
Abstract
We report a simple synthetic scheme for the preparation of several azido-derived analogues of N-acetylglucosamine (GlcNAc). The synthesis of GlcNAc analogues has been achieved through a straightforward approach starting from GlcNAc-OMe via an intermediate C6 azido derivative. Products reported in this work were then obtained respectively by azido-alkyne cycloaddition reactions and reductive derivatizations of the same azido-intermediate. This synthetic pathway presents different possibilities of functionalization that can be exploited for the preparation of novel GlcNAc-based drugs.
Graphical abstract

Similar content being viewed by others


Introduction
N-Acetylglucosamine (GlcNAc) is a common monosaccharide present in various biopolymers structures of significant biological importance. This small-molecule is primarily derived from chitin, a structurally significant polysaccharide that serves as a primary component of cell walls in certain fungi (especially filamentous and mushroom forming fungi), exoskeletons in arthropods, such as crustaceans and insects, radulae, cephalopod beaks, molluscs gladii and in some nematodes and diatoms [1]. GlcNAc is also found in glycosaminoglycans (GAGs), linear oligosaccharides, and cell-surface oligosaccharides (cell surface O/N linked glycans) with a plethora of biological activities and functions, including cell adhesion, regulation of cell growth and proliferation, developmental processes, cell surface binding of lipoprotein lipase and other proteins, angiogenesis, viral invasion, inflammation, and tumor metastasis [2,3,4,5,6]. Therefore, it is not surprising that GlcNAc analogues have been studied with the aim of developing novel substances able to interfere with the pathological processes associated with these complex structures. Not limiting examples are the antitumoral GlcNAc analogues developed by Wasonga et al. [7] as inhibitors for hyaluronan biosynthesis; the C6 acylated derivatives proposed by Sung et al. for the treatment of autoimmune disease such as multiple sclerosis [8]; and also, the GlcNAc oxazoline derivatives proposed by Paiotta et al. [9, 10] as inhibitors of the hexosamine biosynthetic pathway (HBP) with antitumoral activity.
Consequently, versatile and site-selective strategies of GlcNAc functionalization is a topic of great interest in the field of medicinal chemistry for the research of novel derivatives with biological activity. In this study, we propose a straightforward approach for the synthesis of C6 analogues of GlcNAc by exploiting a C6 azido intermediate. We have exploited the high reactivity of the primary hydroxyl group at the C6 position of GlcNAc-OMe to convert it into an azide moiety, which enables the introduction of a wide range of functional groups. We have explored different possible synthetic routes passing through azido-intermediate 2 (Fig. 1), such as reductive amination and further acylation of the resulting amine, and a direct click chemistry approach. Indeed, we propose here the synthesis of some example compounds, but more generally, these strategies can be applied to the preparation of GlcNAc analogues with tailored functional groups at C6 position. We believe that this synthetic route could be suitable for drug discovery and development applications because of its simple adaptability to different functionalization.

Synthetic strategy for GlcNAc analogues design
Results and discussion
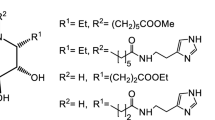
With the aim of obtaining GlcNAc derivatives modified at position C6 and exploiting a common versatile intermediate, we planned to introduce an azido group to GlcNAc-OMe that could be further derivatized in an orthogonal manner. As described in Scheme 1, for the synthesis of compounds 3 and 4, the azido moiety was reduced and converted to the respective products, while for compounds 5, 6, and 7, the copper-catalyzed 1,3-azido-alkyne cycloaddition reaction was performed.

Starting from the known methyl-N-acetyl-α-d-glucosamine 1 [11], our first attempt to introduce the azido group in C6 consisted in the regioselective tosylation of the C6 OH followed by nucleophilic substitution with NaN3. The crude obtained was directly acetylated, but unexpectedly this reaction afforded a complex mixture. Therefore, we changed strategy and adopted the Appel reaction conditions, originally described on other mono-oligosaccharides [12, 13], with N3− from sodium azide as the nucleophile, affording the common precursor 2. From this compound we propose different options of derivatizations, leading to compounds 3–7. Strategies where azido compound 2 was reduced include the reaction with triethylphosphite [14,15,16], which directly afforded phosphoramidite derivative 3 in a yield of 87% and catalytic hydrogenation of compound 2 (H2, Pd/C) followed by acetylation of the intermediate amine to afford compound 4 in 50% yield. On the other hand, compounds 5–7 were obtained by exploiting the copper-catalyzed azido-alkyne Huisgen cycloaddition reaction [17]; in particular, compound 5 resulted from the reaction of azide 2 with 2-octinoic acid and simultaneous decarboxylation in a yield of 86%; reaction with Fmoc-protected propargyl amine afforded compound 6 in a moderate yield of 43%; finally, compound 7 was obtained in discrete yield (64%) using as the alkyne 4-NH-propargyl-7-nitrobenzofurazane.
