
Abstract
The nematode Caenorhabditis elegans is a leading model system in genetics, development and neurobiology; its transparent body and small size make it particularly suitable for fluorescent imaging of cells and neurons within microfluidic setups. Simultaneously recording activity in bilaterally symmetric cells has proved difficult in C. elegans because the worm enters the chip and is then immobilised when it is lying on one side of the body. We developed a side-view rotatable microfluidic device that allows us to image a pair of bilateral neurons in a single focal plane of an epi-fluorescence microscope. We demonstrated the utility of the device by recording the responses of immobilised worms to controlled stimuli, focusing on the responses of two classes of head sensory neurons to changes in NaCl concentration. The results indicate that responses of ASE left and right and ASH left and right sensory neurons are stochastic. Simultaneous recordings of ASH left and right neurons tend to synchronise, pointing to a role of gap junctional connectivity. The anatomy of the C. elegans nerve ring makes this microfluidic approach ideally suited for the study of spatially extended pairs of neurons or larger neuronal circuits that lie within a limited depth of field.
Similar content being viewed by others


1 Introduction
Microfluidic technology offers an excellent set of tools for the manipulation of small animals and measurement of their spontaneous and evoked responses under tightly controlled conditions (Chokshi et al. 2009; Chronis et al. 2007; Chung et al. 2008; Hulme et al. 2007; Krajniak and Lu 2010; Muthaiyan Shanmugam and Subhra Santra 2016; Shi et al. 2008). Microfluidics provide precise flow control so that experiments can be performed with minimal amounts of reagents, thus minimising the risk of chemical interference (Chalasani et al. 2007; Chokshi et al. 2010; Chronis et al. 2007; Gilleland et al. 2010; Gourgou and Chronis 2016; Nekimken et al. 2017; Zimmer et al. 2009).
The nematode C. elegans is an excellent model organism for studying neural dynamics because of its compact and well characterised nervous system (Altun et al. 2002– 2020; Corsi et al. 2015; White et al. 1986). Nematodes and their nervous systems are bilaterally symmetric (White 1985; Brittin et al. 2020). The adult hermaphrodite has 302 neurons, organised in a distributed and highly modular structure. The main neuropil, referred to as the nerve ring, of the nematode encircles the pharynx. Two of the head sensory organs, called amphids, function as the main olfactory and chemosensory organs, and are positioned anteriorly on the left and right sides. Amphids contain 12 left–right pairs of sensory neurons with long cilia that extend to the tip of the nose and are exposed to the outside. Axons of amphid sensory neurons extend into the nerve ring, where they synapse onto other neurons in the sensory integration and decision making circuits (Altun et al. 2009; Chuang et al. 2007). One neuron pair, ASE, is unusual, breaking bilateral symmetry (Poole and Hobert 2006; Suzuki et al. 2008). ASE left (ASEL) and ASE right (ASER) develop similar cell morphologies and synaptic connectivity patterns and both are used by the animal to “taste” its environment, in particular responding to soluble salts. Their asymmetry is evident primarily in their divergent responses to chemical cues, through differently expressed chemoreceptors. Despite their different sensory functions, both neurons mediate salt attractive behaviours. In contrast, the ASH sensory neurons are the dominant hazard sensors of the worm, mediating rapid avoidance in response to a variety of chemical and other repulsive cues. Unlike ASE neurons, ASHL and ASHR respond similarly and are gap junctionally coupled. Gap junctions are clusters of channels which connect the cytoplasm of neighbouring cells (Evans and Martin 2002). Gap junction mediated intercellular communication allows direct transfer of ions and small molecules across the cells (Phelan and Starich 2001; Schumacher et al. 2012). Being known as electrical synapses (Bennett 1997), gap junctions can synchronise electrical activities of the coupled neurons (Bennett and Zukin 2004). While neuronal and, to a lesser extent, sub-circuit functions have been extensively studied in C. elegans (Guo et al. 2015; Larsch et al. 2013; Liu et al. 2018; Shindou et al. 2019; Suzuki et al. 2008), these have almost exclusively been limited to either the left or right side of the body, limiting our understanding of symmetry and asymmetry in sensory responses and information flow. With the exception of whole-brain imaging studies (Nguyen et al. 2016; Schrödel et al. 2013; Tobin et al. 2002), imaging one or more pairs of bilateral neurons simultaneously has largely been avoided since, in most experiments, the animal lies on its left or right side, such that the two cells are not located in the same focal plane of the microscope (Chokshi et al. 2010; Chronis et al. 2007; Gordus et al. 2015; Larsch et al. 2015).
A handful of microfluidic devices with a capability to rotate C. elegans and to simultaneously observe a pair of neurons have been reported. Ardeshiri et al. used a glass capillary to pneumatically grab and rotate a worm within a microchannel (Ardeshiri et al. 2016). Ahmed et al. (2016) reported acoustic field driven microvortices. And, Zhang et al. (2019) achieved a precise bidirectional rotation of the worm using surface acoustic wave. Recently, Reilly et al. fabricated bending microfluidic channels and showed that as the worm passes the bend, its body is rotated by 90° (Reilly et al. 2017). This device was used to simultaneously image a pair of ASH neuron. While imaging bilateral neuron pairs, the worms are physically twisted in these devices during imaging, potentially irritating the animal and affecting its neural responses.
