
Abstract
To investigate the changes of gene expression on the cerebral vasospasm after subarachnoid hemorrhage, we used genome-wide microarray for a canine double-hemorrhage model and analyzed the data by using a network-based analysis. Six dogs were assigned to two groups of three animals: control and hemorrhage. The effects were assessed by the changes in gene expressions in the artery 7 days after the first blood injection. Among 23,914 genes, 447 and 66 genes were up-regulated more than two- and fivefold, respectively, and 332 and 25 genes were down-regulated more than two- and fivefold, respectively. According to gene ontology, genes related to cell communication (P = 5.28E-10), host–pathogen interaction (7.65E-8), and defense–immunity protein activity (0.000183) were significantly overrepresented. The top high-level function for the merged network derived from the network-based analysis was cell signaling, revealing that the subgroup that regulates the quantity of Ca2+ to have the strongest association significance (P = 4.75E-16). Canine microarray analysis followed by gene ontology profiling and connectivity analysis identified several functional groups and individual genes responding to cerebral vasospasm. Ca2+ regulation may play a key role in these gene expression changes and may be involved in the pathogenesis of cerebral vasospasm.
Similar content being viewed by others
Introduction
Although several molecular biological studies have been reported on the pathogenesis of cerebral vasospasm, the complete mechanism of cerebral vasospasm remains unclear. We have speculated that changes in gene expression in the arterial wall, especially inflammatory reactions, contribute to the onset and development of cerebral vasospasm [1, 6, 14–16]. Recent advancements in microarray technology have revolutionized genomics by allowing researchers to study the expression of thousands of genes simultaneously, and it is a very powerful tool to investigate the regulation changes of gene on the complicated pathological change such as cerebral vasospasm.
To date, two articles have been published on gene expression changes seen in intracranial arteries after subarachnoid hemorrhage (SAH): Macdonald et al. [11] examined the time course of gene expression after SAH using one set of arteries for three time points in the primate model of SAH. Vikman et al. [19] investigated early changes in the cerebral arteries of rats that occur after SAH. However, they did not successfully demonstrate conclusive results related to the pathogenesis of cerebral vasospasm.
The most reliable and common model of cerebral vasospasm is the canine double-hemorrhage model. Severe cerebral vasospasm similar to that seen in humans is always obtained in the canine double-hemorrhage model, which has been studied profoundly and well-characterized pharmacologically and pathologically [18]. Therefore, the constitution of microarray in a large animal such as a dog is desirable and essential for the molecular biological knowledge of the cerebral vasospasm.
Nowadays, genetic canine studies are advanced, and specific genome-wide microarrays are available for laboratories. We used genome-wide canine oligonucleotide microarrays to screen for changes in levels of messenger RNA (mRNA) in the canine double-hemorrhage model [2, 8]. Although identification of a list of individual genes that show expression change is important, there is an increasing need to move beyond this level of analysis. Instead of simply enumerating a list of genes, we want to know how they interact as parts of complexes, pathways, and biological networks. For this purpose, the differentially expressed canine genes were automatically converted to human genes and imported into genetic pathway analysis software to identify biological networks and pathways. The networks described functional relationships between gene products based on known interactions in the literature [3].
Materials and methods
Canine double-hemorrhage model
The experimental protocol used for animals was evaluated and approved by the Institutional Animal Care and Use Committee of Tokyo Women’s Medical University. Care of the animals and surgical procedures were performed according to the standards of Tokyo Women’s Medical University Protocol on Laboratory Animals. Six mongrel dogs were divided into two groups of three dogs: control and hemorrhage. Dogs weighing between 7.5 and 17 kg were anesthetized with intravenous sodium pentobarbital (25 mg/kg). In three dogs, the cisterna magna was punctured percutaneously, and 0.3 ml/kg of cerebrospinal fluid was removed by spontaneous egress. After that, 0.5 ml/kg of fresh autologous arterial non-heparinized blood was injected into the cisterna magna at a rate of 5 ml/min. The dogs were tilted with their tail up for 15 min. The catheter was then removed, and the animals were allowed to recover. These animals were killed on day 7 after injections of blood on days 0 and 2. An angiography of the cerebral arteries of each animal was performed on day 0 and before the animal was killed [1, 14, 15].