The examples reported herein represent a starting point for the development of compounds of pharmacological interest. In the present study, the initial ideas underlying the design of example derivatives of GlcNAc included modifications of compound 2 aimed at preserving the structural features of the naturally occurring phosphate groups of GlcNAc-6P, which interfere downstream in HBP. Nevertheless, we believe that the versatility of the proposed synthetic pathway also makes this approach transferrable to the development of other types of inhibitors. The phosphoramide group of compound 3 and amide group of compound 4 can confer drug-like pharmacokinetic properties increasing hydrophobicity and at the same time prevent dephosphorylation reactions in C6 position. The triazole moieties of compound 5–7 can act as phosphate group bioisosteres, preserving crucial H-bond interactions, and can be employed for further functionalization. This strategy can extend the potential application range of these small molecules to the development of multifunctional compounds by exploiting conjugated drugs or imaging agents. In this context, product 6 represents a good precursor after chemoselective Fmoc deprotection, whereas compound 7 contains a fluorescent moiety often used for diagnostic purposes, comparable to the glucose derivative 2-NBDG (2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose) [18].
Conclusion
Some examples of GlcNAc derivatives modified at the C6 position were obtained by starting from a common C6 azido-functionalized precursor. The proposed strategy can be easily implemented for the production of a larger library of compounds, particularly for pharmaceutical applications related to the modulation of biological pathways involving GlcNAc derivatives.
Experimental
When necessary, solvents were dried with 4 Å molecular sieves for at least 24 h prior to use. All reagents and starting materials were purchased from Sigma Aldrich Italy or Carbosynth United UK. Thin layer chromatography (TLC) was performed on silica gel 60 F254 plates (Merck) with detection using UV light when possible, by charring with a solution of conc. H2SO4/EtOH/H2O (5:45:45), or a solution of (NH4)6Mo7O24 (21 g), Ce(SO4)2 (1 g) and conc. H2SO4 (31 cm3) in water (500 cm3). Flash column chromatography was performed on silica gel 230–400 mesh (Merck). The reaction yields have not been optimized. 1H,13C, and 31P NMR spectra were recorded at 25 °C, unless otherwise stated, with a Bruker® 400 MHz instrument. The chemical shift assignments, reported in ppm, were referenced to the corresponding residual solvent peaks. HRMS data were recorded on a Xevo G2-XS qTOF (Waters) using Masslynx 4.2 software for acquisition and processing. Optical rotations were measured at room temperature using an Atago Polax-2L polarimeter and are reported in units of 10–1 deg cm2 g−1.
Methyl 2-acetamido-6-azido-3,4-di-O -acetyl-2,6-didesoxy- α - d-glucopyranoside (2, C13H20N>4O7)>
Compound 1 (1.00 g, 4.25 mmol), PPh3 (2.43 g, 6.80 mmol, 1.6 equiv.), NaN3 (830 mg, 1.28 mmol, 3 equiv.), and CBr4 (2.26 g, 6.80 mmol, 1.6 equiv.) were dissolved in dry DMF (15 cm3) under argon inert atmosphere. The reaction mixture was then stirred for 24 h at 60 °C. The solvent was then removed under reduced pressure, and the residue was filtered through a silica gel pad to remove excess reagents (eluent EtOAc/MeOH 9/1). The obtained reaction crude was dissolved in dry pyridine (8 cm3) and Ac2O (5 cm3) under inert atmosphere and a catalytic amount of DMAP was added. The reaction was kept under magnetic stirring overnight at room temperature. The solvent was then removed under reduced pressure. The crude was purified by flash column chromatography (eluent PE/EtOAc 2/8) to afford compound 2 (600 mg, 1.74 mmol) as the major product (41% over the two steps). 1H NMR (400 MHz, CDCl3): δ = 5.72 (d, J = 9.4 Hz, 1H, N–H), 5.24 (dd, J = 10.6, 9.5 Hz, 1H, H-3), 5.05 (t, J = 9.7 Hz, 1H, H-4), 4.79 (d, J = 3.6 Hz, 1H, H-1), 4.37 (ddd, J = 10.7, 9.6, 3.6 Hz, 1H, H-2), 3.94 (ddd, J = 9.9, 7.0, 2.6 Hz, 1H, H-5), 3.49 (s, 3H, OMe), 3.40 (dd, J = 13.3, 7.0 Hz, 1H, H-6a), 3.30 (dd, J = 13.3, 2.6 Hz, 1H, H-6b), 2.08 (s, 3H, Ac), 2.06 (s, 3H, Ac), 2.00 (s, 3H, Ac) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.31 (C=O), 169.86 (C=O), 169.42 (C=O), 98.09 (C-1), 71.03 (C-3), 69.39 (C-4), 69.11 (C-5), 55.51 (C-2), 51.81 (OMe), 51.16 (C-6), 23.18 (CH3), 20.69 (CH3), 20.61 (CH3) ppm; HRMS: m/z calcd. for [M + H]+ 345.1405, found 345.1406; calcd. for [M + Na]+ 367.1230, found 367.1226.
