
Abstract
Between 2005 and 2011, 23 lineages of circulating vaccine-derived polioviruses (cVDPVs) were detected in Nigeria with nonstructural region (NSR) of non-polio enterovirus C (NPEV-C) origin. However, no information exists on NPEV-C strains recombining with oral poliovirus type 2 vaccine strains (OPV2) to make type 2 cVDPVs (cVDPV2s) in Nigeria. This study was therefore designed to investigate the probable contribution of NPEV-Cs recently isolated in the region to the emergence of cVDPV2s. Eleven enterovirus C (EV-C) strains (8 NPEV-Cs and 3 PV2s) previously isolated by the authors were analysed in this study. All 11 isolates were assayed for cell-line-dependent growth restriction in four cell lines (LLC-MK2, MCF-7, RD and L20B). Subsequently, the isolates were subjected to RT-PCR specific for VP1 and 3Dpol/3′-UTR of EV-C. All PCR products were sequenced, and phylogenetic analysis was performed. All eight NPEV-Cs replicated exclusively in the MCF-7 cell line, while the three PV2s replicated in all four cell lines. The eight NPEV-Cs were identified as CVA13 (7 isolates) and CVA20 (1 isolate) by VP1 analysis, while all 11 isolates were confirmed to be EV-Cs by 3Dpol/3′-UTR analysis. In addition, phylogeny violations suggested that some cVDPVs might have recombined with common ancestors of the NPEV-Cs described in this study. This was confirmed by the scatter plot of divergence in VP1 against that of 3Dpol/3′-UTR sequences for pairs of isolates. The results of this study showed that the NSR of unknown origin found in cVDPVs from the region might have come from NPEV-Cs (e.g., CVA13 and CVA20) circulating in Nigeria.
Similar content being viewed by others


Introduction
The resolution of the World Health Assembly in 1988 to eradicate poliovirus [1] resulted in a reduction in the number of poliomyelitis cases from 350,000 in 1988 to 416 in 2013 (www.polioeradication.org) and the elimination of indigenous poliovirus globally except in Afghanistan, Pakistan and Nigeria [2]. The strategy to accomplish this included a very sensitive poliovirus surveillance programme coupled with massive vaccination campaigns [3]. The inactivated poliovirus vaccine (IPV) and oral poliovirus vaccine (OPV) have been the workhorses of these vaccination campaigns. The OPV has been the vaccine of choice in developing countries because, besides being cheaper and hence more affordable, as opposed to IPV, it confers mucosal immunity to vaccinees, thereby reducing the number of susceptible individuals that can participate in poliovirus transmission [4].
Poliovirus (PV) belongs to the species Enterovirus C, genus Enterovirus, family Picornaviridae. Besides poliovirus, other members of this species include coxsackievirus (CV) A1, CVA13, CVA17, CVA19, CVA20, CVA21, CVA22, CVA24, poliovirus (PV) 1, PV2, PV3, enterovirus (EV) C95, EV-C96, EV-C99, EV-C102, EV-C104, EV-C105, EV-C109, EV-C116, EV-C117 and EV-C118. Enterovirus virions lack a lipid envelope and have an icosahedral capsid with a diameter of 28–30 nm. The ~7,500-nt protein-linked, single-stranded, positive-sense RNA genome has one open reading frame (ORF) flanked on both sides by untranslated regions (5′ and 3′ UTRs) and a 3′ poly A tail. The single ORF is cotranslationally cleaved into 11 proteins, four of which (VP1, 2, 3, and 4) are structural proteins, and the remaining seven are nonstructural proteins. The correlation between sequences of VP1 and enterovirus serotypes has established VP1 as the gene of choice for typing enteroviruses into serotypes [5–13].
Poliovirus has three distinct serological types (serotypes PV1, PV2 and PV3), and prior to this discovery it was impossible to develop vaccines to control the poliomyelitis epidemic [4]. However, subsequently, versions of the three serotypes (OPV1, OPV2 and OPV3) with attenuated virulence were developed and are used to this day for immunization campaigns [4]. On administration, the vaccine remains in circulation for about 8 weeks, after which it ceases to circulate. However, on rare occasions, especially in suboptimally immunised populations, the vaccine virus reverts to wild-type virulence. It thereby continues to circulate in the population longer than the expected 8 weeks [14], at times surviving for over a year [15–20]. Such isolates are referred to as circulating vaccine-derived polioviruses (cVDPVs).
