcoxon rank-sum test. The proposed approach obtained reduced p-values for both datasets, which are less than 1%, suggesting that it has a distinct advantage over the existing SI techniques. As a result, the suggested algorithm statistically differs from the other optimizers considered in this paper.
Secondly, the proposed model is evaluated according to the following standards: accuracy, precision, specificity, sensitivity, and F-score. Major metrics (positive, negative, true, or false) were commonly used as performance measurements. The effectiveness of measurement and assessment metrics is described using the following formulas from a probabilistic perspective:
-
According to Eq 10, the average accuracy is calculated by first computing the accuracy of each class independently, then averaging the results, which represent the accurate number of correspondences between the label of the sample data and the classifier’s output. The following is the best solution for calculating \(AVG_{Acc}\):
$$\begin{aligned} ACC = \frac{{TP + Tn}}{{Tp + Fn + Fp + Tn}} \end{aligned}$$(8)With a multiclass confusion matrix of the form
$$\begin{aligned} C=\text {Actual}\begin{array}{lll} &{} \text {Classifed} &{} \\ c_{11} &{} ... &{} c_{1n}\\ \vdots &{} \ddots &{} \\ c_{n1} &{} &{} c_{nn} \end{array} \end{aligned}$$(9)the confusion elements for each class are given by:
$$\begin{aligned} Tp_{i}&= c_{ii} \nonumber \\ Fp_{i}&= \sum _{l=1}^{n} c_{li} - Tp_{i} \nonumber \\ Fn_{i}&= \sum _{l=1}^{n} c_{il} - Tp_{i} \nonumber \\ Tn_{i}&= \sum _{l=1}^{n} \sum _{k=1}^{n} c_{lk} - Tp_{i} - Fp_{i}- Fn_{i}\nonumber \\ ACC_i&= \frac{{TP_i + Tn_i}}{{Tp_i + Fn_i + Fp_i + Tn_i}} \nonumber \\ ACC&= \frac{1}{n}\sum _{i=1}^{n} ACC_i \nonumber \\ \end{aligned}$$(10)$$\begin{aligned} AVG_{Acc}&= \frac{1}{{{N_r}}}\sum \limits _{k = 1}^{{N_r}}{ACC_{best}^{(k)}} \end{aligned}$$(11)Where the total number of runs is fixed \({N_r=30}\), and n is a number of classes.
-
Average sensitivity \((AVG_{Sn})\): The sensitivity (Sn), which is used to assess the rate of prognosticating positive samples, involves computing the sensitivity of each class separately and then averaging the results, which are determined as follows:
$$\begin{aligned} \begin{aligned} Sn_i&= \frac{{Tp_i}}{{Tp_i + Fn_i}}\\ Sn&= \frac{1}{n}\sum _{i=1}^{n} Sn_i\\ \end{aligned} \end{aligned}$$(12)The \(AVG_{Sn}\) is calculated from the best (x) using
$$\begin{aligned} AV{G_{Sn}} = \frac{1}{{{N_r}}}\sum \limits _{k = 1}^{{N_r}} {Sn_{best}^{(k)}} \end{aligned}$$(13) -
The specificity (Sp): indicates the rate of prognosticating negative samples, involves computing the specificity of each class separately and then averaging the results, and is calculated from:
$$\begin{aligned} \begin{aligned} Sp_i&= \frac{{Tn_i}}{{Fp_i + Tn_i}}\\ Sp&= \frac{1}{n}\sum _{i=1}^{n} Sp_i\\ \end{aligned} \end{aligned}$$(14)The \(AVG_{Sp}\) is determined as follows:
$$\begin{aligned} AV{G_{Sp}} = \frac{1}{{{N_r}}}\sum \limits _{k = 1}^{{N_r}} {Sp_{best}^{(k)}} \end{aligned}$$(15) -
The precision (Pr), which is used to evaluate the effectiveness of the proposed approach, involves computing the precision of each class separately and then averaging the results as follows:
$$\begin{aligned} \begin{aligned} Pr_i&=\left\{ \frac{TP_i}{TP_i+FP_i}\right\} \\ Pr&= \frac{1}{n}\sum _{i=1}^{n} Pr_i\\ \end{aligned} \end{aligned}$$(16)The \(AVG_{Pr}\) is determined as follows:
$$\begin{aligned} AV{G_{Pr}} = \frac{1}{{{N_r}}}\sum \limits _{k = 1}^{{N_r}} {Pr_{best}^{(k)}} \end{aligned}$$(17) -
The F-score (F1) is a measure of the test’s accuracy obtained by computing each class’s precision separately and then averaging the results.
$$\begin{aligned} \begin{aligned} F1_i&=\left\{ \frac{TP_i}{TP_i+FP_i}\right\} \\ F1&= \frac{1}{n}\sum _{i=1}^{n} F1_i\\ \end{aligned} \end{aligned}$$(18)The \(AVG_{F1}\) is determined as follows:
$$\begin{aligned} AV{G_{F1}} = \frac{1}{{{N_r}}}\sum \limits _{k = 1}^{{N_r}} {F1_{best}^{(k)}} \end{aligned}$$(19)
4 Soft computing techniques for biomedical applications
This section introduces soft computing techniques widely used in the biomedical field. Moreover, several biomedical applications will be provided. First, we present the biomedical data analysis-based ML techniques. Then, we present the biomedical data analysis-based swarm intelligence algorithms.
4.1 Machine learning for biomedical data analysis
Gene expression is a fundamental concept in biology, involving the creation of functional gene products that generate proteins. ML techniques can be applied to optimize the selection of genes and propose the best results. However, before applying ML, gene data often requires preprocessing steps. Fig. 13 illustrates several pre-processing steps for gene expression (de Jongh Ronald et al. 2020).

pre-processing step of gene expression
ML techniques have gained significant popularity in biomedical data analysis due to their ability to provide accurate results. In cancer classification, feature selection (FS) plays a crucial role in choosing the most relevant genes from a vast number of microarray genes. Various statistical measures, such as T-statistics, SNR, and F-test values, are used to rank the genes. Swarm intelligence approaches are then employed to select informative genes for classification (Gunavathi and Premalatha 2014).
ML methods have proven effective in diagnosing chronic diseases, which are responsible for a significant portion of global healthcare costs. Predictive models have been developed to aid in the diagnosis and prediction of various diseases, contributing to improved patient care (Gopi et al. 2020).
In the field of medical imaging, ML techniques play a crucial role in identifying and predicting diseases affecting organs such as the liver, breast, brain, heart, and bones. These methods have enabled accurate diagnosis and improved treatment planning (Erickson et al. 2017).
Drug development is a complex and time-consuming process, and ML methods are being employed to enhance its effectiveness. The virtual ligand or structure screening hit identification, optimization, and hit optimization are some of the techniques used. ML methods aid in identifying disease-causing proteins and designing chemical compounds to treat specific diseases (Rajula et al. 2020; Lauv et al. 2020).
Quantitative Structure-Activity Relationship (QSAR) is a computational method used to predict the activity of chemical compounds based on their descriptors. ML techniques such as SVM, kNN, and Deep Learning (DL) are utilized to process chemical data and create predictive models. These models assist in predicting the activity of chemical compounds and optimizing drug design (Hussien et al. 2017; Houssein et al. 2020; Lo et al. 2018).
Chemoinformatics, which encompasses drug design, involves encoding and mapping stages. The encoding stage represents the three-dimensional information of a molecular structure, which is then transformed into a feature vector using various descriptors. The mapping stage utilizes ML techniques to create models that establish relationships between feature vectors and specific properties, facilitating drug design (Akbar et al. 2016; Masand and Rastija 2017).
Fig. 14 illustrates the conversion of structural chemical data to numerical data using ML techniques. The process involves two stages: encoding and mapping. Descriptors of chemical structures are calculated, and then they are transformed into feature vectors. ML methods are subsequently employed to provide accurate results based on the converted data (Lo et al. 2018).

Molecular structure to features
Several ML algorithms have been compared for the accurate identification of drug targets using semantic data from medical resources. Approaches such as self-organizing maps, k-NN, and SVMs have been discussed as tools for drug design. These studies offer step-by-step methodologies for developing ML and statistical approaches in drug design, considering training processes and learning mechanisms (Danger et al. 2010; Gertrudes et al. 2012; Mitchell 2014).
A hybrid Grasshopper Optimization Algorithm (GOA) combined with SVM can optimize SVM parameters and select the optimal set of properties, demonstrating the integration of meta-heuristics and machine learning (Ibrahim et al. 2018). These approaches find application in diverse fields, and the E-Dragon software based on determined descriptors can yield optimal results. Machine learning approaches play a crucial role in classifying chemicals within chemical datasets. These algorithms enable the development of more efficient procedures and are often combined with QSAR techniques. Swarm algorithms, such as the Swarm Search Algorithm (SSA) integrated with k-NN in a QSAR context, provide promising solutions (Grenier et al. 2017; Hussien et al. 2017). Additionally, the integration of Harris’s Hawks Optimization Algorithm and SVMs shows success in classifying popular chemical datasets, such as QSAR biodegradation and MOA, while using hybridized optimization algorithms (Houssein et al. 2020; Houssein et al. 2021). In recent studies, enhancements have been made to optimization algorithms to improve their performance in feature selection. For example, the Harris Hawks Optimization (HHO) algorithm has been modified to incorporate genetic operators like crossover and mutation, aiming to strike a better balance between global and local search. These modifications result in higher classification accuracy and the selection of the most significant molecular descriptors (Houssein et al. 2021).