Vessel caliber assessment
With a calibrated optical micrometer, the diameters of the basilar arteries were measured on the angiographic films at three separate, equally spaced locations along the artery by an experienced person who was blinded to the treatment groups. The average diameter value at each location was used. Comparisons between the two groups were performed using paired or unpaired Student’s t test. Statistical analysis was performed using Statistical Package for the Social Sciences (Tokyo, Japan). Values are expressed as mean ± SD. Differences were considered significant when the p value was <0.05.
RNA isolation from the canine basilar artery
The animals were killed by injecting 100 mg/kg pentobarbital, exsanguinated, and perfused with 2,500 to 3,000 ml physiological saline. Total RNA was isolated from individual basilar arteries of each group using trizol (Gibco BRL) according to the manufacturer’s instructions. Possible traces of genomic DNA contaminating the RNA preparations were removed by DNase I (Promega) digestion.
Canine microarray
An oligonucleotide (25-mer) canine array was used, which was constructed by Affymetrix. A total of 23,836 dog-specific probe sets were tiled on the array, including sequences from GenBank, dbEST, and Lion Bioscience. Fifty nanograms of total RNA was reverse transcribed into double-strand complementary DNA (cDNA) using Megascript T7 Kit and Two-Cycle cDNA Synthesis Kit, and Eukaryotic Poly-A RNA Control Kit. Biotinylated complementary RNA was synthesized using GeneChip Expression 3′-amplification reagent for IVT labeling kit and fragmented before overnight hybridization (45°C, 16 h, 60 rpm) with canine array using GeneChip Hybridization Oven 640. As per the “Affymetrix GeneChip Expression Analysis Technical Manual,” arrays were washed and stained using streptavidin–phycoerythin. Scans were performed using an Affymertrix GeneChip Scanner 3000, and expression value for each gene was calculated by GeneChip Operating Software ver.1.2.
Data analysis
At first, each signal of every treated group was normalized by dividing the signal of one control group, C1, and when the signal of each sample in the hemorrhage group (n = 3) were expressed more than two- or fivefold, or less than half or one fifth of that in the control group (n = 3), these genes were determined as two- or fivefold over- and underexpressed compared to the control group. The more than two- and fivefold differentially expressed values (up and down) address each case and control sample. As an example, twofold up-regulated means that every single expression value of the case samples was at least twofold higher than the highest expression value of the control samples. Expression signal intensities for each gene were averaged. Averaged expression values of the hemorrhage group were compared with those of the control group, and thus, we calculated the expression ratios (H/C). We analyzed and classified these genes on the basis of gene ontology (GO, http://www.geneontology.org/). Functional annotations of the altered transcripts were carried out using GeneSpring GX software (version 7.3, Agilent Technologies, CA, USA), which validated the occurrence of each biological category within a queried group of genes by using the Fisher’s exact test. For the Ingenuity network and pathway analysis, the moderately changed or unregulated genes play an equal role with the highly significantly regulated genes in constructing gene networks and thus result in irrelevant networks. Therefore, a data set containing only the identifiers of the significantly up- or down-regulated genes with their corresponding fold changes (in our case, over twofold) was uploaded as a tab-delimited text file into the Ingenuity software (http://www.ingenuity.com). This web-delivered application makes use of the Ingenuity Pathways Knowledge Base (IPKB) containing large amounts of individually modeled relationships between gene objects (e.g., genes, mRNAs, and proteins) to dynamically generate significant biological networks and canonical pathways, which is a core pathway established for a given molecule in the cell in which molecular interactions occur in a linear and stepwise manner. The submitted genes that are mapped to the corresponding gene objects in the IPKB are called “focus genes.” The focus genes are used as the starting point for generating biological networks. To start building networks, Ingenuity software queries the IPKB for interactions between focus genes and all the other genes stored in the IPKB, and generates a set of networks with a maximum network size of 35 genes. A p value for each network and canonical pathway is calculated according to the fit of the user’s set of significant genes. This is done by comparing the number of focus genes that participate in a given network or pathway, relative to the total number of occurrences of those genes in all networks or pathways stored in the IPKB. The score of the network is displayed as the negative log10 of the p value, indicating the likelihood of the focus genes in a network being found together due to random chance. Therefore, scores of two have at least 99% confidence of not being generated by chance alone. In the current study, a score of 24 or higher was used to select highly significant biological networks playing a role in this model for cerebral vasospasm.