Here, we report a novel microfluidic device, named a side-view device, for simultaneously observing neural activity of bilateral pairs of neurons. The microfluidic device itself is physically rotated while a worm is immobilised inside the device to place a left–right plane parallel to a focal plane. A stimulus delivery channel is integrated to deliver various solutions to a worm that is immobilised in the device. Using this side-view device, we simultaneously observed the activities of left and right ASE and left and right ASH neurons in response to step changes in NaCl concentration. Whereas ASE neurons are known to be functionally asymmetric (Hobert 2014; Johnston Jr and Hobert 2003; Suzuki et al. 2008), ASH neurons are bilaterally symmetric and widely thought to show strong on/off, and deterministic responses to noxious and other aversive stimuli (Thiele et al. 2009). Simultaneous recordings of left and right ASH neurons enabled us to quantitatively test the synchrony of responses of left and right cells as a function of NaCl concentration.
2 Materials and methods
2.1 Fabrication of the rotatable side-view device
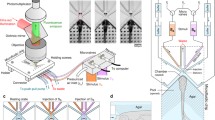
The microfluidic device was fabricated using a standard soft lithography techniques followed by processes for smoothing the side surface of the device (Xia and Whitesides 1998). The device was designed with AutoCAD (Autodesk), which was used to create a master. The master was fabricated by forming a thin layer (40 µm thickness) of a negative photoresist (SU8-2025, Micro Chem) on a silicon wafer with a spinner (model 6000, Electronic Microsystem). To enhance adhesion between the photoresist and the wafer, an adhesion enhancer (OmniCoat, Microchem) was spin-coated on the wafer (500 rpm for 5 s and 3000 rpm for 60 s) and baked in a dry oven at 200°C for 1 min before deposition of the photoresist. The wafer was baked on a hot plate (Model 1000-1, Electronic Microsystem) at 65°C for 5 min and 95°C for 20 min. A direct laser writing system (MicroWriter MLTM, Durham Magneto Optics) was used to illuminate the photoresist with a laser (375 nm) on the photoresist to initiate photo polymerisation. The polymerisation of the photoresist was further progressed by a post-exposure bake at 65°C for 1 min and 95°C for 5 min. The unexposed photoresist was washed off using 2-methoxy-1-methylethyl acetate (Rohm and Haas Electronic Materials) with a gentle agitation. The wafer was baked in a convection oven at 220°C for 10 min. After the development process, the surface of the master was modified by a vapour deposition in a vacuum desiccator for 17 h with 1H,1H,2H,2H-perfluorooctyltriethoxysilane (Sigma Aldrich) to form a hydrophobic layer, which enables an easy detachment of a moulded PDMS from the master. The master surface was exposed to an oxygen plasma (Zepto, Diener electronic, 50 W, 30 s) prior to the vapour deposition. And, liquid PDMS mixture (base: cross-linker = 10:1 in weight, Sylgard 184, Dow Corning) was poured on the master in a petri dish and cured at 75°C for 50 min. The PDMS slab was peeled off the master, and inlet holes were punched using a puncher with 0.8 mm diameter. The PDMS slab was covalently bound to a fully cured thin PDMS slab (2 mm thickness) with an oxygen plasma (50 W, 30 s) treatment. The PDMS device was trimmed as a cuboid so that the worm trap was located at 1 mm away from the device edge. In order to obtain a flat surface on the side of the device, the rough surface was smoothened with liquid PDMS and baked on a flat surface (on a wafer) at 75°C for 30 min. The additionally cured device was detached from the wafer and trimmed (Fig. 1a). The fabrication yield is typically > 90%.

Microfluidic device and experimental setup; a rough surfaces of the device is smoothed (surface A and the opposite side). The moulded PDMS slab is bonded to a thin PDMS slab (2 mm thick) so that the worm trap is close proximity to the edge of the device. We coated the cut surface of the device with uncured PDMS, placed this surface in contact with a blank silicon wafer and baked to bond the new PDMS to the device. The edge of the device is trimmed. b The schematics of the side-view device and an external pressure-controlled solution delivery system. c Top view and side view setup of the side-view device on a microscope. Fluid control and imaging can be carried out with the orientation of either conventional top view (left) or side view (right). d Fluorescence images of an immobilised transgenic worm expressing GCaMP in a target neurons (ASH). One neuron is observed in the top view. Both left and right neurons are observable in the side view configuration. Scale bar indicates 20 μm. e Redirection of the stimulus flow. A fluorescein solution (representing a stimulus) is delivered through channel 2, a buffer in channel 3 and two control buffers in channel 4-1 and 4-2. These images show either a stimulus or a buffer can be controllably delivered to the nose of a worm by an action of the two control channels 4-1 and 4-2. Scale bar indicates 100 μm. f A worm is immobilised in the trap. The trap is designed to fit the shape of the worm's head, which significantly reduces the movement and the escape of the worm. The nose of the worm protrudes into the stimulus delivery channel so that the controlled stimulus in the delivery channel can flow across the nose of the immobilised worm. Scale bar indicates 100 μm
2.2 Fluidic setup and controls of solution delivery
As shown in Fig. 1b, a buffer, a stimulus and a control buffer were connected to the device via a tubing (Tygon® ID 0.02″, OD 0.06″, Cole Palmer) to deliver various solutions to a worm that is immobilised in the worm trap. A 3-way solenoid valve (LFAA0380000C, Lee Company) was used to control an application of the control buffer to switch solutions between a stimulus and a buffer that is introduced to a worm. The solenoid valve was operated by a controller (Genuino 101, Arduino) and custom micromanager plugin (Edelstein et al. 2001). Pressures, applied to the solutions from compressed air, were controlled by a manifold and a regulator. The experiments were carried out with 5–10 psi of pressure. A vacuum pump (50 mbar, Dymax 14, Charles Austen Pump) was connected to an outlet reservoir, which applied a negative pressure to the microfluidic channel.