Methyl 2-acetamido-3,4-di- O -acetyl-2,6-didesoxy-6- N-(diethylphosphoramidyl)- α - d -glucopyranoside (3, C 17 H 31 N 2 O 10 P)
Compound 2 (100 mg, 0.29 mmol) was dissolved under inert atmosphere in DCM dry (5 cm3), then P(OEt)3 (100 mm3, 0.58 mmol, 2 equiv.) was added. The reaction mixture was stirred for 5 days. The solvent was then removed under reduced pressure. The crude was purified by flash column chromatography (eluent EtOAc/MeOH 9/1) to afford 115 mg of compound 3 (87%). 1H NMR (400 MHz, CDCl3): δ = 5.68 (d, J = 9.5 Hz, 1H, N-HAc), 5.20 (dd, J = 10.7, 9.5 Hz, 1H, H-3), 4.93 (t, J = 9.8 Hz, 1H, H-4), 4.69 (d, J = 3.6 Hz, 1H, H-1), 4.27 (ddd, J = 10.7, 9.6, 3.6 Hz, 1H, H-2), 4.12–4.00 (m, 4H, PO(OCH2CH3)2), 3.75 (ddd, J = 9.7, 6.8, 2.7 Hz, 1H, H-5), 3.40 (s, 3H, OMe), 3.17–2.90 (m, 3H, H-6a,6b,NHPO), 2.04 (s, 3H, Ac), 2.01 (s, 3H, Ac), 1.95 (s, 3H, Ac), 1.36–1.28 (m,6H, PO(OCH2CH3)2) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.36 (C=O), 169.90 (C=O), 169.68 (C=O), 98.06 (C-1), 71.13 (C-3), 69.47 (C-4), 69.16 (C-5), 62.54 (CH2 Et), 62.48 (CH2 Et), 55.40 (C-2), 51.98 (OMe), 42.04 (C-6), 23.19 (CH3 Ac), 20.71 (CH3 Ac), 20.66 (CH3 Ac), 16.23 (CH3 Et), 16.16 (CH3 Et) ppm; 31P NMR (162 MHz, CDCl3): δ = 8.76 (P=O) ppm; HRMS: m/z calcd. for [M + H]+ 455.1795, found 455.1792; calcd. for [M + Na]+ 477.1614, found 477.1612; [α]D25 = + 1.4 (c = 0.8, CHCl3).
