
Abstract
The kinetics of oxidation of the antibiotic drug chloramphenicol (CHP) by hexacyanoferrate(III) (HCF) has been investigated spectrophotometrically both in the absence and presence of ruthenium(III) catalyst in aqueous alkaline medium at 25 °C and at constant ionic strength of 1.10 mol dm−3. The stoichiometry is identical in both cases, i.e. [CHP]/[HCF] = 1:2. The oxidation products were identified by TLC and spectral studies such as GC–MS, IR, and 1H NMR. In both catalyzed and uncatalyzed reactions, the order with respect to the concentration of HCF is unity, whereas the order with respect to the concentration of CHP and the concentration of OH− is less than unity over the concentration range studied. The order with respect to the concentration of Ru(III) is unity. The reaction in the presence of Ru(III) is approximately tenfold faster than the uncatalyzed reaction. The active species of oxidant and catalyst are [Fe(CN)6]3− and [Ru(H2O)5(OH)]2+, respectively. On the basis of experimental results suitable mechanisms are proposed. The reaction constants involved in the different steps of the reaction mechanisms were calculated for both cases. The catalytic constant was also calculated for the catalyzed reaction at different temperatures. The activation parameters with respect to the slow step of the mechanism and thermodynamic quantities are also determined.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hexacyanoferrate(III) [HCF(III)] has been widely used to oxidize numerous organic and inorganic compounds in alkaline media. Some authors [1, 2] have suggested that alkaline HCF(III) ion simply acts as an electron-abstracting reagent in redox reactions. However, Speakman and Waters [3] suggested different paths of oxidation of aldehydes, ketones, and nitroparaffins by HCF(III). Singh et al. [4] while discussing the oxidations of formaldehyde, acetone, and ethyl methyl ketone by HCF(III) suggested that the oxidation takes place via an electron transfer process resulting in the formation of a free radical intermediate. HCF(III) is a one-electron oxidant with a redox potential of +0.45 V for the [Fe(CN)6]3−/[Fe(CN)6]4− couple in alkaline medium, leading to its reduction to hexacyanoferrate(II), a stable product [5, 6].
Transition metals are known to catalyze many oxidation–reduction reactions, because they involve multiple oxidation states. In recent years the use of transition metal ions such as ruthenium, osmium, palladium, manganese, chromium, and iridium, either alone or as binary mixtures, as catalysts in various redox processes has attracted considerable interest [7]. Ruthenium(III) acts as a catalyst in the oxidation of many organic and inorganic substrates [8, 9]. Although the mechanism of the catalysis depends on the nature of the substrates, oxidant, and experimental conditions, it has been shown [10] that metal ions acts as catalysts by one of several different paths, such as the formation of complexes with reactants or oxidation of the substrate itself or through the formation of free radicals. Ruthenium(III) catalysis in redox reactions involves different degrees of complexity, owing to the formation of different intermediate complexes and different oxidation states of ruthenium.
Chloramphenicol (2,2-dichloro-N-[(1R,2R)-2-hydroxy-1-(hydroxymethyl)-2-(4-nitrophenyl)ethyl]acetamide, CHP) is a bacteriostatic antimicrobial. Chloramphenicol is effective against a wide variety of Gram-positive and Gram-negative bacteria, including most anaerobic organisms. It is considered a prototypical broad-spectrum antibiotic, alongside the tetracyclines. One important clinical application of chloramphenicol is in the treatment of typhoid. The most serious adverse effect associated with chloramphenicol treatment is bone marrow toxicity. As a result of its extensive usage, chloramphenicol may enter the environment via wastewater effluent and biosolids from sewage treatment plants and via manure and litters from food-producing animal husbandry. The presence and accumulation of chloramphenicol antibiotics in aquatic environments, albeit at low concentrations, may pose threats to the ecosystem and human health by inducing increase and spread of bacteria drug-resistance due to long-term exposure. This necessitates the development of the various advanced oxidation processes for the transformation of chloramphenicol in water [11].