Several outbreaks of poliomyelitis caused by cVDPVs have been reported globally [15–22]. Genomic characterization of cVDPVs recovered from such outbreaks revealed that most cVDPVs are recombinants with a structural region of OPV origin and a nonstructural region derived from (or related to) previously characterized NPEV-C genomes [15–22]. Nigeria suffered the biggest outbreak of cVDPV ever recorded, with 403 confirmed cases between 2005 and 2011 alone [20]. All but seven of the cVDPVs isolated during this period were OPV2/NPEV-C recombinants resolved into 23 independent emergences [20]. Despite this, the NPEV-Cs that recombined with OPV2 strains to make type 2 cVDPVs (cVDPV2s) have not been described.
We recently isolated an array of NPEV-Cs circulating in Nigeria [23]. Here, we show that the indigenous NPEV-Cs might have recombined with OPV2s in Nigeria to make some of the recombinant cVDPV lineages isolated during the outbreak in Nigeria.
Materials and methods
Cell lines
Cell lines used in this study include MCF-7 (human mammary gland adenocarcinoma), LLC-MK2 (rhesus monkey kidney), RD (human rhabdomyosarcoma) and L20B (murine transgenic L cells expressing the poliovirus receptor; CD155). All cells were maintained in medium 199 supplemented with 5 % fetal bovine serum (FBS).
Isolates used in this study
The isolation of the EV-Cs used in this study (Table 1) has been described previously [23]. The eleven isolates analysed in this study were detected in five environmental samples that were collected and concentrated as part of the poliovirus environmental surveillance programme in Nigeria [24]. The concentrates were made available to the authors by the Environmental Surveillance Laboratory and were independently screened by our group at the Department of Virology, University of Ibadan, Oyo State, Nigeria, on MCF-7 and LLC-MK2 cell lines [23].
In this study, 11 isolates were analysed: eight NPEV-Cs and three PV2 isolates. Four of the NPEV-Cs and all three PV2 isolates had been typed previously by VP1 amplification and sequencing [23], while the other four had not been previously identified.
Growth in different cell lines
To assay for growth in different cell lines, all of the cell lines mentioned above were utilised. Two cell culture tubes per cell line (Table 1) of confluent cell monolayers were inoculated with 200 μL of stock of each isolate. The tubes were subsequently incubated at 37 °C and checked for cytopathic effect (CPE) every 24 hours over the course of 72 hours. Cells with classic enterovirus CPE (cell rounding, detachment and death) within 72 hours were considered permissive and susceptible to the test virus.
The three PV2 isolates analysed in this study were first passaged twice in the L20B cell line before they were screened for growth in different cell lines to ensure the serotypic purity of the isolates. Since PVs are the only described EV-Cs to date that replicate in the L20B cell line, this was done to ensure that the three PVs analysed did not contain other NPEV-Cs.
RNA extraction and cDNA synthesis
To extract viral RNA from the isolates, an RNA Mini Kit (QIAGEN, Hilden, Germany) was used in accordance with the manufacturer’s instructions. The RNA extract was then used to synthesize cDNA using a Script cDNA Synthesis Kit (Jena Bioscience, Jena, Germany). Briefly, 12 μL of the RNA extract was added to 8 μL cDNA synthesis mixture containing 4 μL of 5X SCRIPT RT buffer complete, 1 μL of 10 mM dNTP mix, 1 μL of 40 units of RNase inhibitor, 1 μL of 100 mM DTT, 0.5 μL of 100 µM random hexamers and 0.5 μL of 200 units of SCRIPT reverse transcriptase. The mixture was incubated at 42 °C for 10 minutes, followed by 50 °C for 60 minutes in a Veriti Thermal Cycler (Applied Biosystems, California, USA). Subsequently, the cDNA was stored at −80 °C until analysed.
EV-C VP1-region-specific PCR (EV-C VP1 PCR)
EV-C-VP1-PCR was done to identify the four NPEV-Cs that had not been typed. The assay amplifies an ~805-bp fragment spanning the 3′ tail of VP3 and the 5′ two-thirds of VP1 using primers HEV-C-VP3 and HEV-C-VP1 [25]. Primers were made in 50-µM concentrations, and one microliter of each of the primers was added to a 50-μL reaction containing 10 μL of Red Load Taq (Jena Bioscience), 4 μL of cDNA, and 34 μL of RNase-free water. Thermocycling was done as follows; 94 °C for 3 minutes followed by 45 cycles of 94 °C for 30 seconds, 42 °C for 30 seconds, and 60 °C for 60 seconds with ramp of 40 % from 42 °C to 60 °C. This was followed by 72 °C for 7 minutes, and the sample was held at 4 °C until the reaction was terminated. Subsequently, PCR products were resolved on 2 % agarose gels stained with ethidium bromide and viewed using a UV transilluminator.