2.3 Worm loading and calcium imaging
Worms were loaded into the microfluidic device as follows. An individual worm was picked from a plate and released into a droplet of CTX buffer [5 mM KH2PO4/K2HPO4 pH 6, 1 mM CaCl2 and 1 mM MgSO4, syringe filtered (0.2 µm PTFE, VWR)]. The worm, encapsulated in the droplet, was loaded into a syringe and transported to the device. The worm was gently injected into the device via inlet hole by a hand-pressing plunger. It was confirmed under the microscope that the worm entered the worm trap with its head first. The worm was pushed into the worm trap until the amphid protruded into the solution delivery module. When the measurement was completed, the worm was flushed out of the device with an excessive flow provided by the injection syringe.
After immobilisation of the worm, it was incubated for 5 min to settle down. Blue light (480 nm) was illuminated before the application of NaCl to habituate the worm to the blue light as ASH is known to respond to blue light (Ward et al. 2008).
The stimulus was prepared by dissolving NaCl in CTX buffer. For ASE, worms were manitained with a 50 mM NaCl buffer solution, followed by a stimulus: a square pulse with a 60 s duration consisting of an NaCl upstep to 100 mM and downstep back to 50 mM. The start of the upstep was set as 0 s. Fluorescent images were recorded from − 2 to 120 s.
For ASH, 3 s pulses of the NaCl (0 mM to 0, 100, 200, 300, and 500 mM) were applied. Fluorescent images were recorded from − 2 to 10 s.
Optical images were taken using an epi-fluorescence microscope (BIM800F, Bioimager) equipped with a 40 × long working distance lens (BIM5-40xF, NA = 0.6, working distance = 2.2 mm, Bioimager), a mercury lamp (Bum-HBOB, 100 W, Bioimager), a 10% neutral density filter (NE10Bm Thorlabs) and a FITC filter (ex 480 ± 15 nm/em 535 ± 20 nm, Chroma Technology Corporation). Images were captured with an EMCCD camera (IXON3-897, Andor Technology) with an exposure time of 31–100 ms. A custom-made MicroManager plugin was used to coordinate the stimulus delivery and image capture (Edelstein et al. 2001).
2.4 Image analysis
To minimise the signal artefacts due to sudden motions of worms, we used an adaptive correlation filter to track and measure neuronal fluorescence. A rectangular ROI-1 was manually set around both the left and right cell bodies. ROI-1 was used as the tracking window for the Minimum Output Sum of Squared Error (MOSSE) correlation filter (Edelstein et al. 2001). Within OI-1, motion of the cells was greatly reduced. To measure the fluorescence intensity of neurons, we manually defined two rectangular regions (labelled ROI-2), one for each of the bilateral cells. We defined the fluorescence within each ROI-2 as the fluorescence signal F(sig) and fluorescence outside the ROI-2 but still within ROI-1 as the background fluorescence F(bgr). The effective fluorescence of the cell was defined as F = F(sig) − F(bgr). The intensity change of the cell was defined as ΔF = F/F0, where F0 is the mean fluorescence intensity that is averaged over 2 s right before the delivery of stimulus. The neurons were determined to be responsive when their peak intensities exceeded 1.35, and to be hyperpolarised when the intensities lower than 0.81. These threshold values were determined by the observation of ASH in a control experiment. Without any stimulus application in the experiment, ASH cells exhibited a background fluorescent intensity of 1.08 ± 0.09, which was an average of the measurements on seven worms. The error is the standard deviation. The threshold for the responsivity was determined within a range of three standard deviations from the mean intensity.
2.5 Molecular biology and culture
The ASH::GCaMP3 strain was a kind gift from the Bargmann lab (CX10979). To express GCaMP under the flp-6 promoter in both ASEL and ASER, a 3 kb genomic fragment immediately upstream of the flp-6 gene was PCR amplified from wormbank fosmid WRM068aB09 using primers 5′-ACAGGCCGGCCGAAGACTAAGGTGTTCGATCG and 3′-AAACCCGGGCCACGAGAGTTCATATTCTGG. The amplicon was inserted into a pSM:GCaMP3 vector (also a kind gift of the Bargmann lab) using restriction sites Fse1 and Sma1. Germline transformation was carried out by standard microinjection techniques (Mello et al. 1991) flp-6::GCaMP was co-injected with a unc-122:RFP coelomocyte marker at concentrations of 60 ng/μL and 20 ng/μL, respectively, into wild-type (N2 Bristol) worms obtained from the Caenorhabditis Genetics Center (Minneapolis, MN, USA).
2.6 Independent test for synchrony of bilateral neurons
Our null hypothesis is that the left and right neurons respond to the stimulus independently. Under the hypothesis, the joint probability of left and right neuron responses was calculated by multiplying the measured fraction of left and right neuron responses. Then, the probability of synchrony was computed as the sum of the probability that both or neither neurons were responsive (P = pLpR + (1 − pL) (1 − pR), where pL is the fraction of responsive left neurons and pR right neurons). The standard deviation of P was calculated as \(\sqrt {nP\left( {1 - P} \right)}\), where n is the number of worms. Then, measured fraction of synchronous neuron pairs (worms in which both left and right neurons, or neither neuron responded to the stimulus) was compared with the probability of synchrony.
