
Abstract
In present work, we describe the synthesis of graphite intercalation compounds with perrhenic acid (HReO4-GIC) through the anodic oxidation of graphite in aqueous perrhenic acid solution and their thermal exfoliation. Due to electrochemical treatment of graphite in perrhenic acid solution, ReO4− ions are intercalated into interlayer spaces of graphite. Anodic oxidation of graphite in HReO4 solution leads to the formation of 3-stage GIC. Simultaneously, some amount of perrhenic acid becomes deposited on the graphite surface and edges. In the next step, thermal treatment of the previously synthesized GIC was performed, causing both the exfoliation of graphitic structure and transformation of perrhenic acid into rhenium oxides on the surface of graphene layers. The yielded product was exfoliated graphite-ReO2/ReO3 composite. The obtained composite was characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), and Raman spectroscopy. Additionally, specific surface area of the exfoliated materials was measured.
Similar content being viewed by others


Introduction
Due to the presence of relatively weak van der Waals forces between the graphene layers in graphite, ions, molecules, or atoms can be inserted into the interlayer spaces of graphite matrix. This process is called intercalation and the yielded product is graphite intercalation compound (GIC) [1, 2]. The direction of electron transfer between the graphite matrix and intercalate defines the type of synthesized GIC. When intercalate accepts electrical charge from the carbon matrix, an acceptor GIC is synthesized. Contrariwise, if the electrical charge is accepted by carbon matrix, the donor-type GIC is obtained [3]. Most of donor-type intercalates are alkali metals, i.e., Li, Na, K, or ammonium quaternary salts [4]. On the other hand, acceptor GICs commonly contain inorganic acids, i.e., H2SO4, HNO3, HClO4 [5, 6], or metal chlorides [7, 8]. The efficiency of intercalation process is specified by the intercalation stage. It reveals the number of graphene layers separated by the neighboring layers of intercalate, for example, if every intercalant layer is separated by two graphene layers, the pure 2-stage GIC is synthesized [2, 3].
GIC can be synthesized on the way of chemical [9, 10], electrochemical [11,12,13], or photochemical reaction [14]. Chemical methods are divided into these carried out in gas or liquid phase. In the first one, the vapors of intercalate are passed through the bed of graphite, i.e., N2O5, SO3, Br2, or Re2O7 [15,16,17,18], whereas an intercalation in liquid phase is realized in molten salts, i.e., ZnCl2, NiCl2, or FeCl3, or in the dissolved intercalate [19, 20]. However, the commonly used system comprises of inorganic acids, such as sulfuric or perchloric, dissolved in water [5, 10]. To start the chemical intercalation, the addition of reductant or oxidant, depending on the type of prepared GIC, is needed. Electrochemical intercalation is carried out by polarization of graphite in dissolved intercalate, which plays a role of electrolyte. Depending on the electrons flow direction, the electrochemical processes are performed by anodic oxidation or cathodic reduction. Contrary to chemical methods, the electrochemical ones are more controllable and the product with desired properties is easily obtained. Moreover, they are environmental friendly, because no oxidant/reductant agent is used, and the final product has no impurities caused by the presence of oxidation/reduction by-products [21]. On the other hand, chemical methods are cheaper and much more easier to perform. Photochemical methods are relatively rarely used for the GIC preparation. In this case, intercalate is irradiated by UV light generating free radicals, which oxidize graphite matrix thus promoting the intercalation process. The intercalation of graphite by FeCl3 in chloroform medium can be conducted due to photochemical reactions [14].
GIC with Re2O7 was synthesized for the first time by Fuzellier [22]. For this preparation, the melted oxide and vapors were used. In both cases, the prepared GICs were characterized by low content of intercalate. In 1991, Scharff et al. have successfully synthesized Re2O7 -GIC by heating the graphite and dirhenium heptoxide in sealed glass ampoules filled with O2, N2, or Ar [18]. They obtained Re2O7-GIC with mixed third and fourth stages. Fröba et al. investigated the structure of Re2O7-GIC using X-ray adsorption near-edge structure [23, 24]. They noted that charge transfer from graphite to rhenium occurs and between the graphene layers slightly disordered tetrahedral symmetry of ReO4− ion appears. Electrochemical method of HReO4-GIC formation was studied also by Scharff [25]. An anodic oxidation of highly oriented pyrolytic graphite (HOPG) in 65–70% HReO4 by voltammetry technique yielded 3-stage GIC with a ninterlayer distance of 1370 pm.