Although some work on oxidation of CHP by various oxidants has been carried out [11, 12] there is a lack of literature on the oxidation of this drug by HCF(III) and its catalysis by ruthenium(III). We have observed that ruthenium(III) in microamounts catalyzes the oxidation of chloramphenicol by HCF(III) in alkaline medium. Such studies are of much significance in understanding the mechanistic profile of chloramphenicol in redox reactions and provide an insight into the interaction of metal ions with the substrate and its mode of action in biological systems. Also, to determine the active species of HCF(III) and ruthenium(III) catalyst, and to resolve the complicity of the reaction, a detailed study of the reaction becomes important. Hence, the present investigation aimed to establish the reactivity of chloramphenicol towards HCF(III) in both uncatalyzed and ruthenium(III)-catalyzed reactions and to arrive at plausible mechanisms.
Results and discussions
Stoichiometry and product analysis
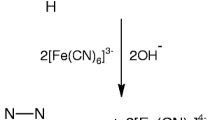
Different sets of reaction mixtures containing varying ratios of HCF(III) to CHP in the presence of constant amounts of OH− and NaClO4 in an uncatalyzed reaction and with a constant amount of Ru(III) in a catalyzed reaction and at constant ionic strength of 1.10 mol dm−3 were allowed to react for about 5 h at 25 °C. The remaining concentration of HCF(III) was assayed by measuring the absorbance at 420 nm. The results indicated 1:2 stoichiometry for both reactions as given in Scheme 1.

After the completion of the reaction, the reaction mixture was acidified, concentrated, and extracted with ether. The reaction product was further recrystallized from aqueous alcohol. The main reaction product was identified as p-nitrobenzaldehyde. This was the only organic product obtained in the oxidation which was confirmed by a single spot on thin-layer chromatography and was characterized by spectral investigations. From the IR (Suppl. Fig. 1), GC–MS (Fig. 1), and NMR (Fig. 2) spectra, the main oxidation product was identified as p-nitrobenzaldehyde. The IR spectrum showed a C=O stretching band for the aldehyde functional group at 1,709 cm−1 and an –NO2 stretching band at 1,349 cm−1 (Suppl. Fig. 1). The presence of p-nitrobenzaldehyde was also confirmed by GC–MS analysis (Fig. 1). The mass spectrum showed a base peak at m/z = 151 which is consistent with a molecular ion of 151 amu (Fig. 1). All other peaks observed in the GC–MS data can be interpreted in accordance with the structure of p-nitrobenzaldehyde. p-Nitrobenzaldehyde was also characterized by its 1H NMR spectra (Fig. 2, DMSO-d 6). Another product, 2-amino-1,2-ethandiol, was confirmed by GC–MS which showed a molecular ion peak at m/z = 77. 2-Chloroacetic acid was identified by spot tests [13]. Its reaction products do not undergo further oxidation under the present kinetic conditions.

GC–MS spectra of the product p-nitrobenzaldehyde showed molecular ion peak and base peak at m/z = 151

1H NMR spectra of p-nitrobenzaldehyde, the product of oxidation of chloramphenicol by HCF(III)
Reaction orders
The reaction orders have been determined from the slopes of k U or k C vs. log(concentration) plots by varying the concentrations of chloramphenicol, OH−, or ruthenium(III), in turn, while keeping the others constant.
Dependence on [HCF(III)]
The oxidant was varied in the absence and presence of catalyst, ruthenium(III), in the concentration range 1.5 × 10−5–2.0 × 10−4 mol dm−3. The pseudo-first-order rate constants (k U and k C) in both cases were almost constant (Tables 1, 2), indicating first-order dependence with respect to HCF(III) concentration. This was also confirmed by the plots of log(absorbance) vs. time which were linear over three half-lives of the reaction for different initial HCF(III) concentrations.