3 Results and discussions
3.1 The side-view device can be used for stimulus delivery to immobilised worms
We developed an all PDMS cuboid microfluidic device in which a single worm can be immobilised and exposed to soluble stimuli (Fig. 1 and Fig S2). The side-view device allows us to observe neuronal activities of multiple neurons in a single dorso-ventral plane (top view) or left–right plane (side view) of the worm, as shown in Fig. 1d.
The microfluidic device consists of a worm channel for trapping and immobilising a worm (hereafter the ‘trap’) and a solution delivery channel for introducing a stimulus to the tip of the immobilised animal (Fig. 1b). The worm trap and stimulus delivery channels were adapted from an existing ‘olfactory chip’ described in Chronis et al. (2007). We optimised trap dimensions to minimise stress caused to the trapped worms due to immobilisation while minimising the worm’s escape from the trap. Our design consists of an entry channel with a fixed cross-sectional area of 40 μm by 40 μm which then leads into a gradually narrowing 1200 μm long channel with a 40 μm × 16 μm cross-section opening onto the stimulus delivery channel at the tip of the trap. The specific channel dimensions were designed for trapping a young adult C. elegans. Using this device, 81% (129 out of 159) of immobilised worms with correct orientation (with the head facing forward) were successfully imaged.
Worms can be loaded into the microfluidic device with a manual syringe-controlled delivery (Chronis et al. 2007) or a semi-automatic or a full automatic delivery, in which worms are injected into the device from a worm reservoir that is connected at constant pressure (Chokshi et al. 2010). When worms were loaded into the device, they were initially randomly oriented with either the head or tail pointing forward. To ensure that the worms reach the trap with their heads first, an array of pillars was fabricated at the entrance of the worm trap, which significantly enhanced the probability for a worm to enter the trap with its head first (channel 1 in Fig. 1b and Fig. S1) (Chokshi et al. 2010). We found that 71% (65/91) of the worms entered with their head first when they were semi-automatically delivered. When manually delivered by a syringe, the plunger can be precisely controlled to orient the head toward the trap such that more than 90% (258/272) of the worms were immobilised in the trap with their head-first orientation.
Immobilised worms in the trap can be easily removed from the device by applying additional pressure to the worm trap, which enables the reuse of the device. We imaged more than 90 worms in a single day with a single device, and typically used a single device for up to 2 weeks.
The stimulus delivery system was integrated with the worm trap in the microfluidic device (Fig. 1d). Worms entered the trap with the head first orientation and were immobilised at the tip of the trap, in which the amphid of the worm protruded into the stimulus delivery channel (Fig. 1e). The flow of the solutions in the channel was controlled by a four-channel module which allowed either the stimulus (channel 2 in Fig. 1b) or a buffer solution (channel 3) to flow onto the nose of the worm. An external three-way valve control the relative pressures (of buffer solution) in channels 4-1 and 4-2, which switches the direction of the stimulus flow toward or away from the immobilised worm (Video S1).
3.2 The side-view device can be used to image multiple neurons in a single lateral plane
The success of the side-view device hinges on the orientation of the worm such that its left–right plane is precisely aligned with a focal plane of the microscope. Any twist of the worm within the trap would shift at least one of the cells out of the focal plane. We exploited the fact that worms crawl by undulating in the dorso-ventral plane, while lying on the left or right side of the body. A wide (700 µm) and shallow (40 μm) entrance channel, located before the worm trap ((1) in Fig. 1b), forced the worm to undulate in the wide plane of the entrance channel. The device is first oriented on the microscope stage to view the trapping from the top view perspective and is then rotated by 90° (with the entrance channel facing down) to allow for neuronal imaging from the side-view perspective.
In order to verify the utility of the side-view device the orientations of immobilised worms were assessed with the aid of a focus knob of the microscope. We found 76% of worms (80/105 worms) were immobilised at an angle of less than 10° relative to the desired lateral orientation (Fig. S3). This level of angular variation did not significantly affect the imaging quality at the desired magnification. The bilateral neuron pairs were clearly visible under fluorescence illumination, allowing for high-quality imaging of calcium signals, using an epi-fluorescence microscope. Worms, showing high angular displacements, were discarded. The rapid manual or semi-automatic loading of worms into the device resulted in a wide distribution of twisting angles compared to the finely controlled manual injection, which was used to conduct the experiments.
3.3 Simultaneous recording of asynchronous stimulus responses in bilaterally asymmetric ASE neurons
Despite their morphological similarity, ASE Left (ASEL) and ASE Right (ASER) neurons express different genes, follow different developmental programmes and exhibit different physiological properties (Chang et al. 2003; Pierce-Shimomura et al. 2001; Suzuki et al. 2008). While both neurons mediate NaCl attraction (Bargmann and Horvitz 1991), their responses are asymmetric: whereas ASEL responds to increases in NaCl concentration, ASER responds to NaCl concentration decreases (Suzuki et al. 2008). We tested the utility of the device by imaging GCaMP3 reporter activity of these well-studied neurons in response to step changes in NaCl concentrations.