Due to redox character of rhenium oxides, they can be applied in different types of catalytic reactions. For example, Re2O7-based catalysts reveal high catalytic activity in olefin metathesis with high efficiency at room temperature [26, 27]. Additionally, catalysts containing ReOx demonstrate high catalytic selectivity and activity for partial oxidation of methanol to methylal [28,29,30].
Exposition of GIC to a high temperature causes evaporation and thermal decomposition of intercalate occupied the interlayer spaces of graphite matrix. Accumulation of gases and increasing pressure lead to the damage of graphene layers. Due to the rapid removal of intercalate, the graphite structure expands up to hundreds of times along the crystallographic c axis yielding exfoliated graphite (EG) [31, 32]. Such a material is characterized by low density, relatively high specific area, hydrophobicity, and high thermal stability [33,34,35]. Generally, the properties of obtained EG depend on the type of GIC used for synthesis as well as conditions employed during exfoliation process. EG finds application as a precursor for flexible graphite foil preparation, filler in composites, material for oil sorption, or as a matrix for composite preparation [32, 33, 35,36,37,38].
The thermal exfoliation of GIC containing intercalates with catalytic activity can lead to transfer of intercalate from GIC matrix to developed surface area of EG yielding EG/catalyst system. Such composites can be used in chemical power sources production as active material for hydrogen storage or methanol electrooxidation.
In this work, we present the preparation of EG-rhenium oxide composites through shock thermal exfoliation of the beforehand synthesized HReO4-GIC. GIC with perrhenic acid was prepared by anodic oxidation of natural graphite in 75–80% HReO4 electrolyte using galvanostatic technique. The synthesized graphitic materials were thermally treated in N2 atmosphere in 700 °C to conduct exfoliation. Structural changes of the gathered samples were investigated by XRD and Raman spectroscopy, whereas morphology was examined by SEM analysis. Surface composition of the synthesized composites was studied using an energy-dispersive X-ray spectroscopy (EDS). The characterization of as prepared materials was completed be the calculation of the Brunauer-Emmet-Teller (BET) surface area.
Experimental
Sample preparation
Natural graphite flakes (170–283 μm, purity 99.5%, Graphit Kropfmühl) were anodically oxidized using galvanostatic technique and current density equal to 40 mA g-1. Graphite (400 mg) was closed in current collector made of platinum gauze (purity 99.9% 120 mesh, Goodfellow). A counter electrode made of platinum wire (purity 99.9%, 1mm diameter, Goodfellow) was used. Both electrodes were immersed in a three-electrode reactor beforehand filled with HReO4 solution (75–80%, Sigma-Aldrich). Anodic and cathodic chambers were separated by glass frit. As a reference electrode, the Hg/Hg2SO4/1 M H2SO4 was used, which was connected with working electrode by salt bridge filled with 1 M H2SO4. Different charge densities ranging from 60 to 1800 °C g−1 were passed through the working electrode yielding samples denoted as ReGIC60, ReGIC120, ReGIC600, and ReGIC1800. After, electrochemical process samples were washed with acetone to prevent the deintercalation of perrhenic acid.
EG-ReO2/ReO3 composites were synthesized using shock thermal treatment under a constant flow of N2. For this purpose, quartz crucible filled with GIC was placed in half of quartz tube which was outside the heating zone of the tube furnace. When the temperature of 700 °C was reached, GIC was rapidly moved to the heating zone causing the simultaneous exfoliation and deposition of rhenium oxides on graphene layers surface. After 4 min, the composite was moved out of the heating zone to cool down until room temperature was reached. Depending on the exfoliated material, obtained materials were named as Ex-ReGIC60, Ex-ReGIC120, Ex-ReGIC600, and Ex-ReGIC1800.
Instrumentation
All electrochemical measurements were performed using potentiostat/galvanostat Autolab PGSTAT 302N. Raman spectra were recorded by inVia Renishaw micro-Raman system with Ar laser and wavelength 514.5 nm. XRD diffractograms were obtained using PANalytical diffractometer with Cu Kα radiation (0.154 nm) with step size of 0.04° and range from 4 to 60°. Morphology and chemical surface composition of synthesized carbon materials were investigated by SEM equipped with EDS detector using S-3400N Hitachi microscope. Specific surface area of exfoliated samples was gained from N2 adsorption isotherms measured at 77 K with 3Flex Micromeritics apparatus.