Dependence on [substrate]
The effect of chloramphenicol was studied for both cases in the concentration range 1.0 × 10−3–10.0 × 10−3 mol dm−3 at a constant concentration of HCF(III), OH−, and a constant ionic strength of 1.10 mol dm−3 in the uncatalyzed reaction and with a constant concentration of Ru(III) in the catalyzed reaction. In both cases, at constant temperature, the k U and k C values increased with the increase in [CHP] (Tables 1, 2). The order with respect to [CHP] was less than unity. This was also confirmed by the plots of k U vs. [CHP]0.40 and k C vs. [CHP]0.39 which were linear, unlike the direct plot of k U vs. [CHP] and k C vs. [CHP] (Fig. 3).

Dependence on [alkali]
The effect of alkali was studied for both cases in the concentration range 0.10–1.0 mol dm−3 at constant concentrations of HCF(III), CHP, and ionic strength in the uncatalyzed reaction and with a constant concentration of Ru(III) in the catalyzed reaction. The rate constants increased with the increase in [alkali] (Tables 1, 2) and the order was found to be less than unity, i.e., 0.57 in the uncatalyzed reaction and 0.64 in the catalyzed reaction.
Dependence on [ruthenium(III)]
Ruthenium(III) concentration was varied from 2.0 × 10−6 to 2.0 × 10−5 mol dm−3, at constant concentrations of HCF(III), CHP, and alkali, and at constant ionic strength. As the concentration of ruthenium(III) increases the rate of reaction also increases (Table 2). The order with respect to concentration of Ru(III) was found to be unity (Fig. 4).

Unit order plot of [Ru(III)] vs. k C
Effect of ionic strength (I) and dielectric constant (D)
In the absence and in the presence of catalyst, constant concentrations of reactants and with other conditions constant, the ionic strength was varied by varying NaClO4 concentration between 0.6, 0.8, 0.9, 1.1, and 1.4 mol dm−3. The rate was found to increase with increase in ionic strength. A plot of logk U or k C vs. I 1/2 was linear with positive slope (Suppl. Fig. 2). The effect of dielectric constant was studied by varying the t-butyl alcohol–water volume fractions from 0 to 30. It was found that as the volume fractions of t-butyl alcohol increased in the reaction medium, the rate of reaction increased in the absence and presence of catalyst (Suppl. Fig. 3). The plot of logk U or k C vs. 1/D was linear with a positive slope.
Effect of initially added products
The initially added products p-nitrobenzaldehyde and hexacyanoferrate(II) did not have any significant effect on the rate of reaction in the absence and presence of catalyst.
Test for free radicals (polymerization study)
For both the uncatalyzed and catalyzed reactions, the intervention of the free radicals in the reaction was examined as follows: the reaction mixture, to which a known quantity of acrylonitrile monomer (scavenger) had been added initially, was kept for 2 h in an inert atmosphere. On diluting the reaction mixture with methanol, a white precipitate was formed, indicating the intervention of free radicals in the reactions. The blank experiments of either [Fe(CN)6]3− or CHP alone with acrylonitrile did not induce any polymerization under the same conditions as those induced for the reaction mixture. Initially added acrylonitrile decreases the rate also indicating the free radical intervention [14].
Effect of temperature
The kinetics were studied at four different temperatures 15, 25, 35, and 45 °C under varying concentrations of chloramphenicol and alkali keeping the other conditions constant for the uncatalyzed reaction. The rate constants (k 1) of the slow step of Scheme 2 were obtained from the intercepts of the plots of 1/k U vs. 1/[CHP] at the four different temperatures. The values are given in Table 3. The energy of activation for the rate-determining step was obtained by the least-squares method of the plot of logk 1 vs. 1/T, and the other activation parameters are calculated and are given in Table 3. For the catalyzed reaction the influence of temperature on the rate of reaction was also studied at 15, 25, 35, and 45 °C. The rate constants (k 2) of the slow step of Scheme 3 were obtained from the intercepts of the plots of [Ru(III)]/k C vs. 1/[CHP] at the four different temperatures. The values are given in Table 4. The energy of activation for the rate determining step was obtained by the least-squares method of the plot of logk 2 vs. 1/T and other activation parameters calculated for the reaction are presented in Table 4.


