
Abstract
Background
A gene or variant has pleiotropic effects, and genetic variant identification across multiple phenotypes can provide a comprehensive understanding of biological pathways shared among different diseases or phenotypes. Discovery of genetic loci associated with multiple diseases can simultaneously support general interventions. Several meta-analyses have shown genetic associations with gastric cancer (GC); however, no study has identified associations with other phenotypes using this approach.
Methods
Here, we applied disease network analysis and gene-based analysis (GBA) to examine genetic variants linked to GC and simultaneously associated with other phenotypes. We conducted a single-nucleotide polymorphism (SNP) level meta-analysis and GBA through a systematic genome-wide association study (GWAS) linked to GC, to integrate published results for the SNP variants and group them into major GC-associated genes. We then performed disease network and expression quantitative trait loci (eQTL) analyses to evaluate cross-phenotype associations and expression levels of GC-related genes.
Results
Seven genes (MTX1, GBAP1, MUC1, TRIM46, THBS3, PSCA, and ABO) were associated with GC as well as blood urea nitrogen (BUN), glomerular filtration rate (GFR), and uric acid (UA). In addition, 17 SNPs regulated the expression of genes located on 1q22, 24 SNPs regulated the expression of PSCA on 8q24.3, and rs7849820 regulated the expression of ABO on 9q34.2. Furthermore, rs1057941 and rs2294008 had the highest posterior causal probabilities of being a causal candidate SNP in 1q22, and 8q24.3, respectively.
Conclusions
These findings identified seven GC-associated genes exhibiting a cross-association with GFR, BUN, and UA.
Similar content being viewed by others


Introduction
Genome-wide association studies (GWASs) are an innovative tool for identifying new single-nucleotide polymorphisms (SNPs) or genes for traits or diseases using powerful genotyping technology [1]. GWASs scan a set of SNPs in multiple individuals to determine possible associations between the variants and traits or diseases [2], with GWAS of a specific disease potentially enabling identification of SNP variants associated with a particular disease.
However, SNP variants for a specific disease may not only be associated with that disease but also with other phenotypes or traits [3, 4]. Previous studies have suggested that many detected genetic loci are associated with multiple phenotypes. Multiple GWASs involving the 8q24 gene desert loci indicate that this region is associated with prostate, breast, colon, ovarian, and bladder cancers, as well as chronic lymphocytic leukemia [5,6,7,8,9]. Additionally, a large GWAS of 42 traits or diseases was performed to identify genetic variants associated with multiple phenotypes [10]. Moreover, some genetic variants can only be partially associated with a subgroup of phenotypes. The Global Lipids Genetics Consortium has identified genetic variants associated with different subgroups of blood lipid properties, including the variants of RSPO3, FTO, VEGFA, and PEPD, which are associated with high-density lipoprotein and triglycerides but not low-density lipoprotein and total cholesterol [11]. Thus, one variant or gene can show pleiotropic characteristics. In general, pleiotropy is defined as a genetic variant associated with multiple phenotypes, and this phenomenon represents a possible underlying cause of a cross phenotype (CP) association [12].
Although GWASs have been performed to identify SNPs associated with one phenotype, the integration of multiple GWAS results is needed when CP effects need to be estimated [13]. Moreover, identification of genetic variants across multiple phenotypes can improve statistical power and provide a comprehensive understanding of the underlying biological pathways and mechanisms common to several different diseases or phenotypes. Furthermore, the discovery of genetic loci associated with multiple diseases can simultaneously support general interventions.
When phenotype data from multiple GWASs are difficult to access at the individual level, summary statistics of reported GWAS results can be utilized. Several meta-analyses based on SNP levels using GWASs have shown associations with gastric cancer (GC) [14,15,16]; however, no studies have identified associations with other phenotypes based on the integration of a gene-based meta-analysis performed through a literature review. Disease network analysis has been proposed to identify genes that are cross-associated between GC and other phenotypes based on interactions related to biological mechanisms [17], whereby the signals of individual SNPs identified in GWASs for GC are integrated into a gene-based analysis (GBA) to identify genetic effects [18, 19]. However, the usefulness of such methods has not yet been verified.
In this study, we identified genes associated with multiple phenotypes through gene-based meta-analysis by performing a systematic review of published GWASs for GC and confirmed the usefulness of these methods for CP selection.
Materials and methods
Ethics statement
This study was approved by the Institutional Review Broad of the Seoul National University Hospital (IRB number 2201–008-1286).
Data source and study population
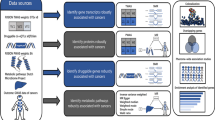
In our previous study [20], we performed a systematic review of GWAS on GC. SNP-level meta-analysis and gene-based analysis (GBA) were performed to identify SNPs and genes significantly associated with GC. Furthermore, SNPs having an effect on the expression level of a given gene were identified through expression quantitative trait loci (eQTL) analysis. Across all 12 eligible studies, 555 SNPs were searched, and finally 12 genes were selected as candidate genes for the cross-phenotype association analysis.
As a sensitivity analysis, the Korean Biobank Array, referred to as the Korean Chip (K-CHIP) consortium, was used to identify SNP-level heritability and the association between SNPs and cross phenotypes. The research by the K-CHIP consortium, which contains approximately 800,000 SNPs identified among the Korean population, has been carried out in accordance with relevant guidelines and regulations [21]. Details of the quality control and collection methods of the K-CHIP consortium have been previously described elsewhere [22, 23]. Moreover, 611 GC cases were excluded from the total 72,298 individuals in the K-chip consortium to identify the association between GC-related SNPs and other phenotypes (BUN, UA, and eGFR) by a genome-wide association study (GWAS) (Fig. 1).