EV-C 3Dpol/3′-UTR-specific PCR (EV-C 3Dpol/3′-UTR PCR)
The EV-C-3Dpol/3′-UTR-PCR assay was used in this study to amplify a ~429-nt fragment spanning the 3′-terminal coding sequence of EC RNA polymerase 3Dpol and the 5′ end of the 3′-UTR of ECs [23]. Primers were made in 50-µM concentrations and one microliter of each of the primers was added to a 50-μL reaction containing 10 μL of Red Load Taq (Jena Bioscience), 4 μL of cDNA and 34 μL of RNase-free water. Thermocycling included 94 °C for 3 minutes followed by 45 cycles of 94 °C for 30 seconds, 42 °C for 30 seconds and 60 °C for 30 seconds with ramp of 40 % from 42 °C to 60 °C. This was followed by 72 °C for 7 minutes, and the sample was held at 4 °C until the reaction was terminated. Finally, PCR products were resolved on 2 % agarose gels stained with ethidium bromide and viewed using a UV transilluminator.
Amplicon sequencing and enterovirus typing
Positive PCR reactions with the required amplicon size were shipped to Macrogen Inc, Seoul, South Korea, where amplicon purification and BigDye chemistry sequencing was done. Sequencing was done using both forward and reverse primers. Subsequently, using the sequence data and the Enterovirus Genotyping Tool version 0.1 [26] isolate genotype and species were determined.
Phylogenetic analysis
For VP1 and 3Dpol/3′-UTR analysis, cVDPV sequences previously reported from Nigeria [20, 27], EV-Cs previously reported by the authors [23], and other reference sequences were retrieved from GenBank and aligned using the CLUSTAL W program in MEGA 5 software with default settings [28]. Subsequently, neighbor-joining trees were constructed using MEGA 5 software with the Kimura 2-parameter model [29] and 1,000 bootstrap replicates. The accession numbers of sequences retrieved from GenBank for phylogenetic analysis are indicated in the sequence names on the phylograms. This strategy was used to find phylogeny violations as evidence of recombination.
To further investigate the “phylogeny violations”, pairwise distances of the VP1 and 3Dpol/3′-UTR sequences were estimated using MEGA 5 software with the Kimura 2-parameter model [29]. This was done for all 11 isolates described in this study and other PV2s from Nigeria [20, 27] whose full genome sequences were available in GenBank. Subsequently, scatter plots were generated. This described the relationship between divergence at two genomic regions (VP1 and 3Dpol/3′-UTR) for each pair of isolates. To accomplish this, the ICTV enterovirus genotype barrier was used to identify isolates that belong to different genotypes (i.e. more than 25 % nucleotide sequence divergence in VP1) but had 3Dpol/3′-UTR sequences with similarity >94 %, a situation that is expected if there is recombination.
Nucleotide sequence accession numbers
The sequences reported in this study have been submitted to the GenBank nucleotide sequence database under the accession numbers KF303091, KF303094, KF303096, KF303100, KM225628, KM233673 – KM233680, KM235192, and KM235193.
Results
Growth in different cell lines
All three PV2 isolates showed CPE in all four cell lines, while the eight NPEV-Cs only showed CPE in the MCF-7 cell line. The eight NPEV-Cs did not show CPE in any of the other cell lines used in this study (Table 1).
EV-C VP1 PCR assay and typing of isolates
The EV-C-VP1-PCR assay was done on only the four yet-to-be-typed NPEV-C isolates (NIEC21–NIEC24), and the expected ~805-bp fragment was successfully amplified for all four isolates. Using the VP1 sequence data, the enterovirus genotyping tool identified the NPEV-Cs as CVA13 (three isolates) and CVA20 (one isolate) (Table 1).
EV-C 3Dpol/3′-UTR PCR assay
All 11 isolates analysed in this study were positive in the EV-C-3Dpol/3′-UTR-PCR assay. The ~429-nt fragment was successfully amplified for all isolates. Furthermore, using the 3Dpol/3′-UTR data, the enterovirus genotyping tool confirmed all 11 isolates to be EV-Cs.
