
Abstract
Porcine epidemic diarrhea virus (PEDV) is a highly contagious enteric pathogen of swine. Acute PEDV outbreaks have continually emerged in most swine-producing Asian countries and, recently, in the United States, causing significant economic losses in the pig industry. The spike (S) protein of PEDV is a type 1 transmembrane envelope glycoprotein and consists of the S1 and S2 domains, which are responsible for virus binding and fusion, respectively. Since the S1 domain is involved in a specific high-affinity interaction with the cellular receptor and induction of neutralizing antibody in the natural host, it is a primary target for the development of effective vaccines against PEDV. In this study, a codon-optimized PEDV S1 gene containing amino acid residues 25–738 was synthesized based on a multiple alignment of the S amino acid sequences of PEDV field isolates and used to establish a stable porcine cell line constitutively expressing the PEDV S1 protein. The purified recombinant S1 protein was found to mediate highly potent antibody responses in immunized rabbits. The antibodies strongly recognized the recombinant S1 protein from cell lysates and supernatants of S1-expressing cells, whereas they bound weakly to the authentic S protein of PEDV vaccine strain SM98-1. Furthermore, a serum neutralization test revealed that the rabbit antisera completely inhibit infection of the PEDV vaccine strain at a serum dilution of 1:16. We then tested the ability of vaccination with the recombinant S1 protein to protect piglets against PEDV. Late-term pregnant sows were inoculated intramuscularly with the purified S1 protein, and the outcome was investigated in passively immunized suckling piglets after a virulent PEDV challenge. The results showed that vaccination with S1 protein efficiently protected neonatal piglets against PEDV. Our data suggest that the recombinant S1 protein shows potential as an effective and safe subunit vaccine for PED prevention.
Similar content being viewed by others

Introduction
Porcine epidemic diarrhea (PED) is a devastating swine disease that is characterized by acute enteritis and lethal watery diarrhea followed by severe dehydration leading to death, with a high mortality rate in piglets [6, 26, 28]. The disease was initially recognized in England in 1971 [23], but the causative agent of this disease, PED virus (PEDV), was later identified in 1978 [25]. PED epidemics were first reported in Asia in 1982, and since then, PED has continued to threaten swine health, causing substantial economic losses in the Asian swine industry [4, 19, 27, 33]. In 2013, PED outbreaks suddenly occurred in the United States and have swept through the pork industry across the country, raising concerns about control measures for PED prevention [21, 30]. In Korea, PEDV appeared in 1992 [14]; however, a retrospective study indicated that the virus had been present as early as 1987 [24]. Although periodic vaccination strategies have been implemented nationwide to control PED in Korean swine herds, PEDV has continually emerged, causing tremendous harm to the productivity of Korean pig farms.
PEDV, a member of the genus Alphacoronavirus within the family Coronaviridae of the order Nidovirales, is a large, enveloped virus possessing a single-stranded, positive-sense RNA genome of approximately 28 kb with a 5′ cap and a 3′ polyadenylated tail [25, 28]. The PEDV genome is composed of the 5′ untranslated region (UTR), at least seven open reading frames (ORF1a, ORF1b, and ORF2 through 6), and the 3′ UTR [13]. The two large ORFs 1a and 1b make up the 5′ two-thirds of the genome and encode the non-structural replicase genes. The remaining ORFs in the 3′ terminal region code for four major structural proteins: the 150–220-kDa glycosylated spike (S) protein, the 20–30-kDa membrane (M) protein, the 7-kDa envelope (E) protein, and the 58-kDa nucleocapsid (N) protein [8, 28].