Workflow for identifying genes associated with the cross phenotype. A Heritability estimation, eQTL analysis, and fine-mapping based on the results of a previous study. B Linear regression and GWAS analysis based on K-CHIP consortium. SNP single-nucleotide polymorphism, eQTL expression quantitative trait loci, eGFR estimated glomerular filtration rate, BUN blood urea nitrogen
Disease network for identifying genes associated with cross phenotypes
A disease network was constructed using candidate genes for GC from a meta-analysis and burden test using DisGeNET [17] to identify CPs. DisGeNET is a discovery platform that contains one of the largest publicly available collections of genes and variants associated with humans. An FDR-corrected p < 0.05 was used to identify to identify significant connections in the disease network.
Expression quantitative trait loci (eQTL) analysis
We performed eQTL (Overlapping Expression Quantitative Trait Locus) analysis to identify SNPs that influence regulatory elements controlling the expression of each gene [24]. The eQTL analysis was based on the eQTLGen Consortium [25], which studies the effect of genetic variation on gene expression in whole blood, and the Genotype-Tissue Expression (GTEx) project [26], which studies the effect of genetic variation on gene expression in whole blood, and the Genotype-Tissue Expression (GTEx) project, which investigates tissue-specific gene expression and regulation. The eQTLGen Consortium measured the strength and direction of the relationship between SNPs and the expression of nearby genes through the z-score of the SNP. The z-score is a standardization of the SNP-gene expression association measure, which represents the number of standard deviations that the SNP-expression association deviates from the mean of a normal distribution. Furthermore, the normalized effect size (NES) based on GTEx project measures the size of the effect of the SNP on gene expression, which is normalized for the variation of gene expression across multiple tissues. Both the z-score and NES indicate a positive association between the SNP and increased gene expression with a positive value, while a negative value indicates the opposite. The magnitude of the value reflects the strength of the association, with a larger value indicating a stronger association.
Fine-mapping analysis
To identify potential causal variants among SNPs [27], we performed fine-mapping using the SuSiE (Sum of Single Effects) method [28]. We selected the lead SNP with the lowest P value among the SNPs located in a single cytoband (17 SNPs on 1q22 and 24 SNPs on 8q24.3). Fine-mapping was not performed for the SNP located on 9q34.2 as only one SNP (rs7849280) was identified in this region. We estimated the posterior inclusion probability (PIP) for each SNP, which represents the probability of including the SNP in the causal relationship, by performing iterative Bayesian stepwise selection based on the linage disequilibrium (LD) structure in the East Asian population of the 1000 Genomes Project [27]. We ranked the SNPs from the highest to the lowest PIP and thus generated a credible set through iterative model fitting. Fine-mapping analysis was performed using the "susieR" R package [28].
Heritability
We used GWAS summary statistics associated with GC, uric acid (UA), estimated glomerular filtration rate (eGFR), and blood urea nitrogen (BUN) to estimate SNP-based heritability, applying summary-level database analysis [29]. For the statistics of GC (8,299 cases, 231,121 controls), meta-results based on BioBank Japan (BBJ) and the K-CHIP consortium were used (https://pheweb.jp/; https://koges.leelabsg.org/) [30, 31]. However, for the statistics of eGFR, UA, and BUN (N = 154,633, 129,405, and 148,767, respectively), for which only single cohort statistics was available, only the results from BBJ statistics (https://pheweb.jp) were used, owing to the greater number of samples available compared to that in the K-CHIP consortium. Based on summary statistics, the association among LD patterns was used to calculate LD score regression (LDSC) using East Asian samples in the 1000 Genomes Project Phase 3 database [29]. We used LDSC to estimate SNP-based heritability across GC-, UA-, eGFR-, and BUN-related genes. Furthermore, heritability can be partitioned to identify key gene sets that have disproportionately high heritability [32]. Therefore, as a sensitivity analysis, we also conducted SNP-based heritability analysis focusing on chromosomes 1, 8, and 9, where CP-associated genes were located.
Association of SNPs on genes related to GC with those related to eGFR, BUN, and UA
After in-silico-based disease network and eQTL analysis, the association between SNPs related to GC located in cross phenotype-related genes (eGFR: TRIM46, MTX1, THBS3, GBAP1, and ABO; BUN: THBS3, MTX1, GBAP1, PSCA, and ABO; UA: TRIM46, MTX1, THBS3, and MUC1) and eGFR, BUN, and UA was analyzed based on the K-CHIP consortium.
Linear regression was used to estimate the odds ratios (ORs) and corresponding 95% confidence intervals (CIs) of SNPs related to GC in additive and dominant models of cross phenotypes.
Genome-wide association study
We performed a GWAS to evaluate the associations of SNPs on TRIM46, MTX1, MUC1, THBS3, GBAP1, PSCA, and ABO with GFR, BUN, and UA under the assumption of an additive genetic model using PLINK version 2.0 [33]. The linkage disequilibrium (LD) clumping (R2 < 0.1 within a 10,00 kb window) was performed based on the 1000 Genomes project (East Asian) phase 3 as the reference panel using the “ieugwasr” R package [34]. The annotation of SNPs from GWAS was conducted by the ANNOtate VARiation (ANNOVAR) [35].