Catalytic activity
Molelwyn-Hughes [15] pointed out that in the presence of catalyst, the uncatalyzed and catalyzed reactions proceed simultaneously, so that
where k T is the total rate constant, k U the pseudo-first-order rate constant for the uncatalyzed path, K C the catalytic constant, and x the order of the reaction with respect to catalyst. In the present investigations the x value for the standard run was found to be unity. Then, the value of K C is calculated using the equation
The values of K C were evaluated for Ru(III) catalyst at four different temperatures (Table 5). Further, plots of logK C vs. 1/T were linear and the values of activation parameters with reference to catalyst were computed. These results are summarized in Table 5.
Mechanism of uncatalyzed reaction
The variation of the concentrations of the oxidant HCF(III), substrate (CHP), and alkali, while keeping others constant, showed that the reaction is first order in oxidant, alkali, and of fractional order in substrate concentrations (Table 1). The reaction between chloramphenicol and Fe[(CN)6]3− has a stoichiometry of 1:2. On the basis of the experimental results, a mechanism can be proposed for which all the observed orders in each constituent, i.e., [oxidant], [reductant], and [OH−], may be well accounted for. Oxidation of chloramphenicol by HCF(III) in NaOH media is a non-complementary reaction with oxidant undergoing equivalent changes.
In the present study, alkali combines first with chloramphenicol to give the anionic form of chloramphenicol (1) in a pre-equilibrium step, which is also supported by the observed fractional order in [OH−] and [CHP]. The HCF(III) species reacts with the anionic form of chloramphenicol to give a complex C1 (2), which decomposes in a slow step to give a free radical 3 derived from the chloramphenicol anion and Fe[(CN)6]4−. This free radical in a subsequent fast step decomposes to give p-nitrobenzaldehyde (4) and another free radical 5. In the next fast step free radical 5 reacts with another mole of HCF in the presence of OH− to form an intermediate 2,2-dichloro-N-(1,2-dihydroxyethyl)acetamide (6). In the further fast steps, 6 undergoes hydrolysis to give the final products 2-amino-1,2-ethandiol (7) and dichloroacetic acid (8) as given in Scheme 2.
Since Scheme 2 is in accordance with the generally well-accepted principle of non-complementary oxidations taking place in a sequence of one-electron steps, the reaction between the substrate and oxidant would afford a radical intermediate. A free radical scavenging experiment revealed such a possibility. Spectroscopic evidence for the complex formation between oxidant and substrate was obtained from UV–Vis spectra of HCF (2.0 × 10−4 mol dm−3), CHP (1.0 × 10−3 mol dm−3), [OH−] (0.5 mol dm−3), and a mixture of both. A bathochromic shift of about 8 nm from 260 to 268 nm in the spectra of HCF to mixture of HCF and CHP was observed. A Michaelis–Menten plot proved the complex formation between oxidant and substrate, which explains the fractional order in [CHP].
Scheme 2 leads to the rate law Eq. (3):
Equation (4) can be rearranged to the following form, which is suitable for verification:
According to Eq. (5), other conditions being constant, plots of 1/k U vs. 1/[CHP], 1/k U vs. 1/[OH−] should be linear and are found to be so (Fig. 5). The slopes and intercepts of such plots lead to the values of K 1, K 2, and k 1 (Table 3).