Another approach involves the modification of the Hunger Games Search Algorithm using fuzzy mutation. This modified algorithm addresses the feature selection problem in ten medical datasets, reducing the number of features while improving classification accuracy by applying an SVM classifier (Houssein et al. 2023). By integrating optimization algorithms and machine learning techniques, researchers continue to explore innovative solutions for feature selection and classification tasks in various domains, leading to improved performance and more effective data analysis.
4.2 Swarm optimization algorithms for biomedical data analysis
The application of optimization algorithms in the biomedical field has proven beneficial, particularly in addressing FS problems. FS can be seen as an optimization problem, and since most biomedical problems are considered NP-hard, optimization algorithms are well-suited for their resolution. FS plays a vital role in data mining and pattern recognition, as it involves filtering and selecting features from training datasets. One advantage of FS is its ability to improve model generalization, eliminate overfitting issues, and enhance performance across various medical domains such as biomedical signal processing, medical images, DNA microarray data, chemical data, and drug development. Due to the high dimensionality of medical data, the successful application of FS in different medical contexts has been demonstrated through relevant research. In ML and data mining, three common FS methods are utilized: hybrid approaches, embedded methods, and filter and wrapper techniques (Wah et al. 2018; Chandrashekar and Sahin 2014). However, FS is often regarded as an NP-hard problem due to many potential solutions, particularly in high-dimensional spaces.
Several metaheuristic algorithms have been employed for FS in biomedical data analysis. These include particle swarm optimization (PSO) (Gupta and Saini 2017), bee colony optimization (BCO) (Hancer et al. 2018), genetic algorithm (GA) (Kennedy and Eberhart 1997), improved multi-operator differential evolution algorithm (IMODE) (Sallam et al. 2020), gravitational search algorithm (GSA) (Rashedi et al. 2009), grey wolf optimizer (GWO) (Seyedali et al. 2014), Harris Hawks optimization (HHO) (Algorithm and applications 2019), whale optimization algorithm (WOA) (Mirjalili and Lewis 2016), and slime mold algorithm (SMA) (Li et al. 2020). Metaheuristic-based techniques offer faster solutions compared to exhaustive search methods. These MH methods effectively determine FS’s most advantageous set of attributes and are time-efficient. However, due to the ”no free lunch” theory, which states that no optimization technique can provide optimal results for every problem, developing more precise techniques becomes necessary (Wolpert and Macready 1997).
Some research in this field is discussed as follows: In Agrawal and Silakari (2015), the utilization of PSO is highlighted in solving various biomedical problems such as RNA secondary structure prediction, gene clustering, energy minimization, and protein modeling.
Motif discovery involves identifying conserved patterns of sequences associated with specific protein or DNA actions, which is a crucial concept in the biological sciences (Zhang et al. 2015). The ABC/DE approach has been developed to tackle the motif discovery challenge (MDP), using local multiple sequence alignments (MSA) with a relative entropy scoring system. Since motif identification is an NP-hard problem, most motif search algorithms are heuristic techniques that provide near-optimal solutions with low computational costs (Cui and Zhang 2014).
The task of discovering new transcription factor binding sites (TFBS) in DNA sequences can be effectively addressed by employing a multi-objective PSO. This approach improves motif detection using a revised linear PSO algorithm that updates the following location and initializes the population using linear numbers. The algorithm selects a single particle known as the ”target motif” in each cycle and determines its fitness by comparing it to other DNA sequences. Depending on the severity of the problem, slower algorithms can still be helpful and necessary (Yang and Li 2013).
Quantum PSO has also been explored as an optimization algorithm for multiple sequence alignment, specifically with selection operations. This approach has been tested on numerous nucleotide and protein sequences, showcasing its potential in the field (Cui and Zhang 2014).
In the realm of drug design, various swarm methods have been employed to accelerate the drug development process in a virtual environment. De novo drug design, a technique within computer-aided drug design (CADD), aims to discover novel drug-like chemical compounds from a vast universe of chemical searches. Swarm optimization algorithms, such as bacterial foraging optimization (BFO) integrated with ligand docking, have demonstrated efficiency and success in drug design (Vasundhara et al. 2015; Jia et al. 2016; Peh and Hong 2016).
4.3 Applications-based soft computing techniques
Soft computing techniques have found numerous applications in the biomedical field, contributing to advancements in healthcare and medical research. Here, we discuss some notable applications in this domain:
-
Digital Diagnosis and Disease Pattern Recognition: ML algorithms have been employed in digital diagnosis to analyze patient electronic medical records and identify patterns associated with specific diseases. By leveraging vast amounts of data, ML algorithms act as a second set of eyes, aiding clinicians in detecting abnormalities and providing valuable insights into patient health (Arima et al. 2020).
-
Collagen Stability Prediction: Collagen, the most abundant structural protein in humans, exhibits significant sequence variation. Deep learning techniques have been applied to large datasets of collagen sequences, along with their corresponding midpoint values, to develop models that predict the stability of collagen triple helices. This approach enables the assessment of the impact of mutations and sequence order on collagen stability, aiding in understanding collagen-related disorders (Chi-Hua et al. 2022).
-
Augmented Reality and AI in Healthcare: The integration of augmented reality (AR) with AI offers exciting applications in healthcare. For example, a HoloLens application combining 3D visualizations and textual information has been developed with input from clinical pharmacists. This technology goes beyond financial value by enabling predictions of future treatment approaches and facilitating effective clinical decision-making (Don et al. 2022).
-
Holography in Medical Training and Research: Using digital image inputs, Holography provides comprehensive visualization of anatomical data and has emerged as a valuable tool for medical training and research. By digitizing and analyzing patient data, holography offers innovative solutions for effective treatment, surgery planning, and medical education. Although it requires substantial data storage and analysis resources, holography demonstrates great potential in addressing various medical challenges (Abid et al. 2020).
-
Parallel Computing for Biomedical Analysis: Parallel computing methods, such as meripseqpipe, have proven beneficial in biomedical data analysis. These methods encompass multiple functional analysis modules, facilitating scalable and reproducible analysis. By leveraging platforms like Docker and Nextflow, meripseqpipe enables efficient processing and analysis of large-scale biomedical datasets (Bao et al. 2022).
-
Personalized Medicine and Precision Drug Discovery: Personalized medicine aims to tailor treatments to individual patients based on their unique characteristics. Machine learning algorithms are crucial in proposing suitable medications by leveraging medical datasets. These algorithms, implemented in silico experimental systems, assist in reducing the burden of major diseases like lung cancer, COVID-19, and cardiovascular disorders. Personalized medicine offers customized and targeted approaches to improve patient outcomes (Lin et al. 2021).
-
Preventive Medicine and Disease Screening: Preventive medicine focuses on understanding health patterns and causes, translating research findings into programs to prevent diseases, and promoting wellness. Through biostatistics, biomedical research, and epidemiology, preventative medicine utilizes bioinformatics to identify disease biomarkers and develop screening tests. By leveraging supervised and unsupervised learning techniques, medical databases derived from electronic medical records enable individualized and preventative healthcare (Cheng-Sheng et al. 2020).
-
Gene Therapy and CRISPR Technology: Gene therapy aims to replace unhealthy genes with functional ones, offering potential treatments for various genetic disorders. Recent advancements in biomedical technology, particularly CRISPR, have accelerated gene therapy research. ANNs and ML methods contribute to developing novel gene therapy tools and techniques, reducing the time and cost associated with this therapeutic approach (Cassandra et al. 2022).
-
Drug Design and Virtual Screening: Computational techniques, such as molecular docking, molecular dynamics simulations, and QSAR modeling, have been employed in drug design and virtual screening. These methods aid in predicting the binding affinity of drug candidates and evaluating their potential efficacy against specific targets, such as naphthofuran derivatives for Alzheimer’s disease or protein-targeted medications for hypothyroidism (Law et al. 2019; Akhil et al. 2019; Diego et al. 2016; Anusha et al. 2015).
-
Adaptive Neuro-Fuzzy Inference System (ANFIS) for Environmental and Health Impact Prediction: The adaptive neuro-fuzzy inference system (ANFIS) utilizes QSAR models to predict the potential impact of chemicals on the environment and human health. Descriptor selection methods, enhanced by the Ant Lion optimizer, address challenges such as time complexity and slow convergence. ANFIS, coupled with appropriate descriptors, offers insights into the environmental and health effects of chemicals, aiding in decision-making processes (Mirjalili 2015; Abd et al. 2018).
By exploring these diverse applications of soft computing techniques in biomedical data analysis, researchers and healthcare professionals can harness the power of these approaches to drive innovation, improve patient care, and advance medical knowledge.