Phylogenetic analysis
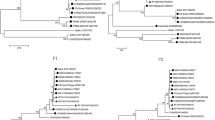
Based on the VP1 sequence data, the identities of the four EV-C isolates according to the phylogram (Fig. 1) was in accordance with the results of the enterovirus genotyping tool. These CVA13s clustered with sequences of other CVA13s from Nigeria obtained from GenBank (Fig. 1). To the best of our knowledge, prior to this study, no CVA20 sequence data had been reported from Nigeria.

Phylogenetic relationships of recovered isolates. The phylogram is based on an alignment of partial VP1 sequences. The newly sequenced strains are indicated by black circles. The GenBank accession number and strain ID are indicated in the tree. Bootstrap values are indicated if ≥70 %. The scale bars represent the evolutionary distance in substitutions per nucleotide
Phylogenetic analysis of 3Dpol/3′-UTR sequences alongside VP1 enables the detection of horizontal gene transfer via recombination events. One of the ways this can be accomplished is through the identification of incongruence between phylograms generated using both genomic regions independently. Another strategy is to determine the relationship between pairs of isolates that diverge in the two different genomic regions. In this study, recombination events were assayed for using both strategies.
For the first strategy, two phylograms were generated independently usingVP1 and 3Dpol/3′-UTR sequences from the 11 isolates described in this study alongside other Nigerian PV2 sequences obtained from GenBank. These phylograms showed several phylogeny violations indicative of recombination events (Fig. 2). Otherwise stated, the isolates did not cluster according to their serotypes in the 3Dpol/3′-UTR phylogram. To confirm these recombination events, the second strategy was employed. Scatter plots of divergence in VP1 and divergence in 3Dpol/3′-UTR for pairs of isolates showed that certain isolates belonging to different serotypes had 3Dpol/3′-UTR sequences with >94 % similarity (Fig. 3). Some of the recombinant isolates included the PV2 isolates described in this study and some Nigerian PV2 isolates described previously with “hitherto unidentified” recombination partners [20, 21] (Table 2). The data also showed that isolates of the same serotype that were recovered from the same sample (Table 1) were usually clones (Figures 2 and 3 and Table 2).

Phylogenetic relationship of selected isolates in the VP1 region and the 3Dpol/3′-UTR. The phylogram is based on an alignment of the VP1 (left) and 3Dpol/3′-UTR (right) sequences. The isolates and/or genomic regions that were newly sequenced in this study are indicated by black circles. Previously reported isolates are indicated by black triangles, and the 2002 VDPV reported previously [21] is indicated by a black diamond. Connecting lines are used to trace the locations of isolates of interest on both phylograms (orange for CVA13 and red for PV2s except for NIE0210766, which is shown in blue alongside CVA20). The GenBank accession number, strain ID and year of isolation are indicated in the 3Dpol/3′-UTR region tree only, if known. Bootstrap values are indicated if ≥70 %. The scale bars represent the evolutionary distance in substitutions per nucleotide

Relationship between sequence divergence (pairwise distance) in the VP1 region (x-axis) and in the 3Dpol/3′-UTR region (y-axis) among PV2, CVA13 and CVA20 sequences. The vertical dashed line (0.25) represents the ICTV threshold for serotype classification. The horizontal dashed line (0.06) represents the divergence threshold set in this study for 3Dpol/3′-UTR gene similarity. The data points in the lower right quadrant (within the green circle with a dashed boundary) represent relationships in which two isolates belong to different serotypes but have 3Dpol/3′-UTR regions with similarity >94 %
Discussion
This study used a combination of phylogenetic and cellular replication restriction studies to investigate the probable contribution of NPEV-Cs recently isolated in Nigeria to the emergence of cVDPV2s within the country. The results of this study showed evidence that the NPEV-C members circulating in Nigeria, including CVA13 and CVA20, had been recombining with Sabin 2 poliovirus (OPV2) in the region. The results therefore show that some of the “hitherto unidentified” NPEV-Cs recombining with OPV2 in Nigeria [20, 27] have been identified and partly characterised.