Results and discussion
Electrochemical oxidation of graphite in 75–80% HReO4
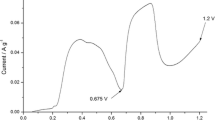
Galvanostatic curve recorded during anodic oxidation of graphite in perrhenic acid solution is presented in Fig. 1. The potential jump to 0.9 V is associated with the formation of electrical double layer. Next, the gentle potential increment to around 1.15 V depicts the formation of HReO4-GIC with higher stages. It cannot be excluded that in parallel the electrochemical deposition of perrhenic acid in form of H(ReO4)(H2O) may also occurs [39]. Potential plateau ranged between the charge densities from 120 to 900 °C g−1 is connected to further deposition of HReO4 on graphite surface as well as filling the interlayer spaces between the graphene layers. The last potential plateau starting at around 1.5 V corresponds to the partial oxidation of graphene layers and water electrolysis leading to deintercalation of HReO4. Measured weights of ReGIC60, ReGIC120, ReGIC600, and ReGIC1800 were equal to 517, 606, 803, and 659 mg, respectively.

Galvanostatic curve recorded during electrochemical oxidation of graphite in 75–80% perrhenic acid
From SEM images (Fig. 2) of graphite electrooxidized in HReO4 solution, it can be noted that during the anodic treatment the deposition of HReO4 forming H(ReO4)(H2O) on graphite surface and edges occurs [39]. The amount as well as size of the deposited H(ReO4)(H2O) increases with the treatment time. For the sample ReGIC1800 (Fig. 2 g, h), the insignificant electrochemical exfoliation of graphite flakes can be observed likely caused by the partial oxidation of graphene layers as well as electrolysis of intercalates closed between the graphene layers.

SEM images of ReGIC60 (a, b), ReGIC120 (c, d), ReGIC600 (e, f), and ReGIC1800 (g, h)
Raman spectra recorded for graphite and synthesized GICs are shown in Fig. 3. Typical bands for carbon materials are observed at 1355, 1580, 2725, and 2910 cm−1. The first one, D band, is associated with the presence of sp3 hybridized carbon deriving from defects, edges, or functional groups [40, 41]. It can be noted that elongation of graphite oxidation causes the increase of D band intensity, due to the formation of oxygen functionalities on the surface of graphene layers. The second one, called as G band, is assigned to the first-order scattering of E2g mode arising from the sp2 bonded carbon [40, 42]. For graphite, the regarded band has one maximum, whereas Raman spectra for HReO4-GICs reveal the second maximum of G band, called E′2g. It evidences the presence of intercalated graphite structure. During the anodic treatment of graphite, the intensity of E′2g band increases compared with E2g band thus indicating the enhanced amount of intercalated HReO4 [43, 44]. Surprisingly, in the case of sample ReGIC1800, the intensity of E′2g band in turn decreases. Such a behavior is caused by the partial degradation of graphitic structure due to oxidation of graphene layers accompanied by the partial deintercalation of ReO4− ions from graphite matrix. 2D band comes from the splitting of π electron dispersion energies and consists of two peaks [40,41,42]. The last band seen in Fig. 3, called as D + G band, is characteristic for oxidized graphene layers [45, 46]. For graphite and ReGIC60, the above-mentioned band is absent. It appears only on Raman spectra for samples ReGIC120, ReGIC600, and ReGIC1800. Its intensity increases with time of electrochemical oxidation. Such a behavior coincides with electrochemical results revealing that prolongation of anodic treatment of graphite leads to the partial oxidation of graphite matrix. Raman bands recorded under 1000 cm−1 can be assigned to the appearance of tetrahedral ReO4− groups [47,48,49,50]. The low-frequency modes at 245 and 341 cm−1 correspond to the bending motion of Re–O bonds [50, 51], whereas modes with higher frequencies at 889, 914, 965, and 988 cm−1 arise from the stretching vibration of Re–O bonds in ReO4− tetrahedra [48, 50, 51]. The most pronounced effect of ReO4− bands is present for sample ReGIC120. Further, electrooxidation causes the diminution in their intensities, which indicate that deintercalation of perhenic ions as well as deposition of H(ReO4)(H2O) on graphite surface takes place.