5 Analysis and discussion
In this section, we examine the findings of previous studies to provide a comprehensive overview of the research conducted in this review. To summarize the existing literature, we present the results in Tables 1, 2, 3, 4, and 5. These tables showcase the various machine learning and metaheuristic algorithms utilized in advancing the medical field.
Table 1 provides an overview of several comprehensive methods proposed in medical research. These methods employ different algorithms and datasets to address various medical analysis and prediction aspects. The first study by Zainudin et al. (2017) focuses on feature selection in QSAR Biodegradation. They utilize a filter-based feature selection method integrating differential evolution (DE) and the relief-f algorithm. By applying this approach, they could identify the most relevant features for accurate prediction, achieving an 85.4% accuracy rate, with only 16 out of 41 features being deemed relevant. Hussien et al. (2017) propose a wrapper feature selection method to predict the actions of chemical compounds (CCA) in the context of MAO. Their approach selects a subset of molecular descriptors (MD) using a swarm search algorithm (SSA) and a k-NN classifier. The results show that with 783 MDs remaining out of 1665 features, the SSA achieved the highest accuracy rate of 87.35%. While HHO-SVM and HHO-kNN, two classification techniques for predicting drug design, are introduced by Houssein et al. (2020). They apply these methods to MAO and QSAR Biodegradation datasets, obtaining promising results. HHO-SVM and HHO-kNN achieved accuracy rates of 97.583% and 97.599%, respectively, for MAO prediction, while for QSAR Biodegradation, they achieved 85.023% and 84.523% accuracy rates. Martinez et al. (2019) propose methods for multiple-objective optimization in QSAR Biodegradation, explicitly targeting the selection of molecular descriptors through feature selection (FS). They achieve an accuracy rate of 84% and a selection ratio of 37%, demonstrating good performance on the QSAR Biodegradation dataset.
In another study by Martinez et al. (2018), a strategy based on bi-clustering is employed to reduce the number of molecular features necessary for predicting the biodegradation of chemical compounds (RF) in QSAR Biodegradation. They compare three classifiers, including Random Committee (RC), Neural Network (NN), and Random Forest (RF), and find that RF achieves the best accuracy of 88.81% with only 19 MDs. Putra et al. (2019) propose a combination of ANN and SVM for QSAR modeling in biodegradation prediction. Their approach achieves an 82% accuracy rate for classification. Dutta et al. (2019) introduce the Hierarchical Graphlet Similarity Embedding (HGSE) method, which utilizes stochastic graphlet embedding (SGE) on various hierarchical configurations to evaluate molecular graph data in the context of MAO. Their approach achieves an accuracy rate of 95.71%. Goh et al. (2018) explore the prediction of chemical activity using a mix of traditional and contemporary neural architectures. They present the DeepBioD+ and DeepBioD models for the QSAR Biodegradation dataset, achieving 90% and 87.5% accuracy rates, respectively.
Additionally, Goh et al. (2018) propose a deep learning model for chemical activity prediction in QSAR Biodegradation, achieving an accuracy rate of 86.7%. Atwood et al. (2016) employ a deep learning architecture, explicitly utilizing the diffusion representation of graph-structured data, to build a model for MAO prediction. Their approach, based on CNN, achieves an accuracy rate of 75.14%.
Table 2 summarizes published literature studies exploring fuzzy logic’s role in biomedical research. Fuzzy logic, which assigns truth values between 0 and 1 to variables, has gained significance in enhancing various optimization methods and finding applications in medical fields like cancer classification (Ozsahin et al. 2020). In the first study by Fauzi et al. (2021), a fuzzy support vector machine with a principal component analysis (PCA) strategy (FSVM) is proposed. The method is applied to microarray cancer datasets, resulting in an accuracy rate of 96.92% while returning only 60 features. Mousavi et al. (2021) introduce the ACTFRO and GATFRO methods, which utilize Tabu Search with Fuzzy Rough Set for selecting optimal properties. These methods are tested on four cancer-related medical datasets and a non-medical dataset. The proposed methods show improvements in f-measure, accuracy, specificity, sensitivity, and positive projected value, achieving gains of 9%, 5%, and 7%, respectively. Moreover, Anter et al. (2020) combine the Chaos Theory-based crow search optimization algorithm and the Fuzzy C-means algorithm (CFCSA) to address ten medical datasets. Their approach demonstrates overall performance improvements across all the medical datasets considered.
In another study by Lin et al. (2014), fuzzy logic is combined with Fisher’s linear discriminant analysis (FDA) on the MIT-BIH database. The accuracy rates achieved using Fisher’s LDA method and fuzzy logic are 94.03% and 93.87%, respectively. Using a hybridized filter-wrapper technique, Chen et al. (2016) propose a fuzzy criterion for multi-objective unsupervised feature selection (FC-MOFS). They evaluate the FC-MOFS approach on six datasets and demonstrate that it provides more precise and feasible outcomes.
Yang et al. (2019) utilize the Fuzzy Support Vector Machine (FSVM) in combination with the Immune Optimization Algorithm (IOA) (FSVM-IOA) on heart disease datasets. Their approach achieves accuracy rates of 95.82% and 96.01% for forward and reverses FSVM-IOA, respectively. Furthermore, Ye et al. (2021) optimize the Fuzzy K-Nearest Neighbor (FKNN) using HHO and refer to it as HHO-FKNN. The methodology is applied to a COVID-19 dataset, enhancing traditional ML techniques regarding prediction accuracy and stability.
Lastly, Hancer et al. (2015) propose an artificial bee colony with multiple objectives recognized as Fuzzy (MOABC). This method is applied to six datasets and is a valuable tool for solving feature selection problems.
In summary, the studies presented in Table 2 demonstrate the utilization of fuzzy logic in various biomedical research scenarios. These methods leverage fuzzy concepts to improve optimization techniques and enhance the accuracy and effectiveness of prediction and classification tasks in different medical domains.
Table 3 presents some research studies that utilize fuzzy logic in conjunction with metaheuristic algorithms. These studies focus on exploring the potential of fuzzy logic in improving the performance and effectiveness of metaheuristic algorithms in various applications. Overall, these studies demonstrate the potential of fuzzy logic in enhancing the performance and effectiveness of metaheuristic algorithms in various applications such as collision control, traffic signal optimization, control system design, and classification tasks. Researchers aim to achieve improved system performance, reduced errors, and enhanced optimization capabilities by integrating fuzzy logic with metaheuristic algorithms.
Table 4 provides a comparative evaluation of several metaheuristic algorithms (MHs) along with their abilities and limitations. The table highlights key characteristics and performance aspects of each algorithm, allowing for a comprehensive understanding of their strengths and weaknesses.
Table 5 presents a summary of several recent publications in the medical field. These publications highlight various trends and advancements in medical research and technology. Each row represents a publication, including the reference, publication year, and a brief description of the related trend or topic discussed in the paper. The table includes five recent publications from the years 2022 and 2023.
5.1 Comparative analysis of literature reviews
This section compares existing literature reviews focusing on soft computing techniques for biomedical data analysis. By examining the scope, methodologies, and contributions of these reviews, we highlight the unique value and superiority of our review in this specialized domain.
Table 6 presents a summary of these comparative studies, aiming to identify the current gaps and underscore the importance of our contributions.
The table provides an overview of four literature reviews conducted in recent years. Each row represents a review, including the reference, publication year, and a brief description of its main contribution. This comparison aimed to demonstrate the unique aspects and added value of our review of the existing studies.
First, Garg and Mago (2021), published in 2021, focuses on various ML methods for medical data analysis. However, it falls short in considering integrating machine learning with other methods or addressing the challenges and future directions in the field.
Second, Zhijun et al. (2019), published in 2019, primarily concentrates on user-generated content (UGC) information from social media and the application of ML techniques. It lacks statistical results and fails to explore a broader range of data sources.
Third, Suganyadevi et al. (2022), published in 2022, discusses the use of deep learning specifically in medical image analysis. It does not encompass other machine learning methods or address the analysis of different types of medical data.
Lastly, Haleem et al. (2022), also published in 2022, focuses on applying self-organizing map (SOM) artificial neural networks in diagnosing COVID-19. It does not consider alternative diagnostic methods utilizing machine learning or provide a comprehensive overview of the broader medical field.
By highlighting the limitations and scope of these existing reviews, our research aims to bridge the gaps and present a comprehensive and innovative analysis of the biomedical field.
6 Limitation and challenges
This section highlights the limitations and challenges encountered in biomedical data analysis. The biomedical field encompasses various computational biology tasks such as gene discovery, multiple alignments, phylogeny building, homology searches, and protein structure prediction. While several methods have been developed to tackle these problems and improve multiresolution structure prediction and functional unit evaluation (Liu and Duan 2020), challenges still exist in achieving efficient multiresolution modeling and incorporating quantum chemical forces into classical molecular dynamics simulations.
Another set of challenges arises in modeling systems, which involves combining data and constructing complex system models across different spatial and temporal scales. This includes simulation modeling, prediction, statistical analysis, data mining, parameter estimation, and handling uncertainty (Keating et al. 2020). The integration of data and the creation of sophisticated system models could be improved in terms of data management, scalability, and accurately representing system dynamics.