Methyl 2,6-diacetamido-3,4-di- O -acetyl-2,6-didesoxy- α - d -glucopyranoside (4, C 15 H 24 N 2 O 8 )
Compound 2 (56 mg, 0.16 mmol) was dissolved in degassed MeOH (2 cm3), then a 3 M solution of HCl in MeOH (50 mm3) and Pd/C catalyst were added. The reaction mixture was stirred overnight under a H2 atmosphere. The catalyst was then removed by filtration through a Celite pad, and the solvent was removed under reduced pressure. The crude was then dissolved in dry DCM (3 cm3) and triethylamine (100 mm3, 5 equiv.), and AcCl (22 mm3, 2 equiv.) were added. The reaction was stirred overnight. The solvent was then removed under reduced pressure. The crude was purified by flash column chromatography (eluent EtOAc/MeOH 9/1) to afford compound 4 (28 mg, 50% over the two steps). 1H NMR (400 MHz, CDCl3): δ = 5.94 (s, 1H, NH), 5.75 (d, J = 9.5 Hz, 1H, NH), 5.18 (dd, J = 10.9, 9.3 Hz, 1H, H-3), 4.92 (t, J = 9.8 Hz, 1H, H-4), 4.68 (d, J = 3.4 Hz, -1H, H-1), 4.27 (dt, J = 2.8, 1.5 Hz, 1H, H-2), 3.85–3.77 (m, 1H, H-5), 3.64–3.54 (m, 1H, H-6a), 3.37 (s, 3H, OMe), 3.36–3.25 (m, 1H, H-6b), 2.04 (s, 3H, Ac), 1.99 (s, 3H, Ac), 1.99 (s, 3H, Ac), 1.94 (s, 3H, Ac) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.32 (C=O), 170.14 (C=O), 170.01 (C=O), 169.81 (C=O), 98.17 (C1), 71.08 (C-3), 68.91 (C-4), 68.14 (C-5), 55.35 (C-2), 52.08 (OMe), 38.99 (C6), 23.19 (CH3 Ac), 23.16 (CH3 Ac), 20.70 (CH3 Ac), 20.66 (CH3 Ac) ppm; HRMS: m/z calcd. for [M + H]+ 361.1611, found 361.1614; calcd. for [M + Na]+ 383.1430, found 383.1432; [α]D25 = + 1.3 (c = 0.9, CHCl3).
Methyl 2-acetamido-3,4-di- O -acetyl-2,6-didesoxy-6- N -(4-pentyltriazol-1-yl)- α - d -glucopyranoside (5, C 20 H 32 N 4 O 7 )
Compound 2 (150 mg, 0.43 mmol) and 2-octinoic acid (287 mg, 2.01 mmol, 5 equiv.) were dissolved in 1:1 mixture of H2O /MeOH (5 cm3), then sodium ascorbate (26 mg, 0.04 mmol, 0.3 equiv.) and CuSO4‧5H2O (11 mg, 0.04 mmol, 0.1 equiv.) were added. The resulting solution was stirred overnight at 60 °C. The reaction mixture was then diluted with H2O and extracted with EtOAc. The organic layer was then dried with anhydrous Na2SO4, filtered and the solvent removed under reduced pressure. The crude was purified by flash column chromatography (eluent EtOAc) affording 162 mg of compound 5 (86%). 1H NMR (400 MHz, CDCl3): δ = 7.38 (s, 1H, HAr), 5.70 (d, J = 9.5 Hz, 1H, NH), 5.19 (dd, J = 10.6, 9.5 Hz, 1H, H-3), 4.91 (t, J = 9.8 Hz, 1H, H-4), 4.65 (d, J = 3.6 Hz, 1H, H-1), 4.54 (dd, J = 14.2, 2.3 Hz, 1H, H-6a), 4.32–4.23 (m, 2H, H-2, H-6b), 4.10 (td, J = 9.8, 2.2 Hz, 1H, H-5), 3.06 (s, 3H, OMe), 2.72–2.68 (m, 2H, H-4’), 2.07 (s, 3H, Ac), 2.00 (s, 3H, Ac), 1.92 (s, 3H, Ac), 1.69–1.60 (m, 2H, H-5’), 1.33–1.28 (m, 4H, H-6’/H-7’), 0.89–0.83 (m, 3H, H-8’) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.14 (C=O), 169.93 (C=O), 169.71 (C=O), 148.27 (CAr), 122.39 (CAr), 97.97 (C-1), 70.90 (C-3), 69.76 (C-4), 68.43 (1C-C5), 55.21 (C-2), 51.93 (1C,OMe), 50.83 (C-6), 31.30 (C1, C-4’), 29.09 (C1, C-5’), 25.43 (C1, C-6’), 23.12 (C1, CH3), 22.33 (C1, C-7’), 20.65 (C1, CH3), 20.63 (C1, CH3), 13.94 (C-8’) ppm; HRMS: m/z calcd. for [M + H]+ 441.2349, found 441.2346; calcd. for [M + Na]+ 463.2169, found 463.2164; [α]D25 = + 0.7 (c = 0.9, CHCl3).