Results
Cross-phenotype-associated genes
Suggestive evidence of association [false discovery rate (FDR) ≤ 0.05] based on disease–gene network analysis was obtained for 12 genes (THBS3, GBAP1, KRTCAP2, TRIM46, HCN3, MUC1, DAP3, EFNA1, MTX1, PRKAA1, PSCA, and ABO) from GBA and eQTL analysis of 12 eligible studies based on our previous study (Fig. 1). Of 12 GC-associated genes, seven were associated with CPs (BUN, eGFR, and UA) (Fig. 2). Figure 2 demonstrates the association of seven genes with hereditary diffuse GC, atrophic gastritis, Helicobacter infection, and Curling ulcer, as well as with BUN, UA, and GFR. In Fig. 2, set size represents the number of genes associated with each phenotype. BUN was associated with five genes (PSCA, ABO, MTX1, THBS3, GBAP), and eGFR was associated with five genes (ABO, MTX1, THBS3, GBAP1, and TRIM46). UA was associated with four genes (MUC1, MTX1, THBS3, and TRIM46), while atrophic gastritis was associated with three genes (PSCA, ABO, MUC1). Hereditary diffuse GC and Helicobacter infection were associated with MUC1 and PSCA, while curling ulcer was associated with PSCA and ABO. In Fig. 2, the interaction size represents the number of genes associated with cross phenotypes of the same kind. As both MTX1 and THBS3 genes are associated with GFR, UA, and BUN, their interaction size is 2, while the remaining genes have a value of 1 because cross phenotypes do not fully match.

Upset and gene interaction network plot summarize between seven cross phenotypes and seven genes. Connected circles indicate a certain intersection of genes between phenotypes. The top bar graph (interaction size) in each panel summarizes the number of genes for each unique or overlapping combination. The bottom left horizontal bar graph labeled “Set Size” shows the total number of genes per phenotypes
Expression quantitative trait loci (eQTL) analysis
Figure 3 presents a network for seven genes associated with various phenotypes, indicating the relationships between SNPs that regulate the expression level of each gene. SNPs located on PSCA at 8q24.3 (rs10216533, rs1045531, rs1045547, rs1045574, rs2294008, rs2585179, rs2717562, rs2920280, rs2920283, rs2920285, rs2920286, rs2976384, rs2976386, rs2976387, rs2976388, rs2976389, rs2976391, rs2976392, rs2976395, rs2976396, rs2978978, rs2978979, rs2978980, rs2978982, rs71509378, and rs71778379) and ABO at 9q34.2 (rs7849280) regulated the expression levels of PSCA and ABO, respectively (Fig. 3; Supplementary Fig. 1). However, THBS3, TRIM46, GBAP1, MTX1, and MUC1 at 1q22 were regulated by multiple SNPs (Fig. 3; Supplementary Fig. 1). rs1057941, rs3814316, rs4971059, rs4971093, rs4971100, and rs4971101 were associated with the regulation of expression levels of GBAP1, MTX1, MUC1, THBS3, and TRIM46 (Supplementary Fig. 1). Additionally, rs4971066 was associated with the regulation of expression levels of GBAP1, MTX1, MUC1, and THBS3 (Supplementary Fig. 1).

Disease-gene network and eQTL analysis. The green diamond represents phenotypes; orange ellipse, genes; blue rectangle, eQTL-SNPs. Circos plots show eQTLs and their associations between genes and SNPs according to loci. Chromosome A 1q22, B 8q24.3, and C 9q34.2. eQTL expression quantitative trait loci, SNP single-nucleotide polymorphism
Fine-mapping analysis
In total, 17 SNPs on 1q22, except for rs28445596, were included in the credible sets (Supplementary Fig. 2). Among them, the SNP with the highest causality strength was rs1057941 (PIP = 0.99). Of 24 SNPs on 8q24.3, 5 SNPs were causality candidates, and among them, rs2294008 had the highest causality strength (PIP = 1.00) (Supplementary Fig. 2). Because only rs7849280 was identified in 9q34.2, fine-mapping analysis could not be conducted for 9q34.2.
Heritability
Based on the total for each chromosome region, the estimates of heritability were 0.187 (SE = 0.016) for BUN, 0.088 (SE = 0.009) for eGFR and 0.080 (SE = 0.011) for UA, respectively (Supplementary Table 1). Furthermore, based on the results for chromosomes 1, 8, and 9, in which CP-associated genes were identified in the sensitivity analysis, the estimates of heritability were 0.147 (SE = 0.031) for BUN, 0.143 (SE = 0.025) for eGFR, and 0.079 (SE = 0.025) for UA, respectively (Supplementary Table 1).
Association of SNPs on genes related to gastric cancer with cross phenotypes
Figure 4a, b, c show the associations between GC-related SNPs and the CPs BUN, eGFR, and UA. Regarding BUN-associated SNPs, the following were observed: rs2974937 T > C on THBS3 gene (Beta [SE]; CC, 0.391 [0.106]; TC, 0.290 [0.035] compared to TT; per C allele), rs760077 T > A on the MTX1 gene (Beta [SE]; AA, 0.547 [0.122]; TA, 0.349 [0.037] compared to TT; per A allele), rs2990223 G > A on the GBAP1 gene (Beta [SE]; AA, 0.534 [0.117]; GA, 0.343 [0.036] compared to GG; per A allele), rs2294008 T > C on the PSCA gene (Beta [SE]; CC, 0.217 [0.043]; TC, 0.062 [0.037] compared to TT; per C allele), and rs7849280 A > G on the ABO gene (Beta [SE]; GG, 0.070 [0.071]; AG, 0.105 [0.032] compared to AA; per G allele), respectively.

a Association of SNPs on genes related to gastric cancer with blood urea nitrogen. One asterisk (*) indicates p value < 0.05. b Association of SNPs on genes related to gastric cancer with eGFR. One asterisk (*) indicates p value < 0.05. eGFR, estimated glomerular filtration rate. c Association of SNPs on genes related to gastric cancer with uric acid. One asterisk (*) indicates p value < 0.05
Regarding eGFR-associated SNPs, the following were observed: rs76872124 C > T on the TRIM46 gene (Beta [SE]; TT, -0.645 [0.855]; CT, -0.512 [0.165] compared to CC; per T allele), rs760077 T > A on the MTX1 gene (Beta [SE]; AA, -1.008 [0.431]; TA, -0.491 [0.129] compared to TT; per A allele), rs423144 T > G on the THBS3 gene (Beta [SE]; GG, -0.660 [0.335]; TG, 0.290 [0.035] compared to TT; per G allele), rs2990223 G > A on the GBAP1 gene (Beta [SE]; AA, − 1.080 [0.413]; GA, − 0.433 [0.128] compared to GG; per A allele), and rs7849280 A > G on the ABO gene (Beta [SE]; GG, 0.482 [0.251]; AG, 0.306 [0.114] compared to AA; per G allele), respectively.