In addition, there are fundamental mathematical challenges in biomedical data analysis, such as formalizing spatial and temporal encoding, and developing theories for systems with stochastic and nonlinear effects, particularly in partially distributed systems (Uçar et al. 2020). Analyzing and visualizing high-dimensional images and utilizing virtual reality (VR) techniques further contribute to the complexity of biomedical data analysis (Lena et al. 2018).
Data management is another critical limitation in biomedical research, encompassing various aspects such as designing data structures, developing efficient query algorithms, modeling heterogeneous data types, process administration, distributed memory, peer-to-peer replication, and data server communication (Leila et al. 2020). The sheer volume and diversity of biomedical data require comprehensive data management solutions to ensure effective storage, retrieval, and analysis.
Identifying drug targets presents a significant challenge, especially for diseases with unknown pathophysiology. The need for confirmed diagnostic and therapeutic biomarkers hinders the objective measurement and detection of biological states. To overcome these challenges, a greater emphasis on human data and integrating biomedical and cheminformatics methods are required (Bender and Brown 2018). The exploration of chemical space, aided by ML methods and optimization algorithms, has played a crucial role in drug target identification and the development of effective treatments.
The availability and accessibility of chemical databases, such as ChEMBL and PubChem, have significantly contributed to biomedical research. However, challenges remain regarding data coverage, standardization, and integration across various databases (Jia et al. 2016). Conventional database techniques often need to be improved for managing and analyzing the vast amount of chemical data, necessitating the application of data mining techniques. Inductive logic programming (ILP) and data mining algorithms are employed to identify frequent substructures, derive probabilistic prediction rules, and enhance the accuracy of chemical data analysis (Cashman et al. 2016).
Protein-ligand docking, a crucial step in drug discovery, involves identifying the structure of the target protein and accurately predicting the binding of ligands. Experimental techniques such as electron microscopy and nuclear magnetic resonance spectroscopy (NMR) aid in determining the 3D macromolecular architectures stored in the Protein Data Bank (PDB) (Wang et al. 2016). However, challenges persist in inter-converting chemical structures and efficiently visualizing macromolecules. Computational modeling techniques, such as those employed in the PyMOL software, facilitate the separation and visualization of ligands and proteins but often require powerful computational resources for high-quality image processing (Shuguang et al. 2016).
Quantitative structure-activity relationship (QSAR) modeling relies on mathematical models to describe the properties and characteristics of chemical compounds. Molecular descriptors, obtained from molecular descriptions and algorithms, play a crucial role in QSAR analysis (Catna and Vijey 2018). However, challenges exist in constructing accurate molecular descriptors, especially for complex molecular graphs. Sophisticated graph theory representations and advanced computational graph theory methods are being explored to overcome these challenges (Werner 2020).
In conclusion, the field of biomedical data analysis faces various limitations and challenges, ranging from computational and mathematical difficulties to data management and integration issues. Addressing these challenges requires innovative approaches, such as integrating soft computing techniques, utilizing advanced data mining algorithms, and the development of robust computational models. Overcoming these limitations will contribute to advancements in biomedical research, ultimately leading to improved understanding, diagnosis, and treatment of diseases.
7 Conclusions
This review provides a comprehensive assessment of various soft computing methods and their application in medical data analysis. It serves as a valuable resource for researchers and practitioners by defining and evaluating these methods and offering insights and guidelines for their effective implementation. One of the key contributions of this review is the extensive collection and analysis of popular medical datasets from diverse resources. Emphasizing the importance of understanding the nature of medical data, the review highlights the significance of extracting relevant and valuable information from these datasets. Additionally, preprocessing methods and techniques for mapping medical data to features are explored, facilitating adequate data preparation for analysis. The review also highlights the significance of optimization algorithms in improving the performance of classification models for medical data analysis. By applying these algorithms, the accuracy and efficiency of classification tasks can be optimized, resulting in improved diagnostic capabilities and decision-making processes. Furthermore, the review delves into the recent advancements in swarm algorithms and machine learning techniques and their applicability to medical data analysis. It discusses how these innovative approaches can effectively solve various medical problems and provides insights into their potential benefits and limitations. Acknowledging the challenges and limitations of using different medical datasets for disease diagnosis or drug proposal is another important aspect covered in this review. By addressing these challenges, researchers better understand the complexities and constraints associated with applying soft computing methods to medical data analysis. Lastly, the review identifies potential future research directions in the field, highlighting areas that require further investigation and advancements. It serves as a valuable reference for researchers seeking to contribute to advancing soft computing techniques in the medical domain. In summary, this review contributes to the existing body of knowledge by comprehensively assessing soft computing methods in medical data analysis, exploring optimization algorithms and swarm algorithms, and addressing challenges and future research directions. It provides a foundation for future studies and advancements in this rapidly evolving field, ultimately improving healthcare outcomes and decision-making processes.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Notes
References
Ab Wahab MN, Nefti-Meziani S, Atyabi A (2015) A comprehensive review of swarm optimization algorithms. PLoS ONE 10(5):e0122827
Abd Elaziz M, Moemen YS, Hassanien AE, Xiong S (2020) Toxicity risks evaluation of unknown FDA biotransformed drugs based on a multi-objective feature selection approach. Appl Soft Comput 97:105509
Abdmouleh Z, Gastli A, Ben-Brahim L, Haouari M, Al-Emadi NA (2017) Review of optimization techniques applied for the integration of distributed generation from renewable energy sources. Renew Energy 113:266–280
Abualigah L, Yousri D, Abd Elaziz M, Ewees AA, Al-Qaness MAA, Gandomi AH (2021) Aquila optimizer: a novel meta-heuristic optimization algorithm. Comput Ind Eng 157:107250
Aggarwal CC, Aggarwal CC (2015) Data classification. Springer, Berlin
Agrawal S, Silakari S (2015) A review on application of particle swarm optimization in bioinformatics. Curr Bioinform 10(4):401–413
Aljarah I, Al-Zoubi AM, Faris H, Hassonah MA, Mirjalili S, Saadeh H (2018) Simultaneous feature selection and support vector machine optimization using the grasshopper optimization algorithm. Cogn Comput 10(3):478–495
Anagnostou P, Barbas P, Vrahatis AG, Tasoulis SK (2020) Approximate kNN classification for biomedical data. In: 2020 IEEE international conference on big data (big data). IEEE, pp 3602–3607
Anand A, Singh AK (2021) Watermarking techniques for medical data authentication: a survey. Multimed Tools Appl 80(20):30165–30197
Andersen JL, Flamm C, Merkle D, Stadler PF (2016) A software package for chemically inspired graph transformation. In: International conference on graph transformation. Springer, pp 73–88
Anter AM, Ali M (2020) Feature selection strategy based on hybrid crow search optimization algorithm integrated with chaos theory and fuzzy c-means algorithm for medical diagnosis problems. Soft Comput 24(3):1565–1584
Anusha CS, Halidha Z, Radha T, Balaji M (2015) Identification of insilico drugs and drug docking studies on hypothyroidism and inferility disorders in human. Int J Novel Trends Pharm Sci 5(3):42–54
Arima A, Tsutsui M, Washio T, Baba Y, Kawai T (2020) Solid-state nanopore platform integrated with machine learning for digital diagnosis of virus infection. Anal Chem 93(1):215–227