Recombination analysis
The presence and circulation of NPEV-Cs in Nigeria had been inferred since 2002 [27] when the first OPV2/NPEV-C recombinant was isolated in the region. This was further suggested by the detection of 23 independent OPV2/NPEV-C lineages between 2005 and 2011 [20] in the same region. Despite the above, there was under-reporting of NPEV-Cs in the region. It was recently demonstrated [23] that the widely used RD–L20B enterovirus isolation protocol might be responsible for the under-reporting as suggested [30], since the introduction of other cell lines such as MCF-7 led to an increase in the detection of NPEV-Cs in the region from zero to 27.66 % [23].
This study investigated the hypothesis [20, 27] that NPEV-Cs were recombining with OPV2 in Nigeria. The results of this study confirm that NPEV-Cs circulating in the region indeed recombined with OPV2 to make both the VDPV and cVDPVs described previously [20, 27]. Prior to this study, Adu et al. [27] suggested that CVA20, which had the most similar (85 % similarity) sequence in GenBank at that time, must have been the serotype that recombined with OPV2 in Nigeria to make the 2002 VDPV2 isolate. However, because of the low similarity score, this was inconclusive. The results of this study however, show conclusively that CVA20 (94 % similarity in the 3Dpol/3′-UTR region) indeed recombined with the 2002 VDPV2 isolate (DQ890385) as suggested [27].
In addition, while Jiang et al., [31] show that engineered PV/CVA20 recombinants are viable, the results of this study showed that natural OPV/CVA20 recombinants exist and such recombination events might have been happening for a while in Nigeria undetected [27].
The findings of this study also showed that a good number of the 2005–2011 cVDPV2 lineages [20] might have recombined with CVA20s and CVA13s circulating in Nigeria. The contribution of CVA13s to the emergence of cVDPVs have been documented [17, 18, 22]. Consequently, in this respect, the situation in Nigeria might be in accordance with what has been described globally [17, 18, 22].
Cell-line-dependent growth restrictions
In this study, to ascertain that the three PV2 isolates analysed (NIEC13, NIEC14 and NIEC19) did not contain NPEV-Cs, the virus stock was passaged twice in the L20B cell line. Given that NPEV-Cs do not replicate in L20B (Table 1), this step aimed to selectively inhibit the replication of any NPEV-Cs that might be present in the PV2 isolates, especially those isolated on MCF-7 cells (NIEC13 and NIEC14). The aforementioned therefore ruled out the possibility that genomes of NPEV-Cs could have been present in the three PV2s analysed. However, the likelihood that the recombination event occured in culture during the initial isolation process for the two PV2s (NIEC13 and NIEC14) isolated on MCF-7 cells (Table 1) cannot be ruled out. The authors, however, emphasise that during the initial isolation process [23], only polioviruses were isolated from the sample (sample 14) that yielded NIEC13 and NIEC14. In the case where the sample (sample 15, Table 1) yielded both poliovirus and NPEV-Cs (NIEC19 [PV2], NIEC16 and NIEC24 [CVA13s]), the PV2 was significantly different from the NPEV-Cs both in the VP1 and 3Dpol/3′-UTR region (Fig. 2). In addition, the observation that all enterovirus isolates of the same serotype from the same sample (Table 1) were molecular clones further validates the conclusion that the recombination events observed did not occur in the laboratory and must have occurred prior to virus isolation (Fig. 2 and Table 2).
Implications for Nigeria
The increased detection rate of NPEV-Cs achieved by changing the isolation protocol has already confirmed their presence and circulation in Nigeria [23]. Furthermore, after the completion of this study, we detected more NPEV-Cs in two states of southwestern Nigeria (unpublished). This further demonstrates their endemicity and geographical spread spanning the entire region. Should the immunity of the population to polioviruses remain lower than is necessary for herd immunity [32, 33], the condition will be perfect for NPEV-C co-infection of vaccinees and their contacts. This, in turn, will provide ideal conditions for the emergence of recombinant OPV/NPEV-C isolates that can regain wild-type pathogenicity and transmissibility [20, 31].
Therefore, as effort is ongoing to increase OPV coverage and consequently immunity, of the population to polioviruses, it is necessary to pay some attention to NPEV-C biology because of the contribution of these viruses to the emergence of cVDPV. This will enable the enterovirus research community to better understand NPEV-Cs in order to develop control strategies that might be needed to enhance polio eradication. It is therefore important to mention that the WHO Global Polio Laboratory Network (GPLN) is best positioned to help in elucidating the global dynamics of NPEV-Cs by including cell lines documented to support NPEV-Cs in their enterovirus isolation protocol. This will greatly enhance the global recovery of NPEV-Cs, making them more available to the research community for further studies, consequently enabling the global community to better understand the evolutionary dynamics of NPEV-Cs and the likelihood of any NPEV-C filling the niche of polioviruses after the global eradication of poliovirus and immunity to polioviruses begins to wane in populations.