The S protein of PEDV is a type I membrane glycoprotein composed of 1,383 to 1,386 amino acids (aa), depending on the strain. It contains a putative signal peptide (aa 1–24), a large extracellular region, a single transmembrane domain (aa 1,334–1,356), and a short cytoplasmic tail. Although PEDV has an uncleaved S protein because it lacks a furin cleavage site, the S protein can be divided into S1 (aa 1–735) and S2 (736–the last aa) domains based on homology with S proteins of other coronaviruses [7, 11, 16, 31]. Like other coronavirus S proteins, the PEDV S protein is known to play a pivotal role, interacting with the cellular receptor to mediate viral entry and inducing neutralizing antibodies in the natural host [2, 3]. More precisely, previous studies have shown that the S1 domain includes the main neutralizing epitopes and the receptor-binding region [17, 32]. Furthermore, along with the full-length S gene, the S1 portion is known to be a suitable region for determining genetic relatedness among the different PEDV isolates and for developing differential diagnostic assays [5, 16]. Considering these molecular and biological features of the S1 domain, it would be an appropriate target for developing effective vaccines against PEDV.
In the present study, therefore, we first synthesized a full-length, codon-optimized PEDV S1 gene and then generated a stable porcine-origin cell line constitutively expressing the recombinant S1 protein. The protective efficiency of a recombinant-S1-protein-based vaccine against PEDV was then evaluated in the natural host. The recombinant S1 protein was capable of inducing an efficient antibody response in immunized rabbits. Moreover, immunization with the PEDV S1 protein was found to elicit specific neutralizing antibodies in sows and to protect passively immunized suckling piglets against PEDV challenge.
Materials and methods
Cells, virus, antibodies, and plasmid
HEK-293T cells (CRL-1573) were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco’s modified Eagle medium (DMEM) with high glucose (Invitrogen, Carlsbad, CA) with 10 % fetal bovine serum (FBS, Invitrogen) and antibiotic-antimycotic solutions (100×; Invitrogen). PK-15 cells were grown in RPMI 1640 medium (Invitrogen) supplemented with 10 % FBS and antibiotic-antimycotic solutions. Vero cells were cultured in alpha minimum essential medium (α-MEM, Invitrogen) with 10 % FBS and antibiotic-antimycotic solutions. The cells were maintained at 37 °C in a humidified 5 % CO2 atmosphere. The PEDV vaccine strain SM98-1 was obtained from the Korean Animal and Plant Quarantine Agency and propagated in Vero cells as described previously [10]. Challenge PEDV was prepared from the small intestine of a 4-day-old suckling piglet orally inoculated with small intestine homogenate containing the field virus. Small intestine tissues were collected and homogenized in a 10 % suspension with α-MEM using a MagNa Lyser (Roche Diagnostics, Mannheim, Germany) with three repetitions of 15 s at a speed of 7,000 rpm, and suspensions were clarified by centrifugation at 4,500 × g (Hanil Centrifuge FLETA5, Incheon, Korea). The clarified supernatant was filtered through a 0.22-μm-pore-size syringe filter (Millipore, Billerica, MA), aliquoted, and stored at −80 °C until use as crude challenge virus. All of the horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The PEDV S-protein-specific monoclonal antibody (MAb) was a kind gift from Sang-Geon Yeo (Kyungpook National University, Daegu, Korea). A plasmid encoding the S1 fragment of severe acute respiratory syndrome coronavirus (SARS-CoV), pCDM8-SARS-CoV-S1-Ig, was kindly provided by Hyeryun Choe (Harvard Medical School, Boston, MA).
Construction of expression plasmids
DNA manipulation and cloning were performed according to standard procedures [29]. The E. coli strain DH5α (RBC Bioscience, Taiwan) was used as the host for general cloning. The plasmid encoding the full-length S1 gene of the PEDV field strain KNU-0801, pCDM8-PEDV-S1-Ig, was described previously [17]. To construct the plasmids expressing S1 and its variants, the consensus sequence of PEDV S1 was identified based on a multiple alignment of the S aa sequences of PEDV field isolates [16] and utilized to synthesize a full-length, codon-optimized PEDV S1 gene (encoding aa 24–735) according to a method described previously [1]. The codon-optimized PEDV S1 gene was cloned into a modified expression vector, pCDM8-Fc, which contains the CD5 signal sequence and the Fc domain of human IgG1 [9], thereby producing a human Fc-tagged fusion protein, rS1-Ig. All of the PEDV S1-truncated variants used in this study were generated using this template with the previously described primer sets [17]. An Fc-tagged PEDV rS1-Ig fragment obtained from pCDM8-Fc-rS1-Ig was then subcloned into a pFB-Neo retroviral vector (Stratagene, La Jolla, CA) using the SalI and XhoI restriction sites to construct a PEDV rS1-Ig gene expression plasmid, pFB-Neo-PEDV-rS1-Ig. All of the constructed plasmids were verified by nucleotide sequencing.