Regarding UA-associated SNPs, the following were observed: rs4971100 A > G on the TRIM46 gene (Beta [SE]; GG, 0.098 [0.031]; AG, 0.044 [0.011] compared to AA; per G allele), rs760077 T > A on the MTX1 gene (Beta [SE]; AA, 0.139 [0.041]; TA, 0.051 [0.012] compared to TT; per A allele), rs2066981 A > G on the THBS3 gene (Beta [SE]; GG, 0.103 [0.035]; AG, 0.043 [0.012] compared to AA; per G allele), and rs4072037 T > C on the MUC1 gene (Beta [SE]; CC, 0.097 [0.035]; TC, 0.044 [0.012] compared to TT; per C allele), respectively.
Genome-wide association study
The most significant SNPs on MTX1, GBAP1, THBS3, PSCA, and ABO associated with BUN were rs760077 on MTX1 (beta = 0.306, SE = 0.031, P value = 2.69E-23), rs2990220 on GBAP1 (beta = 0.244, SE = 0.029, P value = 4.78E-17), rs2974937 on THBS3 (beta = 0.241, SE = 0.029, P value = 8.83E-17), rs2294008 on PSCA (beta = 0108, SE = 0.020, P value = 1.04E-07), and rs635634 on ABO (beta = 0.091, SE = 0.023, P value = 9.39E-05) (Supplementary Table 2).
The most significant SNPs on MTX1, THBS3, MUC1, and TRIM46 associated with UA were rs760077 on MTX1 (beta = 0.051, SE = 0.009, P value = 1.39E-08), rs4072037 on MUC1 (beta = 0.039, SE = 0.008, P value = 3.40E-06), rs2066981on THBS3 (beta = 0.039, SE = 0.008, P value = 4.29E-06), and rs4971101 on TRIM46 (beta = 0.036, SE = 0.008, P value = 5.97E-06) (Supplementary Table 3).
The most significant SNPs on MTX1, GBAP1, THBS3, TRIM46, and ABO associated with eGFR were rs9411372 on ABO (beta = 0.282, SE = 0.076, P value = 1.94E-04), rs2974929 on GBAP1 (beta =—0.360, SE = 0.101, P value = 3.72E-04), rs760077 on MTX1 (beta = − 0.330, SE = 0.099, P value = 9.01E-04), rs76872124 on TRIM46 (beta = − 0.438, SE = 0.135, P value = 1.20E-03), and rs7366775 on THBS3 (beta = -0.237, SE = 0.089, P value = 8.04E-03) (Supplementary Table 4).
Discussion
In this study, we identified seven genes (MTX1, GBAP1, MUC1, TRIM46, THBS3, PSCA, and ABO) linked to CPs based on disease–gene network analysis from SNPs on genes associated with GC identified in our previous meta-analysis.
In addition, based on eQTL analysis, we identified that 17 SNPs regulate the expression levels of 5 genes (MTX1, GBAP1, MUC1, TRIM46, and THBS3) on 1q22, 24 SNPs regulate PSCA on 8q24.3, and rs7849280 regulates the expression level of ABO on 9q34.2.
The identified SNPs led to a decrease in GBAP1, TRIM46, and PSCA expression, while ABO, MTX1, MUC1, and THBS3 showed an increase in expression. All SNPs had a similar direction of effect on gene expression, which is attributed to their high correlation in LD [36].
Therefore, we performed fine-mapping analysis to identify potential causal SNPs (rs1057941 on 1q22 and rs2294008 on 8q24.3) within the LD block. However, there are no previous studies investigating the biological/molecular functions of the 5 genes (GBAP1, MTX1, MUC1, THBS3, and TRIM46) associated with rs1057941 have not been found, and the reported association between rs1057941 and the expression of these 5 genes was estimated in the same manner as in our study using eQTL databases.
Nevertheless, we were able to identify the role of other SNPs that have a high LD with rs1057941 in previous studies. rs4072037, which is in high LD with rs1057941 (R2 = 0.909 based on 1000 Genome project East Asian-JPT), regulates selective splicing of the second exon of MUC1 and modifies gene transcription activity, which may be functional [37,38,39]. Moreover, MUC1 is located downstream of TRIM46 and is part of the THBS3 and MTX1 gene clusters. This is well illustrated in the heatmap produced by the eQTL analysis (Supplementary Fig. 1) [16, 39, 40].
According to previous studies, the expression of GBAP1 is associated with rs2990245 located in the promoter of pseudogene GBAP1; rs2990245 is also in high LD with rs1057941 (R2 = 0.909 based on 1000 Genome project East Asian-JPT) [38, 41]. The function of rs1057941 in suppressing PSCA promoter activity on 8q24.3 was identified based on eQTL analysis, consistent with previous studies that regulate gene expression [42]. Furthermore, one.previous study showed that rs7849280 on 9q34.2 increases ABO expression, which was consistent with the direction of the eQTL analysis [43].
Therefore, we found that expression levels of these 7 genes are regulated by SNPs and are simultaneously linked to GC; furthermore, polymorphisms of these genes are risk factors associated with abnormal levels of eGFR, BUN, and UA.