Results
Assessment of cerebral vasospasm
There was no difference in vessel caliber at the baseline between the control and the hemorrhage groups (unpaired t test). There was a significant difference in vessel caliber between the baseline of the control group and day 7 of the hemorrhage group (p = 0.003, unpaired t test). The mean diameter of the basilar arteries on day 7, as a percentage of that on day 0, was 53.0 ± 7.0% in the hemorrhage group (P = 0.006, paired t test; Fig. 1). The animals that received double injections did not show any clinical symptoms.

Graph showing changes in the vessel caliber of basilar arteries in a canine double-hemorrhage model. Bars represent the mean ± SD (n = 3, each group). There was no difference in vessel caliber at the baseline between groups (unpaired t test). There was a significant difference in vessel caliber between the baseline of the control group and day 7 of the hemorrhage group (p = 0.003, unpaired t test)
Up- and down-regulated genes in the canine basilar artery after subarachnoid hemorrhage
On the canine microarray we used, the number of all genes (probes) was 23,914 and that of named genes was 10,071. Four hundred forty-seven genes were up-regulated more than twofold, of which 206 are named. Sixty-one genes were up-regulated more than fivefold, of which 34 genes are named. Three hundred thirty-two genes were down-regulated more than twofold, of which 148 genes are named. Twenty-five genes were down-regulated more than fivefold, of which 16 genes are named (Table 1).
Classification of up- and down-regulated genes according to the gene ontology
Classification of the 206 up-regulated and 148 down-regulated named genes according to gene ontology is shown in Table 2 (p < 0.01). Based on the analysis using the GeneSpring software, several gene ontology categories were identified as being overrepresented. Among the up-regulated genes, 27 genes were related to cell communication (p = 5.28E-10): 20 to host–pathogen interaction (7.65E-8), 13 to defense–immunity protein activity (0.000183), 14 to peptidase activity (0.000351), ten to response to external stimulus (0.00142), and 30 to catalytic activity (0.0015). Among the down-regulated genes, 12 genes were related to development (p = 0.00219).
More than fivefold up- and down-regulated genes
Table 3 shows the most significantly altered genes among the 34 and 16 named genes (probes) that were up- and down-regulated more than fivefold in the canine basilar artery after subarachnoid hemorrhage. Genes related to inflammation, immunological reaction, and stress response were significantly up-regulated, such as immunoglobulin E receptor alpha chain, pancreatitis-associated protein 1 precursor, interleukin (IL)-1 beta, IL-8, haptoglobin alpha and beta, immunoglobulin-alpha heavy-chain C region, serum amyloid A protein precursor, and IL-6.