The effect of ionic strength and dielectric constant of medium on the rate explains qualitatively the reaction between ions having the same charge, as seen in Scheme 2. The thermodynamic quantities for the different equilibrium steps in Scheme 2 can be evaluated as follows. The [CHP] and [OH−] (Table 1) were varied at four different temperatures. The plots of 1/k U vs. 1/[CHP], 1/k U vs. 1/[OH−] should be linear and are found to be so (Fig. 5). From the slopes and intercepts, the values of K 1, K 2 were calculated at different temperatures (Table 3). A van’t Hoff plot was drawn for the variation of K 1 and K 2 with temperature (logK 1 vs. 1/T and logK 2 vs. 1/T). The values of enthalpy of reaction ΔH, entropy of reaction ΔS, and free energy of reaction ΔG were calculated for the first and second equilibrium steps. These values are given in Table 3. A comparison of the ΔH value (49 kJ mol−1) from K 1 of the first step with that of ΔH # (51 kJ mol−1) obtained for the rate-determining step shows that the reaction before the rate-determining step is fairly fast as it involves low activation energy [16]. A high negative value of ΔS # (−114 J K−1 mol−1) suggests that intermediate complex (C1) is more ordered than the reactants [17].
Mechanism for ruthenium(III)-catalyzed reaction
The variation of the concentrations of the oxidant [Fe(CN)6]3−, substrate (chloramphenicol), Ru(III), and alkali, while keeping others constant, showed that the reaction is first order in oxidant and in Ru(III) and of fractional order in alkali and substrate concentrations (Table 2). The reaction between chloramphenicol and [Fe(CN)6]3− in NaOH in the presence of Ru(III) has a stoichiometry of 1:2. On the basis of the experimental results, a mechanism can be proposed for which all the observed orders in each constituent, i.e., [oxidant], [reductant], catalyst, and [OH−], may be well accounted for. Oxidation of chloramphenicol by HCF(III) in NaOH media is a non-complementary reaction [18].
Ruthenium(III) chloride acts as an efficient catalyst in many redox reactions, particularly in an alkaline medium [19]. In the present study it is quite probable that the [Ru(H2O)5OH]2+ species might assume the general form [Ru(III)(OH) x ]3−x. The x value must always be less than 6 because there are no definite reports of any hexahydroxy ruthenium species. The remainder of the coordination sphere would be filled by water molecules. Hence, under the conditions employed, e.g., [OH−] ≫ [Ru(III)], ruthenium(III) is mostly present [20] as hydroxylated species, [Ru(H2O)5OH]2+.
In earlier reports of Ru(III)-catalyzed oxidation, it was observed [21] that, if there exists a fractional order dependence with respect to [substrate] and [Ru(III)], and with respect to [oxidant], it leads to the formation of Ru(III)–substrate complex. This complex is further oxidized by the oxidant to Ru(III)–substrate complex followed by the rapid redox decomposition with regeneration of Ru(III) catalyst. In another case [22], if the process involves a zero-order dependence with respect to [oxidant], first order with respect to [Ru(III)], and a fractional order with respect to [substrate], it leads to the formation of Ru(III)–substrate complex and further cleaves to Ru(I) species which is rapidly oxidized by the oxidant to regenerate Ru(III) catalyst.
The results indicate that the alkali combines first with chloramphenicol to give the anionic form of chloramphenicol in a pre-equilibrium step, which is also supported by the observed fractional order in [OH−] and [CHP]. The ruthenium(III) species then reacts with the anionic form of chloramphenicol to give a complex C2 (2), which decomposes in the presence of the HCF(III) species in a slow step to give a free radical 3 derived from the chloramphenicol anion and Fe[(CN)6]4− with regeneration of the catalyst, ruthenium(III). K 3 is the equilibrium constant for the equilibrium binding of chloramphenicol to ruthenium(III). This free radical 3 in a subsequent fast step decomposes to give p-nitrobenzaldehyde (4) and another free radical 5. In the next fast step, free radical 5 reacts with another mole of HCF in the presence of OH− to give an intermediate 2,2-dichloro-N-(1,2-dihydroxyethyl)acetamide (6). In a further fast step 6 undergoes hydrolysis to give the final products 2-amino-1,2-ethandiol (7) and dichloroacetic acid (8) as given in Scheme 3.