These results are consistent with the established association between chronic inflammation and GC development, the biological mechanism of which often involves H. pylori infection, which promotes proinflammatory cytokine release and achlorhydria (thereby inducing chronic gastritis). This in turn favors a step-by-step cascade of events involving the transition from metaplasia to dysplasia prior to malignancy [44]. The associations between genetic variants in inflammation-associated genes and GC risk were previously investigated using candidate genetic approaches, and the involvement of MUC1 and PSCA, which was observed in those investigations, was also confirmed in the present study [44, 45]. PSCA was initially identified as a prostate-specific antigen that is overexpressed in prostate cancer. However, subsequent research has revealed that PSCA is expressed in other types of tumors such as those in the bladder, placenta, colon, kidney, and stomach (Supplementary Table 5). The anti-inflammatory properties of MUC1 have been observed in gastric mucosal cell responses to H. pylori infection [46], suggesting that MUC1 might play an inflammatory role in cancer cells but an anti-inflammatory role in infectious diseases (Supplementary Table 5). Additionally, a previous proteomic analysis of urine showed that the urinary excretion of MUC1 is associated with the risk of renal impairment in the general population [47]. Furthermore, medullary cystic kidney disease is caused by MUC1 mutations; however, because MUC1 is produced in many tissues, the authors were only able to detect clinical abnormalities in the kidney [48].
Gastric ulceration is a form of inflammation [37], and chronic H. pylori infection is involved in GC development by inducing chronic inflammation and amplifying GC carcinogenesis [49, 50]. Atrophic gastritis is also a result of chronic inflammation and chronic gastritis, which increase the risk of GC [51]. Given that GC, H. pylori infection, gastric ulcer, and atrophic gastritis share common genes in genetically susceptible individuals who are more susceptible to GC, individuals harboring variants in common genes (i.e., PSCA, ABO, and MUC1) might be at high risk for GC. ABO, whose variation is the basis of the ABO blood group, is associated with infectious and inflammatory status for the onset and progression of immune-mediated diseases (Supplementary Table 5) [52]. A previous study reported that IGA nephropathy patients with blood type O or A showed an increased risk of kidney function deterioration owing to increased inflammatory status [52].
Gastric bleeding and ulceration are common in patients with GC [53]. BUN levels are elevated during upper gastrointestinal bleeding, and during gastric bleeding, ammonia is released from blood hemoglobin in the digestive system [54], which can increase BUN levels [55, 56]]. Based on the results of previous studies, gastric bleeding may act as a mediator in the association between GC and BUN. Additionally, bleeding (especially gastric bleeding) has also been observed in patients with Gaucher disease at diagnosis [57, 58]. MTX1, which encodes a component of a preprotein import complex, and THBS3, which encodes extracellular glycoproteins that mediate cell-to-matrix and cell-to-cell interactions, are associated with Gaucher’s Disease (Supplementary Table 5). GBAP1 mutation also causes accumulation of glucocerebroside in macrophages, which is an important molecular symptom of Gaucher disease (Supplementary Table 5)[41, 59,60,61]. Therefore, in patients harboring mutations in MTX1, THBS3, and/or GBAP1 [41, 59,60,61], those also afflicted with GC or Gaucher disease may be at high risk for the onset of gastric bleeding. Moreover, those patients not afflicted with GC but with elevated BUN levels may also be in a high-risk group with a high probability of developing stomach cancer. However, further studies are required to confirm these possibilities.
Several studies have reported an association between GC and BUN and UA levels. Specifically, UA or urate level is elevated in GC patients [62,63,64] as a result of the rapid proliferation and differentiation of tumor cells, during which nucleotide synthesis and metabolism are also upregulated and undergo rapid catabolism. Because UA is the end product of endogenous nucleotide catabolism, elevated UA levels are observed in GC patients. A previous study reported that gout patients with high levels of UA have a higher risk of various cancers, including GC, than the general population [65]. Notably, previous studies identified TRIM46, which encodes the tripartite motif-containing protein that is a zinc-finger containing protein as associated with serum urate concentrations and gout [66, 67], and this was confirmed in the present study, suggesting a CP association between UA and GC (Supplementary Table 5).
A previous study suggested the role of uric acid as a trigger for GC carcinogenesis, given that elevated uric acid levels increase the rate of gastric cell division and cause their excessive proliferation, thereby promoting GC onset [68, 69]. Another study reported that hyperuricemia contributes to the high-density proliferation of GC cells by acting as a promoter in the proliferation of cancer cell nuclei, thereby contributing to GC [58]. These findings suggest a possible link between UA levels and GC as a result of sharing the same genes associated with nucleotide synthesis and metabolism.
Although previous studies focused on associations between decreased renal function and cancer development [70], the precise biological rationale for the higher risk of digestive cancer in chronic kidney disease (CKD) patients relative to the general population has yet to be clearly explained. However, previous studies have suggested that uremic factors [70] or CKD itself might be implicated as proinflammatory mediators [71]. Renal dysfunction is reportedly associated with the development of certain types of cancers. Previous studies show that both glomerular hyperfiltration and decreased GFR are associated with cancers [72, 73], and recently, an association between glomerular hyperfiltration and digestive cancer was reported based on a nationwide population-based study in Korea [74]. Previous GWASs including Taiwanese populations also discovered that variants in the TRIM46-MUC1-THBS3-MTX1 gene region variants are associated with higher eGFR (including UA levels and the risk of gout) (Supplementary Table 5) [75]. Additionally, a meta-analysis of associations between 67 dietary factors and GC revealed that processed meat and salty food consumption were associated with an increased risk of GC [76]. Protein loading and increasing GFR are well-known factors that lead to glomerular hyperfiltration [77], with this supported by reports suggesting that an increased risk of GC is associated with the consumption of a high-protein diet or unhealthy protein, followed by glomerular hyperfiltration [63].