Raman spectra recorded for pristine graphite and HReO4-GICs
XRD measurements for graphite and oxidized samples provided information on changes in structure caused by electrochemical treatment of a graphite in perrhenic acid (Fig. 4). XRD pattern of graphite shows two main diffraction peaks at 26.6 and 54.4° revealing that the interlayer distance between the graphene layers is equal to 3.35 Å [52]. For sample ReGIC60, a new diffraction peak at 25.7° corresponding to stage-3 HReO4-GIC with average interlayer distance of 13.8 Å can be observed. This result is in agreement with the results presented by Scharff [18, 25]. XRD spectrum of graphite being oxidized using charge density of 120 °C g−1 involves two sharp diffraction peaks at 26.9 and 55.0°. Those peaks confirm the formation of H(ReO4)(H2O) particles on the graphite surface. Further oxidation of graphite in HReO4 solution leads to the degradation of stage-3 GIC and partial oxidation of graphite matrix. For ReGIC1800, an insignificant peak at 10.2° can be observed. It likely means the existence of graphite oxide structure within the deeply oxidized sample [53, 54].

XRD patterns recorded for pristine graphite and HReO4-GICs
Thermal exfoliation of the previously synthesized HReO4-GIC
The morphological changes in GIC structure caused by thermal treatment under ambient atmosphere were investigated using SEM (Fig. 5). As one can see, the shock treatment of GICs performed in N2 atmosphere contributed to the deformation and separation of graphene layers. With increasing intercalation duration, an exfoliation process becomes more efficient. This correlation is clearly connected with the increasing amount of perrhenic acid ions between the graphene layers of exfoliated GIC. Partial transformation of graphite to graphite oxide during the electrochemical overoxidation may be also responsible for the above-mentioned tendency. From the presented SEM images, it can be also noted that all exfoliated materials are covered by rhenium oxides particles. BET measurements reveal that samples Ex-ReGIC60, Ex-ReGIC120, Ex-ReGIC600, and Ex-ReGIC1800 have the surface areas equal to 3.5, 6.2, 18.5, and 19.6 m2 g−1, respectively. EDS mapping shows homogeneous distribution of rhenium oxides over the exfoliated materials (Fig. 6). The highest atomic percent concentration of rhenium in reached for sample Ex-GIC600 (Table 1). Decrease in Re amount in sample Ex-GIC1800 is likely caused by the partial deintercalation during overoxidation of GIC.

SEM images of Ex-ReGIC60 (a, b), Ex-ReGIC120 (c, d), Ex-ReGIC600 (e, f) and Ex-ReGIC1800 (g, h)

SEM and EDS maps of exfoliated graphite-rhenium oxide composites
Raman spectra of exfoliated samples are depicted in Fig. 7. As one can see, due to thermal treatment, the intensity of D band decreased noticeably as compared with HReO4-GIC samples (Fig. 3). It means that thermal treatment under nitrogen atmosphere contributes to the structure ordering. The only sample that exhibits the significant amount of surface defect is Ex-ReGIC1800. In this case, lack of structural ordering likely results the partial electrochemical exfoliation that probably occurs during the overoxidation of graphite in perrhenic acid over potential 1.5 V. It cannot be also excluded that violent removal of oxygen functionalities which formed during overoxidation of graphite generated higher amounts of defects as compared with other exfoliated samples. The presence of oxygen functionalities on the surface of graphene layer of Ex-ReGIC1800 is also confirmed by weak D + G band at 2910 cm−1 (Fig. 7). After the thermal treatment of HReO4-GICs, Raman spectra do not involve bands corresponding to tetrahedral ReO4−. On the other hand, new bands at 141, 166, 198, and 370 cm−1 appeared on the regarded spectra. Bands with maximum at 198 and 370 cm−1 are attributed to ReO3 structure [48, 55]. Other two bands at 141 and 166 cm−1 can be assigned to O = Re = O bonds deriving from ReO2.

Raman spectra recorded for exfoliated graphite-rhenium oxide composites
XRD patterns of thermally EG-rhenium oxide composites are presented in Fig. 8. Due to insignificant amount of intercalate within the ReGIC60 sample, most of graphitic structure is preserved after the thermal treatment process. It is manifested by the presence of sharp peak at 26.6°. On the other hand, relatively weak peaks arising with the appearance cubic ReO3 and rhombohedral ReO2 are also present [56, 57]. Diffraction patterns of Ex-ReGIC120, Ex-ReGIC600, and Ex-ReGIC1800 show that with increasing charge density consumed to anodic oxidation of graphite, the intensity of peaks deriving from the rhenium oxides also increases. It is worth to note that Ex-ReGIC600 sample is composed mostly of EG and ReO3, whereas the graphene layers in the case of sample Ex-ReGIC1800 are covered by ReO3 and ReO2. It means that thermal exfoliation of sample ReGIC1800 with lower concentration of perrhenic acid promotes the ReO2 formation, whereas thermal treatment of ReGIC600 with higher amount of HReO4 leads to the formation of ReO3.