Methyl 2-acetamido-3,4-di- O -acetyl-2,6-didesoxy-6- N -[[4-(9H-fluoren-9-ylmethoxycarbonylamino)methyl]triazol-1-yl]- α - d -glucopyranoside (6, C 31 H 35 N 5 O 9 )
Compound 2 (100 mg, 0.30 mmol) and Fmoc protected propargylamine (161 mg, 0.60 mmol, 2 equiv.) was dissolved in 1:1 mixture of H2O /MeOH (4 cm3), then sodium ascorbate (17 mg, 0.09 mmol, 0.3 equiv.) and CuSO4‧5H2O (7 mg, 0.03 mmol, 0.1 equiv.) were added. The solution was left stirring at 60 °C overnight. The reaction was then diluted with H2O and extracted with EtOAc. The organic layer was then dried with anhydrous Na2SO4, filtered and the solvent removed under reduced pressure. The crude was purified by flash column chromatography (EtOAc/MeOH 19/1) affording 84 mg of compound 6 (43%). 1H NMR (400 MHz, CDCl3): δ = 7.76 (d, J = 7.5 Hz, 2H, HAr), 7.64 (s, 1H, H-triazole), 7.56 (d, J = 7.5 Hz, 2H, HAr), 7.40 (t, J = 7.4 Hz, 2H, HAr), 7.30 (t, J = 7.4 Hz, 2H, HAr), 5.66 (d, J = 9.5 Hz, 1H, NHAc), 5.43 (t, J = 5.5 Hz, 1H, NHFmoc), 5.20 (dd, J = 10.7, 9.5 Hz, 1H, H-3), 4.91 (t, J = 9.8 Hz, 1H, H-4), 4.64 (d, J = 3.5 Hz, 1H, H-1), 4.56 (d, J = 14.2 Hz, 1H), 4.49–4.26 (m, 6H), 4.19 (t, J = 6.9 Hz, 1H), 4.15–4.04 (m, 1H, H-5), 3.04 (s, 3H, OMe), 2.10 (s, 3H, Ac), 2.01 (s, 3H, Ac), 1.94 (s, 3H, Ac) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.19 (C=O), 169.92 (C=O), 169.69 (C=O), 156.33 (C=O FMOC), 145.01 (Ar), 143.81 (Ar), 141.29 (Ar), 127.72 (2C, Ar), 127.04 (2C, Ar), 125.00 (2C, Ar), 120.00 (2C, Ar), 98.02 (C-1), 70.90 (C-3), 69.65 (C-4), 68.36 (C-5), 66.88 (CH2 Fmoc), 55.34 (C-2), 51.82 (OMe), 50.89 (C-6), 47.15 (ArCH2NH), 36.43 (CH2-CH-Fmoc), 23.16 (CH3 Ac), 20.67 (2C, CH3 Ac) ppm; HRMS: m/z calcd. for [M + H]+ 622.2513, found 622.2508; calcd. for [M + Na]+ 644.2332, found 644.2328; [α]D25 = + 1.0 (c = 0.8, CHCl3).
Methyl 2-acetamido-3,4-di- O -acetyl-2,6-didesoxy-6- N -[4-[[(4-nitro-2,1,3-benzoxadiazol-7-yl)amino]methyl]triazol-1-yl]- α - d -glucopyranoside (7, C 22 H 26 N 8 O 10 )
Compound 2 (75 mg, 0.22 mmol) and 4-NH-propargyl-7-nitrobenzofurazane (95 mg, 0.44 mmol, 2 equiv.) was dissolved in 1:1 mixture of H2O /MeOH (6 cm3), then sodium ascorbate (13 mg, 0.07 mmol, 0.3 equiv.) and CuSO4‧5H2O (5 mg, 0.02 mmol, 0.1 equiv.) were added. The solution was left stirring at 60 °C overnight. The reaction was then diluted with H2O and extracted with EtOAc. The organic layer was then dried over anhydrous Na2SO4, filtered, and the solvent was removed under reduced pressure. The crude was purified by flash column chromatography (EtOAc/MeOH 19/1) to afford compound 7 (80 mg, 64% yield). 