References
World Health Organisation, 1988. Global eradication of poliomyelitis by the year 2000 (World Health Assembly resolutionWHA41–28). http://www.polioeradication.org/content/publications/19880513_resolution.pdf
World Health Organisation (2012) Progress towards poliomyelitis eradication, Nigeria, January 2011–September 2012. Wkly Epidemiol Record 87:439–446
World Health Organisation (2004) Polio laboratory Manual, 4th edn, Geneva
Nathanson N, Kew OM (2010) From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 172(11):1213–1229
Oberste MS, Maher K, Kilpatrick DR, Flemister MR, Brown BA, Pallansch MA (1999) Typing of human enteroviruses by partial sequencing of VP1. J Clin Microbiol 37(5):1288–1293
Oberste MS, Maher K, Kilpatrick DR, Pallansch MA (1999) Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol 73(3):1941–1948
Oberste MS, Schnurr D, Maher K, Al-Busaidy S, Pallansch MA (2001) Molecular identification of new picornaviruses and characterization of a proposed enterovirus 73 serotype. J Gen Virol 82(2):409–416
Oberste MS, Nix WA, Maher K, Pallansch MA (2003) Improved molecular identification of enteroviruses by RT-PCR and amplicon sequencing. J Clin Virol 26(3):375–377
Casas I, Palacios GF, Trallero G, Cisterna D, Freire MC, Tenorio A (2001) Molecular characterization of human enteroviruses in clinical samples: comparison between VP2, VP1, and RNA polymerase regions using RT nested PCR assays and direct sequencing of products. J Med Virol 65(1):138–148
Norder H, Bjerregaard L, Magnius LO (2001) Homotypic echoviruses share aminoterminal VP1 sequence homology applicable for typing. J Med Virol 63(1):35–44
Caro V, Guillot S, Delpeyroux F, Crainic R (2001) Molecular strategy for ‘serotyping’ of human enteroviruses. J Gen Virol 82(1):79–91
Thoelen I, Lemey P, Van der Donck I, Beuselink K, Lindberg AM, Van Ranst M (2003) Molecular typing and epidemiology of enteroviruses identified from an outbreak of aseptic meningitis in Belgium during the summer of 2000. J Med Virol 70(3):420–429
Blomqvist S, Paananen A, Savolainen-Kopra C, Hovi T, Roivainen M (2008) Eight years’experience of molecular identification of human enteroviruses. J Clin Microbiol 46(7):2410–2413
Alexander JP Jr, Gary HE Jr, Pallansch MA (1997) Duration of poliovirus excretion and its implications for acute flaccid paralysis surveillance: a review of the literature. J Infect Dis 175:S176–S182
Yang CF, Naguib T, Yang SJ, Nasr E, Jorba J, Ahmed N, Campagnoli R, van der Avoort H, Shimizu H, Yoneyama T, Myamura T, Pallansch M, Kew O (2003) Circulation of endemic type 2 vaccine-derived poliovirus in Egypt from 1983 to 1993. J Virol 77:8366–8377
Blomqvist S, Savolainen C, Laine P, Hirttio P, Lamminsalo E, Penttila E, Joks S, Roivainen M, Hovi T (2004) Characterization of a highly evolved vaccine-derived poliovirus type 3 isolated from sewage in estonia. J Virol 78:4876–4883
Arita M, Zhu SL, Yoshida H, Yoneyama T, Miyamura T, Shimizu H (2005) A sabin 3-derived poliovirus recombinant contained a sequence homologous with indigenous human enterovirus species c in the viral polymerase coding region. J Virol 79:12650–12657
Rakoto-Andrianarivelo M, Guillot S, Iber J, Balanant J, Blondel B, Riquet F, Martin J, Kew O, Randriamanalina B, Razafinimpiasa L et al (2007) Co-circulation and evolution of polioviruses and species c enteroviruses in a district of Madagascar. PLoS Pathogen 3:e191
Rakoto-Andrianarivelo M, Gumede N, Jegouic S, Balanant J, Andriamamonjy SN, Rabemanantsoa S, Birmingham M, Randriamanalina B, Nkolomoni L, Venter M et al (2008) Reemergence of recombinant vaccine-derived poliovirus outbreak in Madagascar. J Infect. Dis 197:1427–1435