XRD patterns recorded for exfoliated graphite-rhenium oxide composites
Conclusions
In our work, we describe the two-step process of EG-rhenium oxide composite preparation. Electrochemical oxidation of graphite in aqueous solution of HReO4 leads to intercalation of ReO4− ion between the interlayer spaces thus yielding of stage-3 GIC. Simultaneously, perrhenic acid is electrodeposited on graphite surface and edges forming H(ReO4)(H2O). Due to long-term oxidation, graphite is partially exfoliated and deintercalation of ReO4− occurs. Thermal treatment in nitrogen atmosphere of oxidized graphite in perrhenic acid causes the separation of graphene layers and simultaneous deposition of rhenium oxides of it surface. The synthesized EGs are characterized by developed specific surface area up to 19 m2 g−1. Taking into account the rhenium oxides on the surface of EG compounds, it should be emphasized that to their formation the deposition of perrhenic acid during electrochemical oxidation desired. XRD patterns of the synthesized composites reveal that thermal treatment of HReO4-GICs leads to the formation of cubic ReO3 and rhombohedral ReO2. The synthesized EG-rhenium oxide composites can be potentially used as a catalysts for methanol oxidation to methylal.
References
Chung DDL (2002) Review: graphite. J Mater Sci 37(8):1475–1489
Forsman WC, Dziemianowicz T, Leong K, Carl D (1983) Graphite intercalation chemistry: an interpretive review. Synth Met 5(2):77–100
Zabel H, Solin S (1990) Graphite intercalation compounds I. Springer, Berlin Heidelberg, Berlin, Heidelberg
Besenhard JO (1976) The electrochemical preparation and properties of ionic alkali metal-and NR4-graphite intercalation compounds in organic electrolytes. Carbon 14(2):111–115
Besenhard JO, Minderer P, Bindl M (1989) Hydrolysis of perchloric acid and sulfuric acid graphite intercalation compounds. Synth Met 34(1-3):133–138
Scharff P, Xu ZY, Stumpp E, Barteczko K (1991) Reversibility of the intercalation of nitric acid into graphite. Carbon 29(1):31–37
Rozmanowski T, Krawczyk P (2019) Electrocatalytic properties of graphite intercalation compound with metal chlorides modified by cathodic treatment. Electrochim Acta 297:735–741
Matsumoto R, Okabe Y (2016) Electrical conductivity and air stability of FeCl3, CuCl2, MoCl5, and SbCl5 graphite intercalation compounds prepared from flexible graphite sheets. Synth Met 212:62–68
Inagaki M, Iwashita N, Kouno E (1990) Potential change with intercalation of sulfuric acid into graphite by chemical oxidation. Carbon 28(1):49–55
Tryba B, Przepiórski J, Morawski AW (2003) Influence of chemically prepared H2SO4-graphite intercalation compound (GIC) precursor on parameters of exfoliated graphite (EG) for oil sorption from water. Carbon 41(10):2013–2016
Krawczyk P, Gurzęda B (2016) Electrochemical properties of exfoliated graphite affected by its two-step modification. J Solid State Electrochem 20(2):361–369
Zhao J, Zou X, Zhu Y, Xu Y, Wang C (2016) Electrochemical intercalation of potassium into graphite. Adv Funct Mater 26(44):8103–8110
Xu J, Dou Y, Wei Z et al (2017) Recent progress in graphite intercalation compounds for rechargeable metal (Li, Na, K, Al)-ion batteries. Adv Sci 4:1700146
Schlögl R, Boehm HP (1988) Photochemical intercalation in graphite. Synth Met 23(1-4):407–413
Lee BJ (2002) Characteristics of exfoliated graphite prepared by intercalation of gaseous SO3 into graphite. Bull Korean Chem Soc 23:1801–1805
Fuzellier H, Lelaurain M, Marêché JF (1989) The graphite nitrate compounds: Graphite-N2O5 system. Synth Met 34(1-3):115–120