1H NMR (400 MHz, CDCl3): δ = 8.49 (d, J = 8.5 Hz, 1H, Ar), 7.76 (s, 1H, Ar), 6.80 (m, 1H, NH), 6.37 (d, J = 8.6 Hz, 1H, Ar), 5.66 (d, J = 9.4 Hz, 1H, NH), 5.21 (dd, J = 10.8, 9.4 Hz, 1H, H-3), 4.90–4.76 (m, 3H, H-4, ArCH2NH), 4.66 (d, J = 3.6 Hz, 1H, H-1), 4.60 (dd, J = 14.5, 2.5 Hz, 1H, H-6a), 4.45 (dd, J = 14.4, 7.6 Hz, 1H, H-6b), 4.24 (ddd, J = 10.8, 9.5, 3.6 Hz, 1H, H-2), 4.16 (ddd, J = 10.3, 7.7, 2.6 Hz, 1H, H-5), 3.12 (s, 3H, OMe), 2.11 (s, 3H, Ac), 2.02 (s, 3H, Ac), 1.94 (s, 3H, Ac) ppm; 13C NMR (101 MHz, CDCl3): δ = 171.23 (C=O), 169.99 (C=O), 169.58 (C=O), 144.30 (C-4’’), 143.64 (C-5’’), 142.93 (C-6’’), 142.20 (C-1’’), 135.95 (C-3’’), 123.65 (C-2’’), 99.60 (C-1), 98.20 (?), 70.66 (C-3), 69.21 (C-4), 68.02 (C-5), 55.43 (C-2), 51.84 (OMe), 50.91 (C-6), 39.44 (?), 29.68 (ArCH2NH), 23.15 (CH3 Ac), 20.64 (CH3 Ac), 20.64 (CH3 Ac) ppm; HRMS: m/z calcd. for [M + H]+ 563.1850, found 563.1850; calcd. for [M + Na]+ 585.1670, found 585.1664; [α]D25 = -0.8 (c = 0.8, DMSO).
Data availability
The NMR data are from the corresponding spectra present in the supplementary information file.
References
Sanjanwala D, Londhe V, Trivedi R, Bonde S, Sawarkar S, Kale V, Patravale V (2022) Expert Opin Drug Delivery 19:1664
Rabenstein DL (2002) Nat Prod Rep 19:312
Helenius A, Aebi M (2001) Science 291:2364
Marth JD, Grewal PK (2008) Nat Rev Immunol 8:874
Jefferis R (2009) Nat Rev Drug Discov 8:226
Lis H, Sharon N (1998) Chem Rev 98:637
Wasonga G, Tatara Y, Kakizaki I, Huang X (2013) J Carbohydr Chem 32:392
Lee S-U, Li CF, Mortales C-L, Pawling J, Dennis JW, Grigorian A, Demetriou M (2019) PLoS ONE 14:e0214253
Paiotta A, D’Orazio G, Palorini R, Zoia L, Votta G, De Gioia L, Chiaradonna F, La Ferla B (2018) Eur J Org Chem 17:1946
Ricciardiello F, Votta G, Palorini R, Raccagni I, Brunelli L, Paiotta A, Tinelli F, D’Orazio G, Valtorta S, De Gioia L, Pastorelli R, Moresco RM, La Ferla B, Chiaradonna F (2018) Cell Death Dis 9:377
Gao F, Yan X, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, Wright GD, Auclair K (2006) J Med Chem 49:5273
Blanco JLJ, Fernández JMG, Gadelle A, Defaye J (1997) Carbohydr Res 303:367
de Raadt A, Stuetz AE (1992) Tetrahedron Lett 33:189
Paulsen H, Pries M, Lorentzen JP (1994) Liebigs Ann Chem 4:389
Cipolla L, Redaelli C, Granucci F, Zampella G, Zaza A, Chisci R, Nicotra F (2010) Carbohydr Res 345:1291
Subratti A, Lalgee LJ, Jalsa KN (2018) Tetrahedron Lett 59:3384
Witczak ZJ, Bielski R (2013) Click chemistry in glycoscience: new development and strategies. Wiley, New York
Yoshioka K, Takahashi H, Homma T, Sato M, Ki Bong O, Nemoto Y, Matsuoka H (1996) Biochim Biophys Acta 1289:5
Acknowledgements
We acknowledge UNIMIB for the financial support (project 2022-ATEQC-0001). B.L.F. acknowledges Paolo Riva for his enthusiastic and excellent work. We acknowledge CORIMAV for the fellowship of M.A.
Funding
Open access funding provided by Università degli Studi di Milano - Bicocca within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alagia, M., Taglietti, L. & La Ferla, B. Synthesis of N-acetylglucosamine analogues modified at C6 position with azido-derived moieties. Monatsh Chem (2024). https://doi.org/10.1007/s00706-024-03198-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00706-024-03198-0