Biological network and pathway analysis
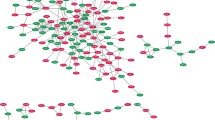
Two highly significant networks with a score ≥24 were identified from the imported data set. After merging the two networks, the pathway contained 88% of all imported genes (29 out of 33; Fig. 2). Four major nodes are evident in this network: IL-8, IL-6, chemokine ligand 2 (CCL2), and hepatocyte growth factor (HGF), two of which are also found in Table 3, depicting the genes that are over fivefold up- or down-regulated (IL-8, IL-6). All major nodes are connected to each other and to other proteins with a large number of interactions. The function with the highest association significance within the merged network was cell signaling. Specifically, the regulation of the quantity of Ca2+ (p = 4.75E-16; Table 4): 19 genes associated with the quantity of calcium regulation were located within the pathway (A4GALT, BDKRB1, CCL2, CCR6, EDG2, EDG4, EDG7, EGF, F2, HGF, IL4, IL6, IL8, MS4A2, NPY, PPY, PTGER3, TGFB1, and TNF; boldfacing indicates genes of the test set, all overexpressed except for EGF, which was underexpressed). If canonical pathways are overlaid on the merged network, the p38MAPK pathway is associated with the highest number of genes present. Four genes of the p38MAPK pathway were located in the merged network (TNF, TGFB1, CREB1, RPSKA4), although these were not over- or underexpressed in the current data set.

Functional network analysis relevant in the vasospasm model. Network map of molecular interactions and subcellular distribution of involved genes composed of two subnetworks that have an overlapping node. Nodes represent genes, with their shapes representing the functional classes of the gene products, and edges indicate the biologic relationship between the nodes, which include physical and functional interactions. The node color indicates up- (red) and down-regulation (green)
Discussion
The genome-wide screening for changes in levels of mRNA of cerebral artery in the canine double-hemorrhage model revealed that the transcript alterations were caused by biological pathways such as cell communication, host–pathogen interaction, and defense immunity protein activity. Genes that belong to these reactions include highly up-regulated molecules associated with inflammation such as IL-8, IL-6, and IL-1β. Immunoglobulin E receptor alpha chain, pancreatitis-associated protein 1 precursor, haptoglobin alpha and beta chains, immunoglobulin-alpha heavy chain C region, and serum amyloid A protein precursor are also significantly up-regulated at the time of vasospasm.
Four major nodes are evident in the merged network: IL-8, IL-6, CCL2, and HGF, all connected to each other and showing a large number of interactions. The top high-level function for the merged network was cell signaling, revealing the subgroup that regulates the quantity of Ca2+ to have the strongest association significance.
There is no direct evidence of persistent increase of intracellular Ca2+ in smooth muscle cells in vasospasm [7, 10, 13]. Although nimodipine and other calcium antagonists showed evidence of reducing poor patient outcome related to cerebral vasospasm, the nimodipine therapy did not show any affect on the vessel caliber seen on angiography. The effects are rather mediated by other factors such as neuroprotection or the promotion of collateral pial circulation [7, 10]. Therefore, a Ca2+-independent mechanism through the inhibition of myosin phosphatase induced by Rho kinase, protein kinase C, and arachidonic acid have been proposed for the main pathogenesis of cerebral vasospasm [5, 13, 17]. Tani and Matsumoto, however, suggested that the characteristic feature of vasospasm is a continuous elevation of intracellular Ca2+ levels in the cerebral artery, as indicated by the continuous activation of μ-calpain and Ca2+/calmodulin-dependent myosin light chain kinase phosphorylation of the myosin light chain [17].
We recently reported that cerebral vasospasm after subarachnoid hemorrhage can be completely prevented by an L-type voltage-dependent calcium antagonist in humans [9, 10]. Tani and Matsumoto speculated that the prevention of intracellular Ca2+ elevation leads to an inhibition of myosin light chain kinase, calpain, and protein kinase C, thereby suppressing the occurrence of cerebral vasospasm [17]. Our genome-wide network-based analysis in the canine cerebral vasospasm model also showed evidence for calcium playing a key role in the changes of gene expression. Our conclusions are limited because the RNA was extracted from the entire artery wall, and the sources of altered genes include not only smooth muscle but endothelial, adventitia, and any inflammatory cells attached to or within the artery. However, the changes in gene expression indicate those of smooth muscle cells, at least in part, because the ratio of smooth muscle cells in the arterial wall represents the majority. Moreover, similar gene expression changes have been found in smooth muscle cells in vitro after the treatment of hemolysate [15].