Tongay S, Hwang J, Tanner DB, Pal HK, Maslov D, Hebard AF (2010) Supermetallic conductivity in bromine-intercalated graphite. Phys Rev B - Condens Matter Mater Phys 81(11):115428
Scharff P, Stumpp E, Höhne M, Wang YX (1991) Upon the intercalation of rhenium heptoxide and rhenium trioxide nitrate into graphite. Carbon 29(4-5):595–597
Ren H, Kang FY, Jiao QJ, Shen WC (2009) Synthesis of a metal chloride-graphite intercalation compound by a molten salt method. New Carbon Mater 24(1):18–22
Rozmanowski T, Krawczyk P (2018) Influence of chemical exfoliation process on the activity of NiCl2-FeCl3-PdCl2-graphite intercalation compound towards methanol electrooxidation. Appl Catal B Environ 224:53–59
Gurzęda B, Krawczyk P (2019) Electrochemical formation of graphite oxide from the mixture composed of sulfuric and nitric acids. Electrochim Acta 310:96–103
Fuzellier H (1974) PhD Thesis, University of Nancy, France
Fröba M, Lochte K, Metz W (1994) XAS and XRD Studies on graphite intercalation compounds of Re2O7. Mol Cryst Liq Cryst Sci Technol Sect A Mol Cryst Liq Cryst 244(1):239–244
Fröba M, Metz W (1997) Phase transitions at room temperature in graphite intercalation compounds of Re2O7. Solid State Ionics 101–103:619–623
Scharff P (1989) Elektrochemische Untersuchungen an Graphitsalzen mit HNO3, HClO4, HReO4 und halogenierten Essigsäuren / electrochemical investigations on graphite salts with HNO3, HClO4, HReO4 and halogenated acetic acids. Zeitschrift für Naturforsch B 44(7):772–777
Mol JC (1999) Olefin metathesis over supported rhenium oxide catalysts. Catal. Today 51(2):289–299
Oikawa T, Ookoshi T, Tanaka T, Yamamoto T, Onaka M (2004) A new heterogeneous olefin metathesis catalyst composed of rhenium oxide and mesoporous alumina. Microporous Mesoporous Mater 74(1-3):93–103
Nikonova OA, Capron M, Fang G, Faye J, Mamede AS, Jalowiecki-Duhamel L, Dumeignil F, Seisenbaeva GA (2011) Novel approach to rhenium oxide catalysts for selective oxidation of methanol to DMM. J Catal 279(2):310–318
Chan ASY, Chen W, Wang H, Rowe JE, Madey TE (2004) Methanol reactions over oxygen-modified re surfaces: influence of surface structure and oxidation. J. Phys. Chem. B 108(38):14643–14651
Yuan Y, Shido T, Iwasawa Y (2000) The new catalytic property of supported rhenium oxides for selective oxidation of methanol to methylal. Chem Commun 1421–1422
Anderson SH, Chung DDL (1984) Exfoliation of intercalated graphite. Carbon 22(3):253–263
Chung DDL (1987) Exfoliation of graphite. J Mater Sci 22(12):4190–4198
Yakovlev AV, Finaenov AI, Zabud’kov SL, Yakovleva EV (2006) Thermally expanded graphite: synthesis, properties, and prospects for use. Russ J Appl Chem 79(11):1741–1751
Skowroński JM, Krawczyk P, Rozmanowski T, Urbaniak J (2008) Electrochemical behavior of exfoliated NiCl2-graphite intercalation compound affected by hydrogen sorption. Energy Convers Manag 49(9):2440–2446
Bian J, Xiao M, Wang SJ, Lu YX, Meng YZ (2009) Novel application of thermally expanded graphite as the support of catalysts for direct synthesis of DMC from CH3OH and CO2. J Colloid Interface Sci 334(1):50–57
Yao T, Zhang Y, Xiao Y, Zhao P, Guo L, Yang H, Li F (2016) The effect of environmental factors on the adsorption of lubricating oil onto expanded graphite. J Mol Liq 218:611–614
Zheng G, Wu J, Wang W, Pan C (2004) Characterizations of expanded graphite/polymer composites prepared by in situ polymerization. Carbon 42(14):2839–2847
Krawczyk P, Rozmanowski T, Osińska M (2016) Electrochemical sorption of hydrogen in exfoliated graphite/nickel/palladium composite. Int J Hydrogen Energy 41(45):20433–20438