Spectroscopic evidence for the complex formation between Ru(III) and CHP was obtained from UV–Vis spectra of [CHP] (1.0 × 10−3 mol dm−3), [Ru(III)] (4.0 × 10−6 mol dm−3), [OH−] (0.5 mol dm−3), and a mixture of both. A bathochromic shift of about 5 nm from 278 to 283 nm in the spectra of Ru(III) to mixture of Ru(III) and CHP was observed. Michaelis–Menten plot proved the complex formation between a catalyst and substrate, which explains the fractional order in [CHP].
From Scheme 3, the rate law Eq. (6) can be derived:
Equation (6) can be rearranged to Eq. (7) and is used for verification:
According to Eq. (7), other conditions being constant, plots of [Ru(III)]/k C vs. 1/[CHP] and 1/[OH−] should be linear and are found to be so (Fig. 6). The slopes and intercepts of such plots lead to the values of K 1, K 3, and k 2 (Table 4).

The effect of ionic strength and dielectric constant of the medium on the rate qualitatively explains the reaction between ions having the same charge, as seen in Scheme 3. The thermodynamic quantities for the different equilibrium steps in Scheme 3 can be evaluated as follows. The [CHP] and [OH−] (Table 2) were varied at four different temperatures. The plots of [Ru(III)]/k C vs. 1/[CHP], [Ru(III)]/k C vs. 1/[OH−] should be linear and are found to be so (Fig. 6). From the slopes and intercepts, the values of K 1 and K 3 were calculated at different temperature (Table 4b). A van’t Hoff plot was drawn for the variation of K 1 and K 3 with temperature (logK 1 vs. 1/T and logK 3 vs. 1/T). The values of enthalpy of reaction ΔH, entropy of reaction ΔS, and free energy of reaction ΔG were calculated for the first and second equilibrium steps. These values are given in Table 4. The negative value of ΔS # (−123 J K−1 mol−1) suggests that the intermediate complex (C2) is more ordered than the reactants [17]. The observed modest enthalpy of activation and higher rate constants for the slow step indicate that the oxidation presumably occurred via an inner sphere mechanism. This conclusion is supported by earlier observations [23, 24]. The activation parameters evaluated for the catalyzed and uncatalyzed reactions explain the catalytic effect on the reaction. The catalyst Ru(III) forms the complex (C2) with substrate, which enhances the reducing property of the substrate relative to that without catalyst. Further, the catalyst Ru(III) modifies the reaction path by lowering the energy of activation.
It is also interesting to note that the transient species involved in both uncatalyzed and Ru(III)-catalyzed reactions is different but leads to formation of the same products. The uncatalyzed reaction in alkaline medium has been shown to proceed via a HCF–CHP complex which decomposes slowly in a rate-determining step to give the products via free radicals in the further steps, whereas, in the catalyzed reaction, it has been shown to proceed via a Ru(III)–CHP complex which further reacts with 1 mole of HCF in the rate-determining step to give the products via free radicals in the further steps. Since in both cases HCF and CHP were involved, the products obtained were the same.
Conclusions
A comparative study of uncatalyzed and Ru(III)-catalyzed oxidation of chloramphenicol by HCF(III) in alkaline medium was performed. The active species of Ru(III) is found to be [Ru(H2O)5OH]2+. The reaction rates are about tenfold faster than those of the uncatalyzed reaction. It becomes apparent that, in carrying out this reaction, the role of reaction medium is crucial. Activation parameters were evaluated for both catalyzed and uncatalyzed reactions. Catalytic constants and activation parameters with reference to the catalyst were also computed. The overall sequence described here is consistent with all experimental findings including the product, spectral, mechanistic, and kinetic studies.