Arrieta AB, Díaz-Rodríguez N, Del Ser J, Bennetot A, Tabik S, Barbado A, García S, Gil-López S, Molina D, Benjamins R et al (2020) Explainable artificial intelligence (XAI): concepts, taxonomies, opportunities and challenges toward responsible AI. Inf Fusion 58:82–115
Atwood J, Towsley D (2016) Diffusion-convolutional neural networks. In: Advances in neural information processing systems, pp 1993–2001
Bao X, Zhu K, Liu X, Chen Z, Luo Z, Zhao Q, Ren J, Zuo Z (2022) MeRIPseqPipe: an integrated analysis pipeline for merip-seq data based on nextflow. Bioinformatics 38(7):2054–2056
Barabási A-L, Gulbahce N, Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nat Rev Genet 12(1):56–68
Baran Y, Bercovich A, Sebe-Pedros A, Lubling Y, Giladi A, Chomsky E, Meir Z, Hoichman M, Lifshitz A, Tanay A (2019) Metacell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol 20(1):1–19
Battineni G, Sagaro GG, Chinatalapudi N, Amenta F (2020) Applications of machine learning predictive models in the chronic disease diagnosis. J Personal Med 10(2):21
Baykasoğlu A, Ozsoydan FB (2015) Adaptive firefly algorithm with chaos for mechanical design optimization problems. Appl Soft Comput 36:152–164
Bender A, Brown N (2018) Cheminformatics in drug discovery. ChemMedChem 13(6):467–469
Bender A, Cortes-Ciriano I (2021) Artificial intelligence in drug discovery: what is realistic, what are illusions? Part 2: a discussion of chemical and biological data. Drug Discov Today 26(4):1040–1052
Brogi S, Ramalho TC, Kuca K, Medina-Franco JL, Valko M (2020) In silico methods for drug design and discovery. Front Chem 8:612
Burkart N, Huber MF (2021) A survey on the explainability of supervised machine learning. J Artif Intell Res 70:245–317
Burley SK, Berman HM, Bhikadiya C, Bi C, Chen L, Di Costanzo L, Christie C, Dalenberg K, Duarte JM, Dutta S et al (2019) RCSB protein data bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res 47(D1):D464–D474
Cashman SA, Meyer DE, Edelen AN, Ingwersen WW, Abraham JP, Barrett WM, Gonzalez MA, Randall PM, Ruiz-Mercado G, Smith RL (2016) Mining available data from the united states environmental protection agency to support rapid life cycle inventory modeling of chemical manufacturing. Environ Sci Technol 50(17):9013–9025
Chandrashekar G, Sahin F (2014) A survey on feature selection methods. Comput Electr Eng 40(1):16–28
Charanasomboon T, Viyanon W (2019) A comparative study of repeat buyer prediction: Kaggle acquired value shopper case study. In: Proceedings of the 2019 2nd international conference on information science and systems, pp 306–310
Chen C, Li M, Sui J, Wei K, Pei Q (2016) A genetic algorithm-optimized fuzzy logic controller to avoid rear-end collisions. J Adv Transp 50(8):1735–1753
Cheng L, Zhao H, Wang P, Zhou W, Luo M, Li T, Han J, Liu S, Jiang Q (2019) Computational methods for identifying similar diseases. Mol Therapy-Nucleic Acids 18:590–604
Choudhuri A, Trompouki E, Abraham BJ, Colli LM, Kock HK, Mallard W, Yang M-L, Vinjamur DS, Ghamari A, Sporrij A et al (2020) Common variants in signaling transcription-factor-binding sites drive phenotypic variability in red blood cell traits. Nat Genet 52(12):1333–1345
Ciociola AA, Cohen LB, Kulkarni P, Kefalas C, Buchman A, Burke C, Cain T, Connor J, Ehrenpreis ED, Fang J, et al (2014) How drugs are developed and approved by the FDA: current process and future directions. Off J Am Coll Gastroenterol| ACG, 109(5):620–623
Clayman CL, Srinivasan SM, Sangwan RS (2020) K-means clustering and principal components analysis of microarray data of 1000 landmark genes. Procedia Comput Sci 168:97–104
Crawford B, Soto R, Astorga G, García J, Castro C, Paredes F (2017) Putting continuous metaheuristics to work in binary search spaces. Complexity. https://doi.org/10.1155/2017/8404231
Cui Z, Zhang Y (2014) Swarm intelligence in bioinformatics: methods and implementations for discovering patterns of multiple sequences. J Nanosci Nanotechnol 14(2):1746–1757
Danger R, Segura-Bedmar I, Martínez P, Rosso P (2010) A comparison of machine learning techniques for detection of drug target articles. J Biomed Inform 43(6):902–913
de Jongh RPH, van Dijk ADJ, Julsing MK, Schaap PJ, de Ridder D (2020) Designing eukaryotic gene expression regulation using machine learning. Trends Biotechnol 38(2):191–201
Derrac J, García S, Molina D, Herrera F (2011) A practical tutorial on the use of nonparametric statistical tests as a methodology for comparing evolutionary and swarm intelligence algorithms. Swarm Evolut Comput 1(1):3–18
Devi RV, Sathya SS, Coumar MS (2015) Evolutionary algorithms for de novo drug design-a survey. Appl Soft Comput 27:543–552
Di Muzio E, Toti D, Polticelli F (2017) Dockingapp: a user friendly interface for facilitated docking simulations with autodock vina. J Comput-Aided Mol Des 31(2):213–218
Diez-Fuertes F, Iglesias-Caballero M, Garcia-Perez J, Monzon S, Jiménez P, Varona S, Cuesta I, Zaballos A, Jimenez M, Checa L et al (2021) A founder effect led early sars-cov-2 transmission in Spain. J Virol 95(3):e01583-20
Doğan E, Akgüngör AP (2016) Optimizing a fuzzy logic traffic signal controller via the differential evolution algorithm under different traffic scenarios. Simulation 92(11):1013–1023
Dutta A, Riba P, Lladós J, Fornés A (2019) Hierarchical stochastic graphlet embedding for graph-based pattern recognition. Neural Comput Appl 32:11579–11596
Eastgate MD, Schmidt MA, Fandrick KR (2017) On the design of complex drug candidate syntheses in the pharmaceutical industry. Nat Rev Chem 1(2):1–16
Elaziz MA, Moemen YS, Hassanien AE, Xiong S (2018) Quantitative structure-activity relationship model for HCVNS5B inhibitors based on an antlion optimizer-adaptive neuro-fuzzy inference system. Sci Rep 8(1):1–17
Emam MM, Samee NA, Jamjoom MM, Houssein EH (2023) Optimized deep learning architecture for brain tumor classification using improved hunger games search algorithm. Comput Biol Med 160:106966
Emam MM, El-Sattar HA, Houssein EH, Kamel S (2023) Modified orca predation algorithm: developments and perspectives on global optimization and hybrid energy systems. Neural Comput Appl 35:15051–15073
Emam MM, Houssein EH, Ghoniem RM (2023) A modified reptile search algorithm for global optimization and image segmentation: case study brain MRI images. Comput Biol Med 152:106404
Erickson BJ, Korfiatis P, Akkus Z, Kline TL (2017) Machine learning for medical imaging. Radiographics 37(2):505
Ershadi MM, Seifi A (2022) Applications of dynamic feature selection and clustering methods to medical diagnosis. Appl Soft Comput 126:109293
Ershadi MM, Rise ZR, Niaki STA (2022) A hierarchical machine learning model based on glioblastoma patients’ clinical, biomedical, and image data to analyze their treatment plans. Comput Biol Med 150:106159
Fauzi IR, Rustam Z, Wibowo A (2021) Multiclass classification of leukemia cancer data using fuzzy support vector machine (FSVM) with feature selection using principal component analysis (PCA). J Phys: Conf Ser 1725:012012
Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ (2016) Computational protein-ligand docking and virtual drug screening with the autodock suite. Nat Protoc 11(5):905
Gambhir S, Malik SK, Kumar Y (2016) Role of soft computing approaches in healthcare domain: a mini review. J Med Syst 40(12):1–20
Gan G, Ma C, Wu J (2020) Data clustering: theory, algorithms, and applications. SIAM
García-Torres M, Gómez-Vela F, Melián-Batista B, Marcos Moreno-Vega J (2016) High-dimensional feature selection via feature grouping: a variable neighborhood search approach. Inf Sci 326:102–118
Garg A, Mago V (2021) Role of machine learning in medical research: a survey. Comput Sci Rev 40:100370
Gasteiger J (2016) Chemoinformatics: achievements and challenges, a personal view. Molecules 21(2):151
Gertrudes JC, Maltarollo VG, Silva RA, Oliveira PR, Honorio KM, Da Silva ABF (2012) Machine learning techniques and drug design. Curr Med Chem 19(25):4289–4297
Goh GB, Sakloth K, Siegel C, Vishnu A, Pfaendtner J (2018) Multimodal deep neural networks using both engineered and learned representations for biodegradability prediction. arXiv preprintarXiv:1808.04456
Goh GB, Siegel C, Vishnu A, Hodas N (2018) Using rule-based labels for weak supervised learning: a ChemNet for transferable chemical property prediction. In: Proceedings of the 24th ACM SIGKDD international conference on knowledge discovery & data mining, pp 302–310
Grenier P-A, Brun L, Villemin D (2017) Chemoinformatics and stereoisomerism: a stereo graph kernel together with three new extensions. Pattern Recogn Lett 87:222–230
Gunavathi C, Premalatha K (2014) A comparative analysis of swarm intelligence techniques for feature selection in cancer classification. Sci World J. https://doi.org/10.1155/2014/693831
Gupta Y, Saini A (2017) A novel fuzzy-PSO term weighting automatic query expansion approach using combined semantic filtering. Knowl-Based Syst 136:97–120