This study has some limitations. Although we identified the association of each SNP with gene expression levels based on the eQTL analysis and the potential causal candidate SNPs found by fine-mapping, the biological/molecular functions of each SNP that regulated gene expression levels remain unknown. Other SNPs in LD have been identified to perform similar functions in previous studies, but further studies on the functional mechanisms of each SNP are needed. In addition, SNPs associated with GC were selected only from the results of published GWASs; because the association between the CPs and genes other than those related to GC were confirmed only in DisGeNET [17], it is possible that other relevant genes or phenotypes were not considered. In the future, phenome-wide association studies (PheWASs) with more phenotypes should be performed to identify additional CPs in GC-related genes. Although we identified the cross-phenotype associations of the 7 genes in the DisGeNET database, according to the results of GWAS for eGFR, BUN, and UA in a Korean general population excluding GC, the most significant SNPs located in PSCA were marginally significant and those in ABO were not highly significant based on the GWAS for BUN. In the GWAS for UA, the most significant SNPs located in MUC1, THBS3, and TRIM46 were not highly significant, neither. According to the GWAS for eGFR, the most significant SNPs located in MTX1, GBAP1, THBS3, TRIM46, and ABO were not highly statistically significant. Nevertheless, as we described before, a previous GWAS based on a Taiwanese population also discovered that variants in the TRIM46-MUC1-THBS3-MTX1 gene region variants are associated with higher eGFR [75]. ABO is associated with an increased risk of renal dysfunction in patients with IgA nephropathy [52]. DisGeNET is a collection of Gene-Disease Associations (GDA) and Variant-Disease Associations (VDA) extracted from scientific literature using text mining [17]. DisGeNET contains a collection of 400,000 publications that include information on GDA and VDA, and 60% of GDA are extracted from scientific literature through text mining. DisGeNET's data source is composed of public databases (e.g., CTD, GWAS catalog, ClinVar, etc.) as well as animal models and literature. Although the associations between genes and diseases derived from DisGeNET are based on previously reported records, there is a possibility that they may not always be replicated in other studies. Therefore, the association between ABO gene and BUN, between five genes (MTX1, GBAP1, THBS3, TRIM46, and ABO) and eGFR, and between three genes (MUC1, THBS3, and TRIM46) and UA can suggest the possibility of association rather than causality. Moreover, additional GWA-studies based on a large cohort consortium are needed between these genes and phenotypes (BUN, UA, eGFR).
Furthermore, CP association has limited ability to reveal a causal inference between GC and biomarkers, such as GFR and/or BUN and UA levels. Therefore, additional analysis, such as bi-directional Mendelian randomization, should be performed based on GWAS results from a large cohort [78].
The strength of this study is that–based on a comprehensive search of publicly available datasets for GC-related genes, as well as of all possible GWAS published in the literature–we proposed a method of cross-phenotype analysis via the identification of in-silico function annotation, which is suitable even when the available raw data contain limited information.
In summary, we identified seven genes (MTX1, GBAP1, MUC1, TRIM46, THBS3, PSCA, and ABO) shared between GC and three biomarkers–GFR and BUN and UA levels–providing evidence for an association between GC and these biomarkers. Further studies using comprehensive GBA and disease–gene network analysis based on published GWAS data for other phenotypes are recommended. We believe that the study design applied here enables acquisition of new knowledge about pleiotropy, which can reveal cross-associations and networks between genes and various phenotypes based on public GWAS statistics. Moreover, it is likely that PheWAS can be similarly analyzed based on the method applied here, to simultaneously support general interventions.
Data availability
The dataset used in this study (K-CHIP consortium) can be provided after review and evaluation of research plan by the Korea National Institute of Health, Korea Centers for Disease Control and Prevention.
References
Wang WY, Barratt BJ, Clayton DG, Todd JA. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet. 2005;6:109–18.
Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–76.
Wagner GP, Zhang J. The pleiotropic structure of the genotype-phenotype map: the evolvability of complex organisms. Nat Rev Genet. 2011;12:204–13.
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6.
Ghoussaini M, Song H, Koessler T, Al Olama AA, Kote-Jarai Z, Driver KE, et al. Multiple loci with different cancer specificities within the 8q24 gene desert. J Natl Cancer Inst. 2008;100:962–6.
Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet. 2010;42:504–7.
Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–7.
Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–8.
Grisanzio C, Freedman ML. Chromosome 8q24-associated cancers and MYC. Genes Cancer. 2010;1:555–9.
Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet. 2016;48:709–17.
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
Solovieff N, Cotsapas C, Lee PH, Purcell SM, Smoller JW. Pleiotropy in complex traits: challenges and strategies. Nat Rev Genet. 2013;14:483–95.
Galesloot TE, van Steen K, Kiemeney LA, Janss LL, Vermeulen SH. A comparison of multivariate genome-wide association methods. PLoS ONE. 2014;9: e95923.
Jin G, Lv J, Yang M, Wang M, Zhu M, Wang T, et al. Genetic risk, incident gastric cancer, and healthy lifestyle: a meta-analysis of genome-wide association studies and prospective cohort study. Lancet Oncol. 2020;21:1378–86.
Yan C, Zhu M, Ding Y, Yang M, Wang M, Li G, et al. Meta-analysis of genome-wide association studies and functional assays decipher susceptibility genes for gastric cancer in Chinese populations. Gut. 2020;69:641–51.
Mocellin S, Verdi D, Pooley KA, Nitti D. Genetic variation and gastric cancer risk: a field synopsis and meta-analysis. Gut. 2015;64:1209–19.
Piñero J, Ramírez-Anguita JM, Saüch-Pitarch J, Ronzano F, Centeno E, Sanz F, et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020;48:D845–55.
Liu JZ, Mcrae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Gene. 2010;87:139–45.
Huang H, Chanda P, Alonso A, Bader JS, Arking DE. Gene-based tests of association. PLoS Genet. 2011;7: e1002177.