Burns CC, Shaw J, Jorba J, Bukbuk D, Adu F, Gumede N, Pate MA, Abanida EA, Gasasira A, Iber J, Chen Q, Vincent A, Chenoweth P, Henderson E, Wannemuehler K, Naeem A, Umami RN, Nishimura Y, Shimizu H, Baba M, Adeniji JA, Williams AJ, Kilpatrick DR, Oberste MS, Wassilak SG, Tomori O, Pallansch MA, Kew OM (2013) Multiple independent emergences of type 2 vaccine-derived polioviruses during a large outbreak in Northern Nigeria. J Virol 87(9):4907–4922
Adu F, Iber J, Bukbuk D, Gumede N, Yang SJ, Jorba J, Campagnoli R, Sule WF, Yang CF, Burns C et al (2007) Isolation of recombinant type 2 vaccine-derived poliovirus (vdpv) from a nigerian child. Virus. Res 127:17–25
Combelas N, Holmblat B, Joffret M, Colbère-Garapin F, Delpeyroux F (2011) Recombination between poliovirus and Coxsackie A viruses of species C: a model of viral genetic plasticity and emergence. Viruses 3:1460–1484
Adeniji JA, Faleye TOC (2014) Impact of cell lines included in enterovirus isolation protocol on perception of Nonpolio enterovirus species C diversity. J Virol Methods. doi:10.1016/j.jviromet.2014.07.016
World Health Organisation (2003) Guidelines for environmental surveillance of poliovirus circulation, Geneva
Oberste MS, Maher K, Williams AJ, Dybdahl-Sissoko N, Brown BA, Gookin MS, Penaranda S, Mishrik N, Uddin M, Pallansch MA (2006) Species-specific RT- PCR amplification of human enteroviruses: a tool for rapid species identification of uncharacterized enteroviruses. J Gen Virol 87:119–128
Kroneman A, Vennema H, Deforche K, Van der Avoort H, Pe narandac S, Oberste MS, Vinjéc J, Koopmans M (2011) An automated genotyping tool for enteroviruses and noroviruses. J Clin Virol 51:121–125
Adu FD, Iber J, Harry T, Burns C, Oyedele O, Adeniji JA, Ossei-Kwasi M, Kilpatrick D, Tomori O, Kew O (2004) Some genetic characteristics of sabin-like poliovirus isolated from acute flaccid paralysis cases in Nigeria. Afr J Biotechnol 2:1684–5315
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16(2):111–120
Sadeuh-Mba SA, Bessaud M, Massenet D, Joffret ML, Endegue MC, Njouom R, Reynes JM, Rousset D, Delpeyroux F (2013) High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J Clin Microbiol 51:759–770
Jiang P, Faase JA, Toyoda H, Paul A, Wimmer E et al (2007) Evidence for emergence of diverse polioviruses from C-cluster coxsackie A viruses and implications for global poliovirus eradication. Proc Natl Acad Sci USA 104:9457–9462
Iliyasu Z, Nwaze E, Verma H, Mustapha AO, Weldegebriel G, Gasasira A, Wanorthern Nigeria emuehler KA, Pallansch MA, Gajida AU, Pate M, Sutter RW (2013) Survey of poliovirus antibodies in Kano, Northern Nigeria. Vaccine 12:1414–1420
Adeniji, J.A., Onoja, A.B., Adewumi, M.O., 2014. Neutralising antibody dynamics among children in a northcentral and southwestern Nigeria State. J Immunoassay Immunochem. doi:10.1080/15321819.2014.893889
Acknowledgments
We thank Dr. M. O. Adewumi for thoughtful critique of our manuscript. We thank Morris Saffold Jones and Erwin Duizer for providing us with the LLC-MK2 cell line. We thank Ayorinde James for providing us with the MCF-7 cell line. We also thank the WHO National Polio Laboratory in Ibadan, Nigeria, for providing us with the L20B and RD cell lines.
TOCF is a PhD student studying the evolutionary dynamics of enteroviruses in Nigeria. This study is a part of his PhD work.
Conflict of interest
The authors declare no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Adeniji, J.A., Faleye, T.O.C. Enterovirus C strains circulating in Nigeria and their contribution to the emergence of recombinant circulating vaccine-derived polioviruses. Arch Virol 160, 675–683 (2015). https://doi.org/10.1007/s00705-014-2322-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-2322-x