Gupta D, Kose U, Khanna A, Balas VE (2022) Deep learning for medical applications with unique data. Academic Press, Cambridge
Gupta D, Borah P, Sharma UM, Prasad M (2022) Data-driven mechanism based on fuzzy Lagrangian twin parametric-margin support vector machine for biomedical data analysis. Neural Comput Appl 34(14):11335–11345
Gusenbauer M, Haddaway NR (2020) Which academic search systems are suitable for systematic reviews or meta-analyses? Evaluating retrieval qualities of google scholar, Pubmed, and 26 other resources. Res Synth Methods 11(2):181–217
Haider Z, Subhani MM, Farooq MA, Ishaq M, Khalid M, Khan RSA, Niazi AK (2020) In silico discovery of novel inhibitors against main protease (Mpro) of sars-cov-2 using pharmacophore and molecular docking based virtual screening from zinc database. Preprint
Halalli B, Makandar A (2018) Computer aided diagnosis-medical image analysis techniques. Breast Imaging 85:85–109
Haleem A, Javaid M, Khan I (2020) Holography applications toward medical field: an overview. Indian J Radiol Imaging 30(03):354–361
Haleem A, Javaid M, Singh RP, Suman R (2022) Medical 4.0 technologies for healthcare: features, capabilities, and applications. Internet Things Cyber-Phys Syst 2:12–30
Han F, Yang C, Wu Y-Q, Zhu J-S, Ling Q-H, Song Y-Q, Huang D-S (2015) A gene selection method for microarray data based on binary PSO encoding gene-to-class sensitivity information. IEEE/ACM Trans Comput Biol Bioinform 14(1):85–96
Han F, Tang D, Cheng Z, Jiang J, Li Q-W et al (2019) A hybrid gene selection method based on gene scoring strategy and improved particle swarm optimization. BMC Bioinform 20(8):1–13
Hancer E, Xue B, Zhang M, Karaboga D, Akay B (2018) Pareto front feature selection based on artificial bee colony optimization. Inf Sci 422:462–479
Hancer E, Xue B, Zhang M, Karaboga D, Akay B (2015) A multi-objective artificial bee colony approach to feature selection using fuzzy mutual information. In: 2015 IEEE congress on evolutionary computation (CEC). IEEE, pp 2420–2427
Hedayati R, Khedmati M, Taghipour-Gorjikolaie M (2021) Deep feature extraction method based on ensemble of convolutional auto encoders: application to alzheimer’s disease diagnosis. Biomed Signal Process Control 66:102397
Heidari AA, Mirjalili S, Faris H, Aljarah I, Mafarja M, Chen H (2019) Harris hawks optimization: algorithm and applications. Future Gener Comput Syst 97:849–872
Hemalatha CN, Muthkumar VA (2018) Application of 3d qsar and docking studies in optimization of perylene diimides as anti cancer agent. Indian J Pharm Educ Res 52:666–75
Houssein EH, Hosney ME, Oliva D (2021) A hybrid seagull optimization algorithm for chemical descriptors classification. In: 2021 international mobile, intelligent, and ubiquitous computing conference (MIUCC). IEEE, pp 1–6
Houssein EH, Sayed A (2023) Dynamic candidate solution boosted beluga whale optimization algorithm for biomedical classification. Mathematics 11(3):707
Houssein EH, Hosney ME, Oliva D, Mohamed WM, Hassaballah M (2020) A novel hybrid Harris hawks optimization and support vector machines for drug design and discovery. Comput Chem Eng 133:106656
Houssein EH, Neggaz N, Hosney ME, Mohamed WM, Hassaballah M (2021) Enhanced Harris hawks optimization with genetic operators for selection chemical descriptors and compounds activities. Neural Comput Appl 33:13601–13618
Houssein EH, Emam MM, Ali AA, Suganthan PN (2021) Deep and machine learning techniques for medical imaging-based breast cancer: a comprehensive review. Expert Syst Appl 167:114161
Houssein EH, Gad AG, Wazery YM, Suganthan PN (2021) Task scheduling in cloud computing based on meta-heuristics: review, taxonomy, open challenges, and future trends. Swarm Evolut Comput 62:100841
Houssein EH, Mahdy MA, Blondin MJ, Doaa S, Mohamed WM (2021) Hybrid slime mould algorithm with adaptive guided differential evolution algorithm for combinatorial and global optimization problems. Expert Syst Appl 174:114689
Houssein EH, Emam MM, Ali AA (2022) An optimized deep learning architecture for breast cancer diagnosis based on improved marine predators algorithm. Neural Comput Appl 34(20):18015–18033
Houssein EH, Çelik E, Mahdy MA, Ghoniem RM (2022) Self-adaptive equilibrium optimizer for solving global, combinatorial, engineering, and multi-objective problems. Expert Syst Appl 195:116552
Houssein EH, Hosney ME, Mohamed WM, Ali AA, Younis EMG (2023) Fuzzy-based hunger games search algorithm for global optimization and feature selection using medical data. Neural Comput Appl 35(7):5251–5275
Hu H, Cui X, Bai Y (2017) Two kinds of classifications based on improved gravitational search algorithm and particle swarm optimization algorithm. Adv Math Phys. https://doi.org/10.1155/2017/2131862
Hunt C, Montgomery S, Berkenpas JW, Sigafoos N, Oakley JC, Espinosa J, Justice N, Kishaba K, Hippe K, Si D et al (2022) Recent progress of machine learning in gene therapy. Curr Gene Ther 22(2):132–143
Hussien AG, Hassanien AE, Houssein EH (2017) Swarming behaviour of salps algorithm for predicting chemical compound activities. In: 2017 eighth international conference on intelligent computing and information systems (ICICIS). IEEE, pp 315–320
Huynh T, Nibali A, He Z (2022) Semi-supervised learning for medical image classification using imbalanced training data. Comput Methods Progr Biomed 216:106628
Ibrahim D (2016) An overview of soft computing. Procedia Comput Sci 102:34–38
Ison MG, Wolfe C, Boucher HW (2020) Emergency use authorization of remdesivir: the need for a transparent distribution process. JAMA 323(23):2365–2366
Jamali AA, Ferdousi R, Razzaghi S, Li J, Safdari R, Ebrahimie E (2016) Drugminer: comparative analysis of machine learning algorithms for prediction of potential druggable proteins. Drug Discov Today 21(5):718–724
Jena L, Nayak S, Swain R (2020) Chronic disease risk (CDR) prediction in biomedical data using machine learning approach. In: Advances in intelligent computing and communication. Springer, pp 232–239
Jhalia V, Swarnkar T (2021) A critical review on the application of artificial neural network in bioinformatics. In: Data analytics in bioinformatics: a machine learning perspective, pp 51–76
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN et al (2016) CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res, p gkw1004
Jiménez-García B, Roel-Touris J, Romero-Durana M, Vidal M, Jiménez-González D, Fernández-Recio J (2018) Lightdock: a new multi-scale approach to protein-protein docking. Bioinformatics 34(1):49–55
Judd CM, McClelland GH, Ryan CS (2017) Data analysis: a model comparison approach to regression, ANOVA, and beyond. Routledge, New York
Kalantari A, Kamsin A, Shamshirband S, Gani A, Alinejad-Rokny H, Chronopoulos AT (2018) Computational intelligence approaches for classification of medical data: state-of-the-art, future challenges and research directions. Neurocomputing 276:2–22
Kamble VH, Dale MP (2022) Machine learning approach for longitudinal face recognition of children. In: Machine learning for biometrics. Elsevier, pp 1–27
Kandel BM, Wolk DA, Gee JC, Avants B (2013) Predicting cognitive data from medical images using sparse linear regression. In: Information processing in medical imaging: 23rd international conference, IPMI 2013, Asilomar, CA, USA, June 28–July 3, 2013. Proceedings 23. Springer, pp 86–97
Karabatak M, Mustafa T (2018) Performance comparison of classifiers on reduced phishing website dataset. In: 2018 6th international symposium on digital forensic and security (ISDFS). IEEE, pp 1–5
Karaboga D, Aslan SE (2016) A discrete artificial bee colony algorithm for detecting transcription factor binding sites in DNA sequences. Genet Mol Res 15(2):1–11
Katsila T, Spyroulias GA, Patrinos GP, Matsoukas M-T (2016) Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J 14:177–184
Kaur P, Kaur R (2020) Comparative analysis of classification techniques for diagnosis of diabetes. In: Advances in bioinformatics, multimedia, and electronics circuits and signals. Springer, pp 215–221
Keating SM, Waltemath D, König M, Zhang F, Dräger A, Chaouiya C, Bergmann FT, Finney A, Gillespie CS, Helikar T et al (2020) SBML level 3: an extensible format for the exchange and reuse of biological models. Mol Syst Biol 16(8):e9110
Kennedy J, Eberhart RC (1997) A discrete binary version of the particle swarm algorithm. In: 1997 IEEE international conference on systems, man, and cybernetics. Computational cybernetics and simulation. IEEE, vol 5, pp 4104–4108
Khan AU et al (2016) Descriptors and their selection methods in QSAR analysis: paradigm for drug design. Drug Discov Today 21(8):1291–1302
Kotsovsky V, Geche V, Batyuk A (2020) Bithreshold neural network classifier. In: 2020 IEEE 15th international conference on computer sciences and information technologies (CSIT), vol 1, pp 32–35. IEEE