Among the up-regulated genes, we should focus on IL-8, IL-6, CCL2, and HGF, which are nodes with most connections in the network analysis. IL-8 and IL-6 also belong to the cell communication and host–pathogen interaction classification of gene ontology. As expected, the genome-wide cDNA microarray confirmed overexpressions of IL-8 and IL-6 in accordance to the results we had obtained in past independent experiments [1, 15]. The production of the protein is regulated by intra-cellular signaling pathways involved in the activation of p38MAPK. The p38MAPK pathway was associated with the highest number of genes present in the current study when canonical pathways were overlaid on the merged network. In a recent publication, we reported that hemolysate induced the prominent up-regulation of IL-1α, IL-1β, and IL-8 in human vascular smooth muscle cells in vitro, which was suppressed by FR167653, a selective inhibitor of p38MAPK. The intravenous administration of FR167653 also prevents cerebral vasospasm by suppression of p38MAPK phosphorylation and inhibition of the overexpression of inflammatory cytokines [15]. The L-type voltage-dependent Ca2+ channel, a main regulator of intracellular Ca2+ in the vascular smooth muscle cells, is tightly regulated by a range of signal transduction pathways in addition to regulation by its intrinsic, voltage-dependent gating processes [4]. As a positive feedback system was reported between cytokine synthesis and cascade phosphorylation of intracellular kinases, there may be a strong relationship between intracellular Ca2+ and cytokines [12, 15].
In conclusion, microarray analysis followed by gene ontology profiling and connectivity analysis identified several functional groups and individual genes responding to canine cerebral vasospasm. Among them, calcium regulation may play a key role on these gene expression changes and may be involved in the pathogenesis of cerebral vasospasm.
References
Aihara Y, Kasuya H, Onda H, Hori T, Takeda J (2001) Quantitative analysis of gene expression related to inflammation in canine spastic artery after subarachnoid hemorrhage. Stroke 32:212–217
Asakura M, Takashima S, Asano Y, Honma T, Asanuma H, Sanada S, Shintani Y, Liao Y, Kim J, Ogita H, Node K, Minamino T, Yorikane R, Agai A, Kitamura S, Yomoike H, Hori M, Kitazake M (2003) Canine DNA array as a potential tool for combing physiology and molecular biology. Circ J 67:788–792
Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF (2005) A network-based analysis of systemic inflammation in humans. Nature 437:1032–1037
Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16:521–555
Dietrich HH, Dacey RG Jr (2000) Molecular keys to the problems of cerebral vasospasm. Neurosurgery 46:517–530
Dumont AS, Dumont RJ, Chow MM, Lin CI, Calisaneller T, Ley KF, Kassell NF, Lee KS (2003) Cerebral vasospasm after subarachnoid hemorrhage: Putative role of inflammation. Neurosurgery 53:123–135
Feigin VL, Rinkel GJE, Algra A, Vermeulen M, van Gijn J (1998) Calcium antagonists in patients with aneurysmal subarachnoid hemorrhage: a systemic review. Neurology 50:876–883
Higgins MA, Berridge BR, Mills BJ, Shultze AE, Gao H, Searfoss GH, Baker TK, Ryan TP (2003) Gene expression analysis of the acute phase response using a canine microarray. Toxicol Sci 74:470–484
Kasuya H, Onda H, Takeshita M, Okada Y, Hori T (2002) Efficacy and safety of nicardipine prolonged-release implants for preventing vasospasm in humans. Stroke 33:1011–1015
Kasuya H, Onda H, Sasahara A, Takeshita M, Hori T (2005) Application of nicardipine prolonged-release implants: analysis of 97 consecutive patients with acute subarachnoid hemorrhage. Neurosurgery 56:895–902
Macdonald RL, Zhang ZD, Ono S, Komuro T (2002) Up-regulation of parathyroid receptor in cerebral arteries after subarachnoid hemorrhage in monkeys. Neurosurgery 50:1083–1093