Experimental
All chemicals were of analytical reagent grade and millipore water was used throughout the work. The solution of chloramphenicol (SISCO CHEM) was prepared by dissolving known amounts of the samples in millipore water. The purity of the sample was checked by their melting point (150 °C). Solutions of chloramphenicol were always freshly prepared before use. A stock solution of the oxidant, HCF(III), was prepared by dissolving K3Fe(CN)6 (SISCO CHEM) in millipore water and standardizing the solution iodometrically [25]. The ruthenium(III) solution was prepared by dissolving RuCl3 (s. d. fine) in HCl (0.20 mol dm−3) and Hg was added to the Ru(III) stock solution to reduce any Ru(IV) formed during the preparation of the Ru(III) stock solution, and was set aside for 24 h. The Ru(III) concentration was assayed by EDTA titration [26]. Hexacyanoferrate(II) solution was prepared by dissolving a known amount of K4Fe(CN)6 (s. d. fine) in water. NaOH (Merck) and NaClO4 (BDH) were used to provide the required alkalinity and to maintain ionic strength, respectively.
For kinetic measurements, a Peltier Accessory (temperature control) attached to a Varian CARY 50 Bio UV–Vis spectrophotometer (Varian, Victoria-3170, Australia) was used. For product analysis, the QP-2010S Shimadzu GC–MS system, Nicolet 5700-FT-IR spectrometer (Thermo, USA), and Bruker 300 MHz 1H NMR spectrophotometer (Bruker, Switzerland) were used.
Procedure and kinetic measurements
The oxidation of chloramphenicol by HCF(III) was followed under pseudo-first-order conditions where [CHP] > [HCF(III)] in both the uncatalyzed and catalyzed reactions at 25.0 ± 0.1 °C, unless otherwise specified. In the absence of catalyst, the reaction was initiated by mixing HCF(III) with the CHP solution, which also contained the required concentrations of NaClO4 and NaOH. The reaction in the presence of the Ru(III) catalyst was initiated by mixing HCF(III) with the CHP solution which also contained the required concentrations of NaClO4, NaOH, and Ru(III) catalyst. The progress of the reaction was followed by measuring absorbance of Fe[(CN)6]3− in the reaction mixture at 420 nm in a 1-cm cell placed in the cell compartment of an Varian carry 50 Bio UV–Vis spectrophotometer. Application of Beer’s law under the reaction conditions had been verified at 420 nm (ε = 1,070 ± 10 dm3 mol−1 cm−1) [27]. In uncatalyzed and catalyzed cases, the kinetics was followed to more than 90 % completion of the reaction and good first-order kinetics were observed. During the kinetic studies it was observed that under the present experimental conditions in the absence of catalyst ruthenium(III), the oxidation of chloramphenicol by Fe[(CN)6]3− occurs very slowly, but in a measurable quantity. Hence, during the calculation of pseudo-first-order rate constants, k C, in the presence of catalyst, the uncatalyzed rate has also been taken into account. Therefore in each ruthenium(III)-catalyzed kinetic run, a parallel kinetic run under similar conditions in the absence of ruthenium(III) was also carried out. In both cases the pseudo-first-order rate constants (k U and k C) were obtained from the plots of log(absorbance) vs. time. The pseudo-first-order plots were linear over three half-lives. Thus, the total rate constant (k T) is equal to the sum of the rate constants in the absence (k U) and in the presence k C of catalyst, i.e.,
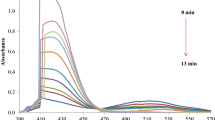
The rate constants k U and k C values are shown in Tables 1 and 2. The spectral changes during the chemical reaction for the standard condition at 25 °C are shown in Suppl. Fig. 4. It is evident from the figure that the concentration of HCF(III) decreases at 420 nm. It was also observed that there was almost no interference from other species in the reaction mixture at this wavelength. Similar results were obtained for the degradation of CHP by measuring COD values at different time.