Kumar A, Srivastava G, Negi AS, Ashok S (2019) Docking, molecular dynamics, binding energy-MM-PBSA studies of naphthofuran derivatives to identify potential dual inhibitors against bace-1 and gsk-3\(\beta\). J Biomol Struct Dyn 37(2):275–290
Lauv P, Shukla T, Huang X, Ussery DW, Wang S (2020) Machine learning methods in drug discovery. Molecules 25(22):5277
Lavecchia A (2015) Machine-learning approaches in drug discovery: methods and applications. Drug Discov Today 20(3):318–331
Law TR, Hancox J, Wright SA, Jarvis SA (2019) An algorithm for computing short-range forces in molecular dynamics simulations with non-uniform particle densities. J Parallel Distrib Comput 130:1–11
Leila I, Materwala H, Karduck AP, Adem A et al (2020) Requirements of health data management systems for biomedical care and research: scoping review. J Med Internet Res 22(7):e17508
Li S, Chen H, Wang M, Heidari AA, Mirjalili S (2020) Slime mould algorithm: a new method for stochastic optimization. Future Gener Comput Syst 111:300–323
Li J, Tong X-Y, Zhu L-D, Zhang H-Y (2020) A machine learning method for drug combination prediction. Front Genet 11:1000
Lilla AD, Khan MA, Barendse P (2013) Comparison of differential evolution and genetic algorithm in the design of permanent magnet generators. In: 2013 IEEE international conference on industrial technology (ICIT). IEEE, pp 266–271
Lin H, Siu SWI (2018) A hybrid cuckoo search and differential evolution approach to protein-ligand docking. Int J Mol Sci 19(10):3181
Lin L-C, Yeh Y-C, Chu T-Y (2014) Feature selection algorithm for ECG signals and its application on heartbeat case determining. Int J Fuzzy Syst 16(4):483–496
Lin Y, Hu Q, Liu J, Zhu X, Wu X (2021) MULFE: multi-label learning via label-specific feature space ensemble. ACM Trans Knowl Discov Data (TKDD) 16(1):1–24
Liu H, Duan Z (2020) Corrected multi-resolution ensemble model for wind power forecasting with real-time decomposition and bivariate kernel density estimation. Energy Convers Manag 203:112265
Liu Y, Zhao L, Li W, Zhao D, Song M, Yang Y (2013) FIPSdock: a new molecular docking technique driven by fully informed swarm optimization algorithm. J Comput Chem 34(1):67–75
Liu J, Xing Y, Li Y (2018) A gravitational search algorithm with adaptive mixed mutation for function optimization. Int J Perform Eng 14(4):681
Lo Y-C, Rensi SE, Torng W, Altman RB (2018) Machine learning in chemoinformatics and drug discovery. Drug Discov Today 23(8):1538–1546
Lynch I, Dawson KA (2020) Protein–nanoparticle interactions. In: Nano-enabled medical applications, pp 231–250
Ma C-Y, Liao C-S (2020) A review of protein-protein interaction network alignment: from pathway comparison to global alignment. Comput Struct Biotechnol J 18:2647–2656
Ma Y, Li H-L, Chen X-B, Jin W-Y, Zhou H, Wang R-L (2018) 3D QSAR pharmacophore based virtual screening for identification of potential inhibitors for CDC25B. Comput Biol Chem 73:1–12
Mafarja M, Thaher T, Too J, Chantar H, Turabieh H, Houssein EH, Emam MM (2023) An efficient high-dimensional feature selection approach driven by enhanced multi-strategy grey wolf optimizer for biological data classification. Neural Comput Appl 35(2):1749–1775
Mahmoodabadi MJ, Danesh N (2018) Gravitational search algorithm-based fuzzy control for a nonlinear ball and beam system. J Control Decis 5(3):229–240
Maier-Hein L, Eisenmann M, Reinke A, Onogur S, Stankovic M, Scholz P, Arbel T, Bogunovic H, Bradley AP, Carass A et al (2018) Why rankings of biomedical image analysis competitions should be interpreted with care. Nat Commun 9(1):1–13
Martínez MJ, Dussaut JS, Ponzoni I (2018) Biclustering as strategy for improving feature selection in consensus QSAR modeling. Electron Notes Discret Math 69:117–124
Martínez MJ, Razuc M, Ponzoni I (2019) Modesus: a machine learning tool for selection of molecular descriptors in qsar studies applied to molecular informatics. BioMed Res Int. https://doi.org/10.1155/2019/2905203
Masand VH, Rastija V (2017) Pydescriptor: a new pymol plugin for calculating thousands of easily understandable molecular descriptors. Chemom Intell Lab Syst 169:12–18
Maseleno A, Sabani N, Huda M, Ahmad R, Jasmi KA, Basiron B (2018) Demystifying learning analytics in personalised learning. Int J Eng Technol 7(3):1124–1129
Maulud D, Abdulazeez AM (2020) A review on linear regression comprehensive in machine learning. J Appl Sci Technol Trends 1(4):140–147
Maurer TS, Smith D, Beaumont K, Di L (2020) Dose predictions for drug design. J Med Chem 63(12):6423–6435
Mauri A, Consonni V, Pavan M, Todeschini R (2006) Dragon software: an easy approach to molecular descriptor calculations. MATCH Commun Math Comput Chem 56:237–248
Meng T, Jing X, Yan Z, Pedrycz W (2020) A survey on machine learning for data fusion. Inf Fusion 57:115–129
Mirjalili S (2015) The ant lion optimizer. Adv Eng Softw 83:80–98
Mirjalili S, Lewis A (2016) The whale optimization algorithm. Adv Eng Softw 95:51–67
Mirjalili S, Mirjalili SM, Lewis A (2014) Grey wolf optimizer. Adv Eng Softw 69:46–61
Mitchell J (2014) Machine learning methods in chemoinformatics. Wiley Interdiscip Rev: Comput Mol Sci 4(5):468–481
Mohanty B, Tripathy S (2016) A teaching learning based optimization technique for optimal location and size of dg in distribution network. J Electr Syst Inf Technol 3(1):33–44
Moriwaki H, Tian Y-S, Kawashita N, Takagi T (2018) Mordred: a molecular descriptor calculator. J Cheminform 10(1):4
Mousavi SM, Abdullah S, Niaki STA, Banihashemi S (2021) An intelligent hybrid classification algorithm integrating fuzzy rule-based extraction and harmony search optimization: medical diagnosis applications. Knowl-based Syst 220:106943
Ng MCK, Fong S, Siu SWI (2015) PSOVina: the hybrid particle swarm optimization algorithm for protein-ligand docking. J Bioinform Comput Biol 13(03):1541007
Ning J, Zhang C, Sun P, Feng Y (2019) Comparative study of ant colony algorithms for multi-objective optimization. Information 10(1):11
Norrby M, Grebner C, Eriksson J, Bostrom J (2015) Molecular rift: virtual reality for drug designers. J Chem Inf Model 55(11):2475–2484
Osama S, Shaban H, Ali AA (2022) Gene reduction and machine learning algorithms for cancer classification based on microarray gene expression data: a comprehensive review. Expert Syst Appl 213:118946
Oughtred R, Rust J, Chang C, Breitkreutz B-J, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F et al (2021) The biogrid database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 30(1):187–200
Ozsahin DU, Uzun B, Ozsahin I, Mustapha MT, Musa MS (2020) Fuzzy logic in medicine. In: Biomedical signal processing and artificial intelligence in healthcare. Elsevier, pp 153–182
Peh SCW, Hong JL (2016) Bacteria foraging optimization for drug design. In: International conference on computational science and its applications. Springer, pp 322–331
Ponce P, Soriano LA, Molina A, Garcia M (2018) Optimization of fuzzy logic controllers by particle swarm optimization to increase the lifetime in power electronic stages. In: Electric machines for smart grids applications-design, simulation and control
Prada-Gracia D, Huerta-Yépez S, Moreno-Vargas LM (2016) Application of computational methods for anticancer drug discovery, design, and optimization. Boletín Médico Del Hospital Infantil de México (English Edition) 73(6):411–423
Putra RID, Maulana AL, Saputro AG (2019) Study on building machine learning model to predict biodegradable-ready materials. In: AIP conference proceedings. AIP Publishing LLC, vol 2088, p 060003
Rajula HSR, Verlato G, Manchia M, Antonucci N, Fanos V (2020) Comparison of conventional statistical methods with machine learning in medicine: diagnosis, drug development, and treatment. Medicina 56(9):455
Rashedi E, Nezamabadi-Pour H, Saryazdi S (2009) GSA: a gravitational search algorithm. Inf Sci 179(13):2232–2248
Rem BS, Käming N, Tarnowski M, Asteria L, Fläschner N, Becker C, Sengstock K, Weitenberg C (2019) Identifying quantum phase transitions using artificial neural networks on experimental data. Nat Phys 15(9):917–920
Renaud J-P, Chung C, Danielson UH, Egner U, Hennig M, Hubbard RE, Nar H (2016) Biophysics in drug discovery: impact, challenges and opportunities. Nat Rev Drug Discov 15(10):679
Rodríguez-Perez R, Vogt M, Bajorath J (2017) Support vector machine classification and regression prioritize different structural features for binary compound activity and potency value prediction. ACS omega 2(10):6371–6379
Roosan D, Chok J, Baskys A, Roosan MR (2022) PGxKnow: a pharmacogenomics educational hololens application of augmented reality and artificial intelligence. Pharmacogenomics 23(4):235–245
Sa Lakshmi K, Vadivu G (2017) Extracting association rules from medical health records using multi-criteria decision analysis. Procedia Comput Sci 115:290–295