Lee S, Yang HK, Lee HJ, Park DJ, Kong SH, Park SK. Systematic review of gastric cancer-associated genetic variants, gene-based meta-analysis, and gene-level functional analysis to identify candidate genes for drug development. Front Genet. 2022;13: 928783.
Moon S, Kim YJ, Han S, Hwang MY, Shin DM, Park MY, et al. The Korea biobank array: design and identification of coding variants associated with blood biochemical traits. Sci Rep. 2019;9:1382.
Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics. 2015;31:782–4.
Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;9:179–81.
Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philosop Transact R Soc B. 2013;368:20120362.
Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. (2018) Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. BioRxiv.. doi.:447367.
Carithers LJ, Moore HM. The genotype-tissue expression (GTEx) Project. Biopreserv Biobank. 2015;13:307–8.
Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018;19:491–504.
Wang G, Sarkar A, Carbonetto P, Stephens M. A simple new approach to variable selection in regression, with application to genetic fine-mapping. BioRxiv. 2018;25:1.
Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, et al. A global atlas of genetic associations of 220 deep phenotypes. MedRxiv. 2021;10:4393.
Nam K, Kim J, Lee S. Genome-wide study on 72,298 Korean individuals in Korean biobank data for 76 traits identifies hundreds of novel loci. Medrxiv. 2022;53:1415.
Luo Y, Li X, Wang X, Gazal S, Mercader JM, Neale BM, et al. Estimating heritability and its enrichment in tissue-specific gene sets in admixed populations. Hum Mol Genet. 2021;30:1521–34.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015. https://doi.org/10.1186/s13742-015-0047-8.
Elsworth B, Lyon M, Alexander T, Liu Y, Matthews P, Hallett J, et al. The MRC IEU OpenGWAS data infrastructure. BioRxiv. 2020;35:99.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010. https://doi.org/10.1093/nar/gkq603.
Slatkin M. Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 2008;9:477–85.
Saeki N, Saito A, Choi IJ, Matsuo K, Ohnami S, Totsuka H, et al. A functional single nucleotide polymorphism in mucin 1, at chromosome 1q22, determines susceptibility to diffuse-type gastric cancer. Gastroenterology. 2011;140:892–902.
Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics. 2015;31:3555–7.
Liu P, Zeng M. Role of MUC1 rs4072037 polymorphism in gastric cancer: a meta-analysis. Int J Clin Exp Pathol. 2020;13:465–72.
Wen R, Gao F, Zhou CJ, Jia YB. Polymorphisms in mucin genes in the development of gastric cancer. World J Gastrointest Oncol. 2015;7:328–37.
Ma G, Liu H, Du M, Zhang G, Lin Y, Ge Y, et al. A genetic variation in the CpG island of pseudogene GBAP1 promoter is associated with gastric cancer susceptibility. Cancer. 2019;125:2465–73.
Saeki N, Ono H, Yanagihara K, Aoyagi K, Sasaki H, Sakamoto H, et al. rs2294008T, a risk allele for gastric and gallbladder cancers, suppresses the PSCA promoter by recruiting the transcription factor YY1. Genes Cells. 2015;20:382–91.
Tanikawa C, Kamatani Y, Toyoshima O, Sakamoto H, Ito H, Takahashi A, et al. Genome-wide association study identifies gastric cancer susceptibility loci at 12q24.11-12 and 20q11.21. Cancer Sci. 2018;109:4015–24.
Ajani JA, Lee J, Sano T, Janjigian YY, Fan D, Song S. Gastric adenocarcinoma. Nat Rev Disease Primers. 2017;3:1–19.
McLean MH, El-Omar EM. Genetics of gastric cancer. Nat Rev Gastroenterol Hepatol. 2014;11:664.
Guang W, Ding H, Czinn SJ, Kim KC, Blanchard TG, Lillehoj EP. Muc1 cell surface mucin attenuates epithelial inflammation in response to a common mucosal pathogen. J Biol Chem. 2010;285:20547–57.
Zhang Z-Y, Ravassa S, Pejchinovski M, Yang W-Y, Zürbig P, López B, et al. A urinary fragment of mucin-1 subunit α is a novel biomarker associated with renal dysfunction in the general population. Kidney Int Rep. 2017;2:811–20.
Bleyer AJ, Kmoch S, Antignac C, Robins V, Kidd K, Kelsoe JR, et al. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol. 2014;9:527–35.
Jang J, Cho EJ, Hwang Y, Weiderpass E, Ahn C, Choi J, et al. Association between body mass index and gastric cancer risk according to effect modification by helicobacter pylori infection. Cancer Res Treat. 2019;51:1107–16.
Jang J, Wang T, Cai H, Ye F, Murphy G, Shimazu T, et al. The U-shaped association between body mass index and gastric cancer risk in the Helicobacter pylori Biomarker Cohort Consortium: a nested case-control study from eight East Asian cohort studies. Int J Cancer. 2020;147:777–84.
Hu Y-R, Yu Y, Li J-X, Lu W-C, Shu Q-J, Yu Y-C. The progression of atrophic gastritis to gastric cancer: a retrospective study in 61,810 cases. J Cancer Ther. 2017;8:553.
Yang M, Xie J, Ouyang Y, Zhang X, Shi M, Li X, et al. ABO blood type is associated with renal outcomes in patients with IgA nephropathy. Oncotarget. 2017;8:73603–12.
Kim YI, Choi IJ. Endoscopic management of tumor bleeding from inoperable gastric cancer. Clin Endosc. 2015;48:121–7.
An Y, Jang J, Lee S, Moon S, Park SK. Sex-specific associations between serum hemoglobin levels and the risk of cause-specific death in korea using the national health insurance service-national health screening cohort (NHIS HEALS). J Prev Med Public Health. 2019;52:393–404.
Tomizawa M, Shinozaki F, Hasegawa R, Togawa A, Shirai Y, Ichiki N, et al. Reduced hemoglobin and increased C-reactive protein are associated with upper gastrointestinal bleeding. World J Gastroenterol. 2014;20:1311–7.