Vargas-Uscategui A, Mosquera E, López-Encarnación JM, Chornik B, Katiyar RS, Cifuentes L (2014) Characterization of rhenium compounds obtained by electrochemical synthesis after aging process. J Solid State Chem 220:17–21
Ferrari AC (2007) Raman spectroscopy of graphene and graphite: disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun 143(1-2):47–57
Pimenta MA, Dresselhaus G, Dresselhaus MS, Cançado LG, Jorio A, Saito R (2007) Studying disorder in graphite-based systems by Raman spectroscopy. Phys Chem Chem Phys 9(11):1276–1291
Malard LM, Pimenta MA, Dresselhaus G, Dresselhaus MS (2009) Raman spectroscopy in graphene. Phys Rep 473(5-6):51–87
Alsmeyer DC, McCreery RL (1992) In situ raman monitoring of electrochemical graphite intercalation and lattice damage in mild aqueous acids. Anal Chem 64(14):1528–1533
Chacón-Torres JC, Wirtz L, Pichler T (2014) Raman spectroscopy of graphite intercalation compounds: charge transfer, strain, and electron-phonon coupling in graphene layers. Phys Status Solidi 251(12):2337–2355
Kudin KN, Ozbas B, Schniepp HC, Prud'homme RK, Aksay IA, Car R (2008) Raman spectra of graphite oxide and functionalized graphene sheets. Nano Lett 8(1):36–41
Dimiev AM, Tour JM (2014) Mechanism of graphene oxide formation. ACS Nano 8(3):3060–3068
Thevenet A, Marie C, Tamain C, Amendola V, Miljkovic A, Guillaumont D, Boubals N, Guilbaud P (2020) Perrhenate and pertechnetate complexation by an azacryptand in nitric acid medium. Dalt Trans 49(5):1446–1455
Andriopoulou C, Anastasiou I, Boghosian S (2019) Di-oxo and tri-oxo Re(VII)-oxosulfato complexes in the Re2O7-K2S2O7 molten system. Molecular structure, vibrational properties and temperature-dependent interconversion. Vib Spectrosc 100:14–21
Lacheen HS, Cordeiro PJ, Iglesia E (2007) Isolation of rhenium and ReOx species within ZSM5 channels and their catalytic function in the activation of alkanes and alkanols. Chem - A Eur J 13(11):3048–3057
Purans J, Kuzmin A, Cazzanelli E, Mariotto G (2007) Disorder-induced Raman scattering in rhenium trioxide (ReO3). J Phys Condens Matter 19(22):226206–226214
Kon Y, Araque M, Nakashima T, Paul S, Dumeignil F, Katryniok B (2017) Direct Conversion of glycerol to allyl alcohol over alumina-supported rhenium oxide. ChemistrySelect 2(30):9864–9868
Li ZQ, Lu CJ, Xia ZP, Zhou Y, Luo Z (2007) X-ray diffraction patterns of graphite and turbostratic carbon. Carbon 45(8):1686–1695
Nakajima T, Matsuo Y (1994) Formation process and structure of graphite oxide. Carbon 32(3):469–475
Krishnamoorthy K, Veerapandian M, Yun K, Kim SJ (2013) The chemical and structural analysis of graphene oxide with different degrees of oxidation. Carbon 53:38–49
Beattie IR, Ozin GA (1969) Vibrational spectrum of gaseous, liquid, and solid Re2O7. J Chem Soc A Inorganic, Phys Theor Chem:2615–2619
Biswas K, Muthu DVS, Sood AK, Kruger MB, Chen B, Rao CNR (2007) Pressure-induced phase transitions in nanocrystalline ReO3. J Phys Condens Matter 19(43):436214
Shcheglov PA, Drobot DV (2006) Heterogeneous equilibria in the rhenium-oxygen system. Russ J Phys Chem A 80(11):1819–1825
Funding
This research was financially supported by the National Science Centre, Poland (2018/02/X/ST5/02765). This research was financially supported by the Ministry of Science and Higher Education.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gurzęda, B., Buchwald, T. & Krawczyk, P. Thermal exfoliation of electrochemically synthesized graphite intercalation compound with perrhenic acid. J Solid State Electrochem 24, 1363–1370 (2020). https://doi.org/10.1007/s10008-020-04642-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10008-020-04642-x