To test the responses of animals to NaCl increases, worms were pre-exposed to 50 mM NaCl for 5 min followed by a 60 s 100 mM NaCl pulse. We confirmed that ASEL responded to the NaCl upstep while ASER responded to NaCl downstep at the end of the pulse (Video S2, Fig. 2a, b) similarly to Suzuki et al. (2008). Raster plots of all ASE neuron responses of individual worms are presented in Fig. 2e (upstep) and 2f (downstep). As shown in the raster plots, the ASER baseline response was either suppressed (indicative of membrane hyperpolarisation) or non-responsive (unchanged in response to the NaCl concentration upstep), and ASEL exhibited no significant responses to the downstep. The mean peak of ASEL responses was observed at 9.8 s after the upstep whereas the mean peak of ASER responses was observed at 3.8 s after the downstep. We note that both ASEL and ASER responded to the stimulus stochastically with a probability of about 80%. We found no correlation between the response probability of ASEL to the upstep and the response of ASER to the downstep.

Neuronal activity of ASEL and ASER in response to upsteps and downsteps of NaCl concentration, between 50 and 100 mM for 60 s duration and was recorded for 120 seconds a, c and e for the upstep, b, d and f for the downstep. a, b ASEL responded to upstep of the stimulus and ASER to downstep. Both ASEL and ASER neurons were simultaneously imaged. 14 out of 18 ASELs were responsive to the upstep and 13 out of 18 ASER responded to the downstep. And, 5 out of 18 ASERs were hyperpolarised in response to the upstep. Peak intensity of ASEL response was observed at 9.8 s after the upstep (1.96 ± 0.14), whereas for ASER at 3.8 s after the downstep (1.97 ± 0.29). Mean peak ASER hyperpolarisation was observed 20.0 s after the upstep (0.73 ± 0.06). Error bars are calculated with ± \({\upsigma }/\sqrt n\), where σ is standard deviation and n is the number of worms. c, d A concentration profile of NaCl which was introduced to the amphid of an immobilised worm in the side-view device. The concentration of NaCl was changed from 50 to 100 mM for 60s. And, subsequently, it is changed back to 50 mM. e, f Raster plots show the simultaneously measured transient of ASEL and ASER from individual worms. ASEL and ASER respond to the stimulus stochastically. Numerical data are presented in Table S2
3.4 Simultaneous recording of synchronous stimulus responses reveals a functional connectivity between left and right ASH neurons
ASH neurons are the principal nociceptive sensory neurons in C. elegans and respond to mechanical, osmotic and chemical stimuli (Chronis et al. 2007; Hart et al. 1999; Hilliard et al. 2005; Kaplan and Horvitz 1993; Kato et al. 2014; Larsch et al. 2013; Sambongi et al. 1999; Troemel et al. 1999), including high concentrations of NaCl. Both ASHL and ASHR neurons respond to NaCl increases (Thiele et al. 2009). These sensory neuron responses are typically assumed to be reliable and highly synchronised (Thiele et al. 2009), supported by the direct gap junctional coupling between the two neurons (White 1985). C. elegans sensory neurons have been described as reliable, or invariably responding to stimuli. However, a growing body of work indicates that at least some sensory neuron responses appear to be conditional or stimulus-dependent. For example, Kato et al. (2014), Gordus et al. (2015) and Itskovits et al. (2018) describe reliable responses of AWC to isoamyl alcohol and diacetyl removal, respectively, but Biron et al. (2008) reports stochastic responses of AWC to temperature, and Iwanir et al. (2019) describes stochastic responses of AWC to a trimetylthiazole gradient (Biron et al. 2008; Gordus et al. 2015; Itskovits et al. 2018; Iwanir et al. 2019; Kato et al. 2014). ASH responses have previously been assessed in response to diacetyl or glycerol addition and showed consistent and reliable calcium transients (Kato et al. 2014; Larsch et al. 2015). However, the synchrony between ASHL and ASHR response has not been directly assessed.
To determine whether ASH neurons are functionally synchronised or not, we used our sideview device to image the calcium transients of ASH neurons in response to different NaCl concentrations (100, 200, 300 and 500 mM). We measured the propensity and amplitude of responses as a function of the concentrations of the NaCl stimulus. As expected, all worms showed a strong, synchronous response to 500 mM NaCl (Video S3) and none responded to 0 mM (control) or 100 mM NaCl steps. However, worms responded stochastically to steps of intermediate concentrations with 62% and 75% of worms responding to 200 and 300 mM, respectively (Fig. 3 and Table S3). The average peak intensity of responding neurons also increased as a function of the applied NaCl concentration (Fig. 3a, d and Table S1). There were no significant differences in the profiles of calcium transients between ASHL and ASHR (Fig. 3a). To test the synchrony between left and right ASH neurons, we looked at the propensity of the neurons in their response to applications of stimuli.

Neuronal activity of ASHL and ASHR in response to a 3-s pulse of NaCl. Various concentrations of NaCl were delivered to immobilised worms to find out the concentration range in which the neuron shows stochastic response. a The average traces of ASH responses to the application of a range of NaCl concentrations (0, 100, 200, 300 and 500 mM). The grey area and profiles in b represent the pulses of stimuli applied to the worms. The fluorescence signals in the neuron rapidly increase when the stimulus pulse starts, and rapidly decrease when the pulse stops. There is no significant difference between the average intensity of ASHL and ASHR (error bar ± \({\upsigma }/\sqrt n\), where σ is standard deviation and n is the number of the worms). c Fluorescence intensities observed from individual worms. ASH responded stochastically to 200 and 300 mM of NaCl; 62% for 200 mM and 67% for 300 mM. Numerical data in Table S3. As shown in d and e, the peak intensity and the fraction of the responsive neuron increased as a function of NaCl concentrations. Numerical data in Table S1 and S3.