Safi SJ, Tezcan SS, Eke I, Farhad Z (2018) Gravitational search algorithm (GSA) based pid controller design for two area multi-source power system load frequency control (LFC). Gazi Univ J Sci 31(1):139–153
Sallam KM, Elsayed SM, Chakrabortty RK, Ryan MJ (2020) Improved multi-operator differential evolution algorithm for solving unconstrained problems. In: 2020 IEEE congress on evolutionary computation (CEC). IEEE, pp. 1–8
Satapathy R, Cambria E, Hussain A (2017) Sentiment analysis in the bio-medical domain, vol 11. Springer, Cham, p 6630
Schoch CL, Ciufo S, Domrachev M, Hotton CL, Kannan S, Rogneda K, Leipe D, Mcveigh R, O’Neill K, Robbertse B et al (2020) NCBI taxonomy: a comprehensive update on curation, resources and tools. Database 2020
Sen PC, Hajra M, Ghosh M (2020) Supervised classification algorithms in machine learning: a survey and review. In: Emerging technology in modelling and graphics: proceedings of IEM graph 2018. Springer, Berlin, pp 99–111
Shehabeldeen TA, Abd Elaziz M, Elsheikh AH, Hassan OF, Yin Y, Ji X, Shen X, Zhou J (2020) A novel method for predicting tensile strength of friction stir welded aa6061 aluminium alloy joints based on hybrid random vector functional link and henry gas solubility optimization. IEEE Access 8:79896–79907
Shen D, Jiang T, Chen W, Shi Q, Gao S (2015) Improved chaotic gravitational search algorithms for global optimization. In: 2015 IEEE congress on evolutionary computation (CEC). IEEE, pp 1220–1226
Shi B, Heidari AA, Chen C, Wang M, Huang C, Chen H, Zhu J (2020) Predicting di-2-ethylhexyl phthalate toxicity: hybrid integrated Harris hawks optimization with support vector machines. IEEE Access 8:161188–161202
Singh N, Houssein EH, Singh SB, Dhiman G (2022) Hssahho: a novel hybrid Salp swarm-Harris hawks optimization algorithm for complex engineering problems. J Ambient Intell Humaniz Comput 14:11569–11605
Sörensen K, Sevaux M, Glover F (2018) A history of metaheuristics. In: Handbook of heuristics. Springer, pp 791–808
Stevens H (2015) The politics of sequence: data sharing and the open source software movement. Inf Cult 50(4):465–503
Suganyadevi S, Seethalakshmi V, Balasamy K (2022) A review on deep learning in medical image analysis. Int J Multimed Inf Retr 11(1):19–38
Sughasiny M, Rajeshwari J (2018) Application of machine learning techniques, big data analytics in health care sector—a literature survey. In: 2018 2nd international conference on I-SMAC (IoT in social, mobile, analytics and cloud)(I-SMAC) I-SMAC (IoT in social, mobile, analytics and cloud) (I-SMAC), 2018 2nd international conference on. IEEE, pp 741–749
Tarle B, Tajanpure R, Jena S (2016) Medical data classification using different optimization techniques: a survey. Int J Res Eng Technol 5:101–108
Telikani A, Gandomi AH, Shahbahrami A (2020) A survey of evolutionary computation for association rule mining. Inf Sci 524:318–352
Toropova MA, Veselinović AM, Veselinović JB, Stojanović DB, Toropov AA (2015) QSAR modeling of the antimicrobial activity of peptides as a mathematical function of a sequence of amino acids. Comput Biol Chem 59:126–130
Torres PHM, Sodero ACR, Jofily P, Silva-Jr FP (2019) Key topics in molecular docking for drug design. Int J Mol Sci 20(18):4574
Uçar MK, Nour M, Sindi H, Polat K (2020) The effect of training and testing process on machine learning in biomedical datasets. Math Probl Eng. https://doi.org/10.1155/2020/2836236
Van Engelen JE, Hoos HH (2020) A survey on semi-supervised learning. Mach Learn 109(2):373–440
Wah YB, Ibrahim N, Hamid HA, Abdul-Rahman S, Fong S (2018) Feature selection methods: case of filter and wrapper approaches for maximising classification accuracy. Pertanika J Sci Technol 26(1):329–340
Wang Z, Sun H, Yao X, Li D, Xu L, Li Y, Tian S, Hou T (2016) Comprehensive evaluation of ten docking programs on a diverse set of protein-ligand complexes: the prediction accuracy of sampling power and scoring power. Phys Chem Chem Phys 18(18):12964–12975
Werner F (2020) Graph-theoretic problems and their new applications
Willighagen E (2021) Bacting: a next generation, command line version of bioclipse. J Open Source Softw 6(62):2558
Winkel DJ, Heye T, Weikert TJ, Boll DT, Stieltjes B (2019) Evaluation of an AI-based detection software for acute findings in abdominal computed tomography scans: toward an automated work list prioritization of routine CT examinations. Investig Radiol 54(1):55–59
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z et al (2018) Drugbank 5.0: a major update to the drugbank database for 2018. Nucleic Acids Res 46(D1):D1074–D1082
Wolpert DH, Macready WG (1997) No free lunch theorems for optimization. IEEE Trans Evolut Comput 1(1):67–82
Woźniak M, Grana M, Corchado E (2014) A survey of multiple classifier systems as hybrid systems. Inf Fusion 16:3–17
Yang B (2019) Dynamic risk identification safety model based on fuzzy support vector machine and immune optimization algorithm. Saf Sci 118:205–211
Yang P (2023) Data visualization and prediction for telecom customer churn. Highlights Sci Eng Technol 39:1080–1085
Yang W, Li G (2013) Application an hybird artificial fish swarm algorithm in motif detecting problem. J Bionanosci 7(6):703–706
Yang M, Derbyshire MK, Yamashita RA, Marchler-Bauer A (2020) NCBI’s conserved domain database and tools for protein domain analysis. Curr Protoc Bioinform 69(1):e90
Yap TA, Smith AD, Ferraldeschi R, Al-Lazikani B, Workman P, De Bono JS (2016) Drug discovery in advanced prostate cancer: translating biology into therapy. Nat Rev Drug Discov 15(10):699–718
Ye C, Ji G, Li L, Liang C (2014) detectIR: a novel program for detecting perfect and imperfect inverted repeats using complex numbers and vector calculation. PLoS ONE 9(11):e113349
Ye H, Wu P, Zhu T, Xiao Z, Zhang X, Zheng L, Zheng R, Sun Y, Zhou W, Fu Q et al (2021) Diagnosing coronavirus disease 2019 (covid-19): efficient Harris hawks-inspired fuzzy k-nearest neighbor prediction methods. IEEE Access 9:17787–17802
Yin Z, Sulieman LM, Malin BA (2019) A systematic literature review of machine learning in online personal health data. J Am Med Inform Assoc 26(6):561–576
Yu W, MacKerell AD (2017) Computer-aided drug design methods. Antibiotics. Springer, Berlin, pp 85–106
Yu C-S, Lin Y-J, Lin C-H, Lin S-Y, Wu JL, Chang SS et al (2020) Development of an online health care assessment for preventive medicine: a machine learning approach. J Med Internet Res 22(6):e18585
Yu C-H, Khare E, Narayan OP, Parker R, Kaplan DL, Buehler MJ (2022) ColGen: an end-to-end deep learning model to predict thermal stability of de novo collagen sequences. J Mech Behav Biomed Mater 125:104921
Yuan S, Chan HCS, Filipek S, Vogel H (2016) PyMOL and Inkscape bridge the data and the data visualization. Structure 24(12):2041–2042
Zainudin M, Sulaiman M, Mustapha N, Perumal T, Nazri A, Mohamed R, Manaf S (2017) Feature selection optimization using hybrid relief-f with self-adaptive differential evolution. Int J Intell Eng Syst 10(3):21–29
Zhang H-M, Kuang S, Xiong X, Gao T, Liu C, Guo A-Y (2015) Transcription factor and microrna co-regulatory loops: important regulatory motifs in biological processes and diseases. Briefings Bioinform 16(1):45–58
Zhang JF, Paciorkowski AR, Craig PA, Cui F (2019) BioVR: a platform for virtual reality assisted biological data integration and visualization. BMC Bioinform 20(1):1–10
Zhang L, Song J, Kong L, Yuan T, Li W, Zhang W, Hou B, Lu Y, Du G (2020) The strategies and techniques of drug discovery from natural products. Pharmacol Ther 216:107686
Zhang H, Liu C-T, Mao J, Shen C, Xie R-L, Mu B (2020) Development of novel in silico prediction model for drug-induced ototoxicity by using naïve bayes classifier approach. Toxicol Vitro 65:104812
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
Essam H. Houssein: Supervising, Methodology, Conceptualization, Formal analysis, Investigation, Visualization, Writing - review & editing. Mosa E. Hosney: Resources, Writing - original draft. Marwa M. Emam: Methodology, Conceptualization, Formal analysis, Investigation, Visualization, Writing - review & editing. Eman M.G. Younis: Methodology, Formal analysis, Investigation, Data Curation, Writing - review & editing. Waleed M. Mohamed: Conceptualization, Formal analysis, Investigation, Data Curation, Writing - review & editing. Abdelmgeid A. Ali: Supervision, Formal analysis. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest. The authors declare no competing interests. Non-financial competing interests. Funding information is not available.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Houssein, E.H., Hosney, M.E., Emam, M.M. et al. Soft computing techniques for biomedical data analysis: open issues and challenges. Artif Intell Rev 56 (Suppl 2), 2599–2649 (2023). https://doi.org/10.1007/s10462-023-10585-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10462-023-10585-2