Generation of a stable PK-15 cell line expressing PEDV rS1-Ig
The retrovirus gene transfer system (Stratagene) was applied to generate a cell line constitutively expressing the recombinant PEDV rS1-Ig gene or an empty retroviral vector as described elsewhere [16, 22]. Antibiotic-resistant continuous cell clones were examined by RT-PCR to verify the presence of the full-length rS1-Ig gene, and the positive clones (PK-rS1-Ig) were then amplified for subsequent analyses.
Immunofluorescence assay (IFA)
PK-rS1-Ig cells were grown on microscope coverslips placed in 6-well tissue culture plates. At 48 h post-seeding, the cells were fixed with 4 % paraformaldehyde for 10 min at room temperature (RT) and permeabilized with 0.2 % Triton X-100 in PBS at RT for 10 min. The cells were subsequently blocked with 1 % bovine serum albumin (BSA) in PBS for 30 min at RT and then incubated with a goat anti-human IgG antibody conjugated with fluorescein isothiocyanate (FITC) (Santa Cruz Biotechnology). Finally, the cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma, St. Louis, MO), and cell staining was visualized using a fluorescent Leica DM IL LED microscope (Leica, Wetzlar, Germany).
Fluorescence-activated cell sorting (FACS) analysis
rS1 expression in PK-rS1-Ig cells was analyzed by flow cytometry. Briefly, cells were trypsinized at 48 h post-seeding and centrifuged at 250 × g (Hanil Centrifuge FLETA 5) for 5 min. The cell pellet was washed with cold washing buffer (1 % BSA and 0.1 % sodium azide in PBS) and 1 × 106 cells were resuspended in 1 % formaldehyde solution in cold wash buffer and fixed at 4 °C in the dark for 30 min, followed by incubation in 0.2 % Triton X-100 in PBS for permeabilization at 37 °C for 15 min. Following centrifugation, the cell pellet was resuspended in normal mouse IgG1 antibody (Santa Cruz Biotechnology) and incubated at 4 °C for 30 min. The cells were washed and reacted with FITC-conjugated anti-human or anti-mouse IgG secondary antibody at 4 °C for 30 min in the dark. The stained cells were washed again and analyzed by FACScan flow cytometry.
Western blot analysis
HEK 293T cells were transfected with each plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol and solubilized in lysis buffer at 48 h post-transfection as described previously [22]. Cell lysates were also prepared from Vero cells infected with PEDV SM98-1 at a multiplicity of infection (MOI) of 0.1 at the indicated time points using lysis buffer. The protein concentrations of the cell lysates were determined using a BCA protein assay (Pierce, Rockford, IL). PK-rS1-Ig cells were grown at 5 × 105 cells/well in a 6-well tissue culture plate, and the protein-containing culture supernatants were harvested on days 1, 2, and 3. Soluble proteins were immunoprecipitated with protein A Sepharose CL-4B beads (GE Healthcare, Piscataway, NJ) in the presence of protease inhibitors at 4 °C for 16 h. The beads were collected by centrifugation at 5,000 × g (Eppendorf centrifuge 5415R, Hamburg, Germany) for 5 min at 4 °C and washed three times with 0.5 M NaCl in PBS. The cell lysates or the beads were mixed with 4× NuPAGE sample buffer (Invitrogen) and boiled at 70 °C for 10 min. The proteins were separated on a NuPAGE 4–12 % gradient Bis-Tris gel (Invitrogen) under reducing conditions, and electrotransferred onto Immunobilon-P (Millipore). The membranes were then blocked with 3 % powdered skim milk (BD Biosciences, Belford, MA) in TBS (10 mM Tris-HCl [pH 8.0], 150 mM NaCl) with 0.05 % Tween-20 (TBST) at 4 °C for 2 h and reacted directly with the goat anti-human IgG HRP-conjugated secondary antibody, the anti-S1 rabbit serum or the anti-PEDV S MAb, followed by the corresponding HRP-labeled secondary antibody at a dilution of 1:5,000 for 2 h at 4 °C. Finally, the proteins were visualized using enhanced chemiluminescence (ECL) reagents (GE Healthcare) according to the manufacturer’s instructions.