We found that the likelihood of a synchronised response by both neurons was significantly higher than would be expected by chance (red dots in Fig. 4 and Table S3) and similarly when one neuron failed to respond, the probability for the second neuron to respond was significantly diminished. Thus, the ability to record both neurons at the same time allows us to conclude that ASHL and ASHR synchrony is likely enhanced by their gap junctional coupling.

The measured fraction of the synchronous neuron pairs compared with the calculated probability of synchrony. The fraction of synchronous neuron pairs is obtained by dividing the number of worms that are either both-neuron responsive or no-neuron responsive by the total number of assessed worms. The probability of synchrony (P) is calculated as P = pLp + (1 − p)L(1 − p)R, where pL and pR are the number of the left and right neuron being responsive out of total number of the worm. For ASH in the intermediate concentrations (200 mM, 300 mM), the fractions of synchronous neuron pairs are higher than the probability of synchrony. The error bar was calculated as \(\sqrt {n{\text{P}}\left( {1 - {\text{P}}} \right)} /n\), where n is the total number of worms. Conversely, in ASE, the fraction of synchronous neuron pairs is not significantly different from the probability of synchrony
4 Conclusions
This paper describes a microfluidic approach that enables us to observe simultaneously bilateral head neurons in C. elegans. The rotatable side-view device allows one to immobilise individual worms and image them through the side of the device, thus simultaneously observing those cells located in the same lateral plane. We demonstrated the utility of the device by recording the responses of ASE and ASH neuron pairs in response to stepwise changes of NaCl concentration. We also present experimental evidence that ASHL and ASHR are functionally coupled. While assessing synchrony typically requires genetic or pharmacological manipulation, here we exploited the probabilistic responses of ASH neurons to intermediate concentrations of NaCl. Our finding of synchrony supports the conjecture that the gap junctional coupling between ASHL and ASHR synchronises their responses (Liu et al. 2011). Our approach does not require any potentially irritating manipulations such as applications of glues to immobilise worms, which enables one to study relatively natural behaviours of worms in response to the application of stimuli. The prospective value of this approach lies in the capability of a microfluidic platform to immobilise a worm, control microenvironments, and observe their neural activity. This approach can be further developed to study the functional correlations of activities of sensory neurons, interneurons and motor neurons in conjunction with simultaneous observation of the animal’s behaviour. Such experiments have the potential to reveal the detailed dynamics of small, bilateral neural circuits and relate them to behaviours in C. elegans at the cellular level.
References
Ahmed D et al (2016) Rotational manipulation of single cells and organisms using acoustic waves. Nat Commun. https://doi.org/10.1038/ncomms11085
Altun ZF, Herndon LA, Wolkow CA, Crocker C, Lints R, Hall DH (2002-2020) WormAtlas. https://www.wormatlas.org. Accessed 23 Jun 2020
Altun ZF, Chen B, Wang ZW, Hall DH (2009) High resolution map of Caenorhabditis elegans gap junction proteins. Dev Dyn 238:1936–1950. https://doi.org/10.1002/dvdy.22025
Ardeshiri R, Mulcahy B, Zhen M, Rezai P (2016) A hybrid microfluidic device for on-demand orientation and multidirectional imaging of C. elegans organs and neurons. Biomicrofluidics 10:064111. https://doi.org/10.1063/1.4971157
Bargmann CI, Horvitz HR (1991) Chemosensory neurons with overlapping functions direct chemotaxis to multiple chemicals in C. elegans. Neuron 7:729–742. https://doi.org/10.1016/0896-6273(91)90276-6
Bennett MVL (1997) Gap junctions as electrical synapses. J Neurocytol 26:349–366. https://doi.org/10.1023/A:1018560803261
Bennett MVL, Zukin RS (2004) Electrical coupling and neuronal synchronization in the mammalian. Brain Neuron 41:495–511. https://doi.org/10.1016/S0896-6273(04)00043-1
Biron D, Wasserman S, Thomas JH, Samuel ADT, Sengupta P (2008) An olfactory neuron responds stochastically to temperature and modulates Caenorhabditis elegans thermotactic behavior. Proc Natl Acad Sci 105:11002–11007. https://doi.org/10.1073/pnas.0805004105
Brittin CA, Cook SJ, Hall DH, Emmons SW, Cohen N (2020) Beyond the connectome: a map of a brain architecture derived from whole-brain volumetric reconstructions. https://doi.org/10.1101/2020.05.24.112870
Chalasani SH, Chronis N, Tsunozaki M, Gray JM, Ramot D, Goodman MB, Bargmann CI (2007) Dissecting a circuit for olfactory behaviour in Caenorhabditis elegans. Nature 450:63. https://doi.org/10.1038/nature06292
Chang S, Johnston RJ, Hobert O (2003) A transcriptional regulatory cascade that controls left/right asymmetry in chemosensory neurons of C. elegans. Genes Dev 17:2123–2137. https://doi.org/10.1101/gad.1117903