González-González JA, García-Compean D, Vázquez-Elizondo G, Garza-Galindo A, Jáquez-Quintana JO, Maldonado-Garza H. Nonvariceal upper gastrointestinal bleeding in patients with liver cirrhosis. Clinical features, outcomes and predictors of in-hospital mortality. A prospective study. Ann Hepatol. 2011;10:287–95.
Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18020441.
Elstein D, Rosenmann E, Reinus C, Paz J, Altarescu G, Zimran A. Amyloidosis and gastric bleeding in a patient with Gaucher disease. J Clin Gastroenterol. 2003;37:234–7.
Kawasaki H, Suzuki T, Ito K, Takahara T, Goto-Inoue N, Setou M, et al. Minos-insertion mutant of the Drosophila GBA gene homologue showed abnormal phenotypes of climbing ability, sleep and life span with accumulation of hydroxy-glucocerebroside. Gene. 2017;614:49–55.
Armstrong LC, Saenz AJ, Bornstein P. Metaxin 1 interacts with metaxin 2, a novel related protein associated with the mammalian mitochondrial outer membrane. J Cell Biochem. 1999;74:11–22.
Mosher DF, Adams JC. Adhesion-modulating/matricellular ECM protein families: a structural, functional and evolutionary appraisal. Matrix Biol. 2012;31:155–61.
Hu J-D, Tang H-Q, Zhang Q, Fan J, Hong J, Gu J-Z, et al. Prediction of gastric cancer metastasis through urinary metabolomic investigation using GC/MS. World J Gastroenterol: WJG. 2011;17:727.
Xiao S, Zhou L. Gastric cancer: metabolic and metabolomics perspectives. Int J Oncol. 2017;51:5–17.
Yu L, Aa J, Xu J, Sun M, Qian S, Cheng L, et al. Metabolomic phenotype of gastric cancer and precancerous stages based on gas chromatography time-of-flight mass spectrometry. J Gastroenterol Hepatol. 2011;26:1290–7.
Chen C, Yen J, Chang S. Gout patients have an increased risk of developing most cancers, especially urological cancers. Scand J Rheumatol. 2014;43:385–90.
Köttgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45:145–54.
Dong Z, Zhou J, Jiang S, Li Y, Zhao D, Yang C, et al. Effects of multiple genetic loci on the pathogenesis from serum urate to gout. Sci Rep. 2017;7:43614.
Kuo C-F, Luo S-F, See L-C, Chou I-J, Fang Y-F, Yu K-H. Increased risk of cancer among gout patients: a nationwide population study. Joint Bone Spine. 2012;79:375–8.
Wang W, Xu D, Wang B, Yan S, Wang X, Yin Y, et al. Increased risk of cancer in relation to gout: a review of three prospective cohort studies with 50,358 subjects. Media inflam. 2015. https://doi.org/10.1155/2015/680853.
Wong G, Hayen A, Chapman JR, Webster AC, Wang JJ, Mitchell P, et al. Association of CKD and cancer risk in older people. J Am Soc Nephrol. 2009;20:1341–50.
Tonelli M, Sacks F, Pfeffer M, Jhangri GS, Curhan G. Biomarkers of inflammation and progression of chronickidney disease. Kidney Int. 2005;68:237–45.
Hjorth L, Wiebe T, Karpman D. Hyperfiltration evaluated by glomerular filtration rate at diagnosis in children with cancer. Pediatr Blood Cancer. 2011;56:762–6.
Kwatra NS, Meany HJ, Ghelani SJ, Zahavi D, Pandya N, Majd M. Glomerular hyperfiltration in children with cancer: prevalence and a hypothesis. Pediatr Radiol. 2017;47:221–6.
Kim Y, Lee S, Lee Y, Kang MW, Park S, Park S, et al. Glomerular Hyperfiltration and Cancer: a nationwide population-based study. Cancer Epidemiol Biomarkers Prev. 2020;29:2070–7.
Teng MS, Wu S, Hsu LA, Chou HH, Ko YL. Pleiotropic Effects Of Functional MUC1 variants on cardiometabolic, renal, and hematological traits in the taiwanese population. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms221910641.
Fang X, Wei J, He X, An P, Wang H, Jiang L, et al. Landscape of dietary factors associated with risk of gastric cancer: a systematic review and dose-response meta-analysis of prospective cohort studies. Eur J Cancer. 2015;51:2820–32.
Helal I, Fick-Brosnahan GM, Reed-Gitomer B, Schrier RW. Glomerular hyperfiltration: definitions, mechanisms and clinical implications. Nat Rev Nephrol. 2012;8:293–300.
Zheng J, Baird D, Borges MC, Bowden J, Hemani G, Haycock P, et al. Recent developments in mendelian randomization studies. Curr Epidemiol Rep. 2017;4:330–45.
Acknowledgements
This study was conducted with bioresources from National Biobank of Korea, the Korea Disease Control and Prevention Agency, Republic of Korea (KBN-2022-056).
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) [No. NRF-2016R1A2B4014552]; the Korean Foundation for Cancer Research [No. CB-2013–01].
Author information
Authors and Affiliations
Contributions
SL and SKP conceptualized and designed the study. SL analyzed the data. SL, HKY, HJL, DJP, SHK, and SKP interpreted data. SL and SKP. drafted the article. HKY, HJL, DJP, and SHK revised the article critically for important intellectual content. All authors approved the final manuscript for submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lee, S., Yang, HK., Lee, HJ. et al. Cross-phenotype association analysis of gastric cancer: in-silico functional annotation based on the disease–gene network. Gastric Cancer 26, 517–527 (2023). https://doi.org/10.1007/s10120-023-01380-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-023-01380-7