References
Kelson EP, Phengsy PP (2000) Int J Chem Kinet 32:760
Vovk AI, Muraveva IV, Kukhar VP, Baklan VF (2000) Russ J Gen Chem 70:1108
Speakman PT, Waters WA (1955) J Chem Soc 1955:40
Singh VN, Gangwar MC, Saxena BBL, Singh MP (1969) Can J Chem 47:1051
Farokhi SA, Nandibewoor ST (2003) Tetrahedron 59:7595
Nowdari A, Adari KK, Gollapalli NR, Parvataneni V (2009) Eur J Chem 6:93
Das AK (2001) Coord Chem Rev 213:307
Shivananda KN, Lakshmi B, Jagadeesh RV, Puttaswamy, Mahendra KN (2007) Appl Catal A Gen 326:202
Shetti NP, Malode SJ, Nandibewoor ST (2011) J Mol Catal A Chem 30:1785
Singh AK, Srivastava S, Srivastava J, Srivastava R, Singh P (2007) J Mol Catal A Chem 278:72
Hosahalli RV, Byadagi KS, Nandibewoor ST, Chimatadar SA (2011) Z Phys Chem 225:79
Milano JC, Losteberdot P, Vernet JL (1995) Environ Technol 16:1101
Feigl F (1956) Spot tests in organic analysis, 5th edn. Elsevier, Amsterdam, p 391
Jagadeesh RV, Puttaswamy (2008) J Phys Org Chem 21:844
Molelwyn-Hughes EA (1947) Kinetics of reaction in solutions. Oxford University Press, London, p 297
Bilehal DC, Kulkarni RM, Nandibewoor ST (2001) Can J Chem 79:1926
Weissberger A, Lewis ES (eds) (1974) Investigations of rates and mechanism of reactions in techniques of chemistry, vol 4. Wiley, New York, p 421
Hiremath MI, Nandibewoor ST (2004) Transit Met Chem 29:703
Connick RE, Fine DA (1960) J Am Chem Soc 82:4187
Chimatadar SA, Kini AK, Nandibewoor ST (2005) Inorg React Mech 5:231
Sondu S, Sethuram B, Rao TN (1983) J Indian Chem Soc 60:198
Abbar JC, Malode SJ, Nandibewoor ST (2009) J Mol Catal A Chem 313:88
Martinez M, Pitarque MA, Eldik RV (1996) J Chem Soc Dalton Trans 2665
Farokhi SA, Nandibewoor ST (2003) Tetrahedron 59:7595
Jeffery GH, Bassett J, Mendham J, Denny RC (1996) Vogel’s text book of quantitative chemical analysis, 5th edn. ELBS, Longman, Essex, p 339
Reddy CS, Vijay Kumar T (1995) Indian J Chem 34A:615
Farokhi SA, Nandibewoor ST (2009) Catal Lett 129:207
Acknowledgments
The authors thank UGC, New Delhi for providing Major Research Project, Project No: F. NO. 39-833/2010.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Appendix
Derivation of rate law for uncatalyzed reaction
According to Scheme 2
The total concentration of [CHP]T is given by
where T and f refer to total and free concentrations
In view of the low concentration of [OH−], the numerator term K 1 K 2[OH][Fe(CN) 3−6 ] in the above equation is neglected:
Similarly,
In view of the low concentration of [CHP], the numerator term 1 + K 1 [CHP] in the above equation is neglected.
Therefore the total concentration of OH− is given by
Similarly,
Substituting Eqs. (11), (12), and (13) into Eq. (9) and omitting t and f we get
The term K 21 K 2[CHP][OH−] in the denominator of Eq. (14) is negligibly small compared to unity in view of the low concentration of substrate [CHP] used. Therefore Eq. (14) can be written as
The rate law for the ruthenium(III)-catalyzed reaction was derived similarly.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Meti, M.D., Byadagi, K.S., Nandibewoor, S.T. et al. Mechanistic studies of uncatalyzed and ruthenium(III)-catalyzed oxidation of the antibiotic drug chloramphenicol by hexacyanoferrate(III) in aqueous alkaline medium: a comparative kinetic study. Monatsh Chem 145, 1561–1573 (2014). https://doi.org/10.1007/s00706-014-1208-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-014-1208-7