New L, Han J (1998) The p38 kinase pathway and its biological function. Trends Cardiovasc Med 8:220–229
Nishizawa S, Laher I (2005) Signaling mechanisms in cerebral vasospasm. Trends Cardiovasc Med 15:24–34
Onda H, Kasuya H, Takakura K, Hori T, Imaizumi T, Takeuchi T, Inoue I, Takeda J (1999) Identification of genes differentially expressed in canine vasospastic cerebral arteries after subarachnoid hemorrhage. J Cereb Blood Flow Metab 19:1279–1288
Sasaki T, Kasuya H, Onda H, Sasahara A, Goto S, Hori T, Inoue I (2004) Role of p38 mitogen-activated protein kinase on cerebral vasospasm after subarachnoid hemorrhage. Stroke 35:1466–70
Sercombe R, Tran Dinh YR, Gomis P (2002) Cerebrovascular inflammation following subarachnoid hemorrhage. Jpn J Pharmacol 88:227–249
Tani E, Matsumoto T (2004) Continuous elevation of intracellular Ca2+ is essential for the development of cerebral vasospasm. Curr Vasc Pharmacol 2:13–21
Varsos VG, Liszczak TM, Han DH, Kistler JP, Vielma J, Black PM, Heros RC, Zervas NT (1983) Delayed cerebral vasospasm is not reversible by aminophylline, nifedipine, or papaverine in a “two-hemorrhage” canine model. J Neurosurg 58:11–17
Vikman P, Beg S, Khurana T, Hansen-Schwartz J, Edvinsson L (2006) Gene expression and molecular changes in cerebral arteries following subarachnoid hemorrhage in the rat. J Neurosurg 105:438–444
Acknowledgment
This work was supported in part by a grant-in-aid for Scientific Research (C; to HK) and Scientific Research on Medical Genome Science (to II) from the Japanese Ministry of Education, Culture, Sports, Science, and Technology.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Comments
Ute Felbor, Würzburg, Germany
The pathogenesis of cerebral vasospasm after subarachnoid hemorrhage remains unknown. Sasahara et al. have used genome-wide canine oligonucleotide microarray analyses to investigate alterations in gene expression in the basilar artery wall 7 days after induction of hemorrhage in a canine model. A decrease in vessel caliber of basilar arteries was accompanied by an up- and down-regulation of diverse genes. Network analyses demonstrated a significant association with molecules belonging to Ca2+ cell signaling and the p38 MAPK stress response pathway. Although the approach chosen may have documented late-stage changes of the disease process rather than early, disease-causing pathomechanisms, the results support an important role for intracellular Ca2+ homeostasis given the inconclusive effects of calcium antagonists on cerebral vasospasm.
Comments
Kazuhiko Nozaki, Shiga, Japan
The authors investigated the changes of gene expression on cerebral vasospasm in canine subarachnoid hemorrhage model. They used genome-wide microarray and analyzed the data by focusing on up-regulated and down-regulated genes, and using a network-based analysis. They concluded that Ca signaling might be a candidate for clarifying the pathogenesis of cerebral vasospasm. This paper provides us with useful implications for further studies in terms of molecular networks and related genes. On the other hand, there are some limitations. They used non-injected day 0 animals as a control. The data from animals that received plasma or Ringer’s solution may be a good counterpart to analyze gene expression in delayed vasospasm. They only analyzed animals on day 7 (when vasospasm reaches its peak). The time course of gene expression might be important to analyze the role of each gene and network, and clarify the pathogenesis of cerebral vasospasm.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sasahara, A., Kasuya, H., Krischek, B. et al. Gene expression in a canine basilar artery vasospasm model: a genome-wide network-based analysis. Neurosurg Rev 31, 283–290 (2008). https://doi.org/10.1007/s10143-008-0135-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10143-008-0135-7