Chokshi TV, Ben-Yakar A, Chronis N (2009) CO2 and compressive immobilization of C. elegans on-chip. Lab Chip 9:151–157. https://doi.org/10.1039/B807345G
Chokshi TV, Bazopoulou D, Chronis N (2010) An automated microfluidic platform for calcium imaging of chemosensory neurons in Caenorhabditis elegans. Lab Chip 10:2758–2763. https://doi.org/10.1039/C004658B
Chronis N, Zimmer M, Bargmann CI (2007) Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat Methods 4:727–731. https://doi.org/10.1038/nmeth1075
Chuang C-F, VanHoven MK, Fetter RD, Verselis VK, Bargmann CI (2007) An innexin-dependent cell network establishes left-right neuronal asymmetry in C. elegans. Cell 129:787–799. https://doi.org/10.1016/j.cell.2007.02.052
Chung K, Crane MM, Lu H (2008) Automated on-chip rapid microscopy, phenotyping and sorting of C. elegans. Nat Methods 5:637–643. https://doi.org/10.1038/nmeth.1227
Corsi AK, Wightman B, Chalfie M (2015) A Transparent window into biology: a primer on Caenorhabditis elegans. Genetics 200:387–407. https://doi.org/10.1534/genetics.115.180133
Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N (2010) Computer control of microscopes using µManager. Curr Protoc Mol Biol 92:14.20.1–14.20.17. https://doi.org/10.1002/0471142727.mb1420s92
Evans WH, Martin PEM (2002) Gap junctions: structure and function (review). Mol Membr Biol 19:121–136. https://doi.org/10.1080/09687680210139839
Gilleland CL, Rohde CB, Zeng F, Yanik MF (2010) Microfluidic immobilization of physiologically active Caenorhabditis elegans. Nat Protoc 5:1888–1902. https://doi.org/10.1038/nprot.2010.143
Gordus A, Pokala N, Levy S, Flavell Steven W, Bargmann Cornelia I (2015) Feedback from network states generates variability in a probabilistic olfactory circuit. Cell 161:215–227. https://doi.org/10.1016/j.cell.2015.02.018
Gourgou E, Chronis N (2016) Chemically induced oxidative stress affects ASH neuronal function and behavior in C. elegans. Sci Rep 6:38147. https://doi.org/10.1038/srep38147
Guo M et al (2015) Reciprocal inhibition between sensory ASH and ASI neurons modulates nociception and avoidance in Caenorhabditis elegans. Nat Commun 6:5655. https://doi.org/10.1038/ncomms6655
Hart AC, Kass J, Shapiro JE, Kaplan JM (1999) Distinct signaling pathways mediate touch and osmosensory responses in a polymodal sensory neuron. J Neurosci 19:1952–1958. https://doi.org/10.1523/JNEUROSCI.19-06-01952.1999
Hilliard MA, Apicella AJ, Kerr R, Suzuki H, Bazzicalupo P, Schafer WR (2005) In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. EMBO J 24:63–72. https://doi.org/10.1038/sj.emboj.7600493
Hobert O (2014) Development of left/right asymmetry in the Caenorhabditis elegans nervous system: from zygote to postmitotic neuron. Genesis 52:528–543. https://doi.org/10.1002/dvg.22747
Hulme SE, Shevkoplyas SS, Apfeld J, Fontana W, Whitesides GM (2007) A microfabricated array of clamps for immobilizing and imaging C. elegans. Lab Chip 7:1515–1523. https://doi.org/10.1039/B707861G
Itskovits E, Ruach R, Zaslaver A (2018) Concerted pulsatile and graded neural dynamics enables efficient chemotaxis in C. elegans. Nat Commun 9:2866. https://doi.org/10.1038/s41467-018-05151-2
Iwanir S, Ruach R, Itskovits E, Pritz CO, Bokman E, Zaslaver A (2019) Irrational behavior in C. elegans arises from asymmetric modulatory effects within single sensory neurons. Nat Commun 10:3202. https://doi.org/10.1038/s41467-019-11163-3
Johnston RJ Jr, Hobert O (2003) A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature 426:845. https://doi.org/10.1038/nature02255
Kaplan JM, Horvitz HR (1993) A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans. Proc Natl Acad Sci USA 90:2227–2231. https://doi.org/10.1073/pnas.90.6.2227
Kato S, Xu Y, Cho Christine E, Abbott LF, Bargmann Cornelia I (2014) Temporal responses of C. elegans chemosensory neurons are preserved in behavioral dynamics. Neuron 81:616–628. https://doi.org/10.1016/j.neuron.2013.11.020
Krajniak J, Lu H (2010) Long-term high-resolution imaging and culture of C. elegans in chip-gel hybrid microfluidic device for developmental studies. Lab Chip 10:1862–1868. https://doi.org/10.1039/C001986K
Larsch J, Ventimiglia D, Bargmann CI, Albrecht DR (2013) High-throughput imaging of neuronal activity in Caenorhabditis elegans. Proc Natl Acad Sci 110:E4266–E4273. https://doi.org/10.1073/pnas.1318325110
Larsch J, Flavell Steven W, Liu Q, Gordus A, Albrecht Dirk R, Bargmann Cornelia I (2015) A circuit for gradient climbing in C. elegans chemotaxis. Cell Rep 12:1748–1760. https://doi.org/10.1016/j.celrep.2015.08.032
Liu P, Chen B, Wang Z-W (2011) Gap junctions synchronize action potentials and Ca2+ transients in Caenorhabditis elegans body wall muscle. J Biol Chem 286:44285–44293. https://doi.org/10.1074/jbc.M111.292078
Liu Q, Kidd PB, Dobosiewicz M, Bargmann CI (2018) C. elegans AWA olfactory neurons fire calcium-mediated all-or-none action potentials. Cell 175(1):57–70.e17 https://doi.org/10.1016/j.cell.2018.08.018
Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10(12):3959–3970. https://doi.org/10.1002/j.1460-2075.1991.tb04966.x
Muthaiyan Shanmugam M, Subhra Santra T (2016) Microfluidic devices in advanced Caenorhabditis elegans research. Molecules 21:1006
Nekimken AL et al (2017) Pneumatic stimulation of C. elegans mechanoreceptor neurons in a microfluidic trap. Lab Chip 17:1116–1127. https://doi.org/10.1039/C6LC01165A
Nguyen JP et al (2016) Whole-brain calcium imaging with cellular resolution in freely behaving Caenorhabditis elegans. Proc Natl Acad Sci 113:E1074–E1081. https://doi.org/10.1073/pnas.1507110112
Phelan P, Starich TA (2001) Innexins get into the gap. BioEssays 23:388–396. https://doi.org/10.1002/bies.1057
Pierce-Shimomura JT, Faumont S, Gaston MR, Pearson BJ, Lockery SR (2001) The homeobox gene lim-6 is required for distinct chemosensory representations in C. elegans. Nature 410:694–698. https://doi.org/10.1038/35070575
Poole RJ, Hobert O (2006) Early embryonic programming of neuronal left/right asymmetry in C. elegans. Curr Biol 16:2279–2292. https://doi.org/10.1016/j.cub.2006.09.041
Reilly DK, Lawler DE, Albrecht DR, Srinivasan J (2017) Using an adapted microfluidic olfactory chip for the imaging of neuronal activity in response to pheromones in male C. elegans head neurons. J Vis Exp JoVE. https://doi.org/10.3791/56026
Sambongi Y, Nagae T, Liu Y, Yoshimizu T, Takeda K, Wada Y, Futai M (1999) Sensing of cadmium and copper ions by externally exposed ADL, ASE, and ASH neurons elicits avoidance response in Caenorhabditis elegans. Neurology 10:753–757. https://doi.org/10.1097/00001756-199903170-00017
Schrödel T, Prevedel R, Aumayr K, Zimmer M, Vaziri A (2013) Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light. Nat Methods 10:1013. https://doi.org/10.1038/nmeth.2637
Schumacher JA et al (2012) Intercellular calcium signaling in a gap junction-coupled cell network establishes asymmetric neuronal fates in C. elegans. Development 139:4191–4201. https://doi.org/10.1242/dev.083428
Shi W, Qin J, Ye N, Lin B (2008) Droplet-based microfluidic system for individual Caenorhabditis elegans assay. Lab Chip 8:1432–1435. https://doi.org/10.1039/B808753A
Shindou T, Ochi-Shindou M, Murayama T, Saita E, Momohara Y, Wickens JR, Maruyama IN (2019) Active propagation of dendritic electrical signals in C. elegans. Sci Rep 9(1):3430. https://doi.org/10.1038/s41598-019-40158-9
Suzuki H, Thiele TR, Faumont S, Ezcurra M, Lockery SR, Schafer WR (2008) Functional asymmetry in Caenorhabditis elegans taste neurons and its computational role in chemotaxis. Nature 454:114. https://doi.org/10.1038/nature06927
Thiele TR, Faumont S, Lockery SR (2009) The neural network for chemotaxis to tastants in Caenorhabditis elegans is specialized for temporal. Differentiation 29:11904–11911. https://doi.org/10.1523/JNEUROSCI.0594-09.2009
Tobin DM et al (2002) Combinatorial expression of TRPV channel proteins defines their sensory functions and subcellular localization in C. elegans. Neurons Neuron 35:307–318. https://doi.org/10.1016/S0896-6273(02)00757-2
Troemel ER, Sagasti A, Bargmann CI (1999) Lateral signaling mediated by axon contact and calcium entry regulates asymmetric odorant receptor expression in C. elegans. Cell 99:387–398. https://doi.org/10.1016/s0092-8674(00)81525-1
Ward A, Liu J, Feng Z, Shawn Xu XZ (2008) Light-sensitive neurons and channels mediate phototaxis in C. elegans. Nat Neurosci 11:916–922. https://doi.org/10.1038/nn.2215
White GJ (1985) Neuronal connectivity in Caenorhabditis elegans. Trends Neurosci 8:277–283. https://doi.org/10.1016/0166-2236(85)90102-X
White JG, Southgate E, Thomson JN, Brenner S (1986) The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci 314:1–340. https://doi.org/10.1098/rstb.1986.0056
Xia Y, Whitesides GM (1998) Soft lithography. Ann Rev Mater Sci 28:153–184. https://doi.org/10.1002/(SICI)1521-3773(19980316)37:5<550:AID-ANIE550>3.0.CO;2-G
Zhang J et al (2019) Surface acoustic waves enable rotational manipulation of Caenorhabditis elegans. Lab Chip 19:984–992. https://doi.org/10.1039/C8LC01012A
Zimmer M et al (2009) Neurons detect increases and decreases in oxygen levels using distinct guanylate cyclases. Neuron 61:865–879. https://doi.org/10.1016/j.neuron.2009.02.013
Acknowledgements
The ASH::GCaMP3 strain was a kind gift from Bargmann lab at Rockefeller University (CX10979). We thank the CGC for providing worm strains. NC was funded by the EPSRC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file2 (AVI 12890 kb)
Supplementary file3 (AVI 15228 kb)
Supplementary file4 (AVI 3296 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chung, J., Brittin, C.A., Evans, S.D. et al. Rotatable microfluidic device for simultaneous study of bilateral chemosensory neurons in Caenorhabditis elegans. Microfluid Nanofluid 24, 56 (2020). https://doi.org/10.1007/s10404-020-02362-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10404-020-02362-2