Protein purification
PK-rS1-Ig cells were grown at 5 × 105 cells/well in a 6-well tissue culture plate in serum-free medium (OptiPRO SFM; Invitrogen). At 72 h post-seeding, the protein-containing culture supernatants were harvested and soluble proteins were immunoprecipitated with protein A Sepharose CL-4B beads in the presence of protease inhibitors at 4 °C for 16 h. The beads were collected and washed as described above. The samples were subsequently eluted with 50 mM sodium citrate/50 mM glycine (pH 2.0) and neutralized with 1 M Tris-HCl (pH 8.0). The purified proteins were concentrated with Amicon Ultra centrifugal filters 100K (Millipore). Protein concentration was measured using a BCA protein assay (Pierce, Rockford, IL) and the final products were analyzed by western blotting to confirm target protein purification.
Immunization of rabbits
Two New Zealand white rabbits were immunized intradermally with 250 μg of purified rS1-Ig resuspended in PBS in the presence of Freund’s complete adjuvant and boosted four times with a freshly prepared emulsion of 250 μg immunogen and Freund’s incomplete adjuvant at 2-week intervals. Pre-immune sera were collected before starting the immunization, and antisera were collected at each boost.
Pig inoculation studies
The in vivo swine experiments described here were performed at the Choongang Vaccine Laboratory Animal Facility under the guidelines established by its Institutional Animal Care and Use Committee. A total of eight pregnant sows were obtained from a pig farm with no outbreaks or vaccination with PEDV and randomly divided into four groups of two sows. All animals were determined to be free of antibodies to PEDV. The design for the present immunogenicity study involving eight pregnant sows is outlined in Table 1. The sows in group 1 were immunized intramuscularly with attenuated PEDV live vaccine and inactivated PEDV vaccine obtained from Choongang Vaccine Laboratory, in order at 2-week intervals prior to farrowing. The pigs in group 2 were immunized intramuscularly with PEDV live vaccine and 400 μg of purified rS1-Ig resuspended in PBS in the presence of an oil-in-water adjuvant, in order at 2-week intervals before farrowing. Both the PEDV live and inactivated vaccines were administrated according to the manufacturers’ manuals. The sows in group 3 and group 4 were inoculated intramuscularly three times at 2-week intervals with 400 μg of purified rS1-Ig mixed with the oil adjuvant or with cell culture medium as a negative control. Paired blood samples and colostrum were collected at 3-week intervals before farrowing and at farrowing, at delivery, respectively. One 4- to 5-day-old piglet (8 piglets in total) was selected randomly from each farrowing sow in the immunized and control groups for challenge exposure with virulent PEDV. Piglets from all groups were challenged orally with 1 ml of small intestine homogenate containing 105 TCID50/ml of PEDV field virus, equivalent to 109.8 viral genome copies per ml, determined using real-time RT-PCR as described previously [12]. Clinical signs of diarrhea and death in challenged piglets were monitored daily throughout the study. Stool samples from all groups were collected every day with 16-inch cotton-tipped swabs and subjected to RT-PCR using a TGE/PED Detection Kit (iNtRON Biotechnology, Seongnam, Korea) according to the manufacturer’s protocol. All piglets from the vaccinated and control groups were euthanized at 5 days after challenge for post-mortem examination. Small-intestinal tissue specimens collected from each piglet (<3 mm thick) were fixed with 10 % formalin for 24 h at RT and embedded in paraffin according to standard laboratory procedures. The formalin-fixed paraffin-embedded tissues were cut into 5- to 8-μm-thick sections on a microtome, floated on a 40 °C water bath containing distilled water, and transferred onto glass slides. The tissues were then deparaffinized in xylene for 5 min and washed in decreasing concentrations of ethanol (100, 95, 85, 70, and 50 %) for 3 min each. The deparaffinized intestinal tissues sections were subjected to immunofluorescence assay using an N-specific MAb and Alexa Fluor 594–conjugated goat anti-mouse antibody as described above.
Serum neutralization
The presence of PEDV-specific neutralizing antibodies in serum and colostrum samples collected from sows in all groups was determined using a serum neutralization (SN) test in 96-well microtiter plates using PEDV vaccine strain SM98-1 as described previously [15]. Briefly, individual virus stocks were diluted in serum-free α-MEM to make 200 PFU in a 50 μl volume. The diluted virus was then mixed with 50 μl of twofold dilutions of individual inactivated sera in 96-well plates and incubated at 37 °C for 1 h. Next, the mixture was incubated at 37 °C for 1 h, approximately 1 × 104 Vero cells in 100 μl of α-MEM were added to each well, and the mixture was maintained at 37 °C in a 5 % CO2 incubator for 3 to 4 days. The neutralization titer was calculated as the reciprocal of the highest dilution of serum that inhibited the virus-specific cytopathic effect in both of the duplicate wells. Results were also visualized by staining the wells with a crystal violet-formaldehyde staining reagent (0.013 % crystal violet, 2.5 % ethanol, and 10 % formaldehyde in 0.01 M PBS) for 1 h at RT.
Statistical analysis
The Student’s t test was used for all statistical analyses, and p-values of less than 0.05 were considered statistically significant.
Results
Generation of stable porcine-origin cell lines expressing the full-length, codon-optimized S1 protein
We have previously constructed a series of expression plasmids encoding the full-length S1-Ig (S125–738). Although expression of this gene could be detected in transiently transfected cells by western blot analysis, its expression level was relatively low and insufficient for further protein purification study (Fig. 1, lane 2). Thus, to enhance S1 protein expression, we first synthesized the full-length, codon-optimized S1 gene and constructed a panel of expression plasmids encoding the codon-optimized rS1-Ig (S125–738) and two C-terminally truncated S1 variants: rS125–543-Ig and rS125–432-Ig. These constructs were independently transfected into HEK-293T cells, and expression of each construct was robustly detectable by western blot analysis using an anti-human IgG secondary antibody (Fig. 1, lanes 3 to 5), compared with a normal S1 protein (lane 2). This result indicates that codon optimization greatly increases the expression level of S1 upon transient transfection. Subsequently, to easily produce preparative amounts of the S1 protein, sublines of PK-15 cells were established to stably express the recombinant codon-optimized S1 under the control of a retroviral LTR promoter. Ten generated cell clones were initially collected and subjected to RT-PCR and western blotting to identify S1 gene expression at the mRNA and protein level, respectively (data not shown). Based on the results of western blot analysis, one PK-rS1-Ig cell clone that consistently expressed the highest level of S1 was chosen for subsequent studies.

Expression of the codon-optimized S1 protein. HEK 293T cells were transfected with plasmid DNA encoding codon-optimized SARS-CoV rS1 (lane 1), PEDV S1 (lane 2), codon-optimized PEDV rS1 (lane 3), truncated rS125–543 (lane 4), or truncated rS125–432 (lane 5). Whole-cell lysates were prepared at 48 h post-transfection, resolved in a 4–12 % gradient Bis-Tris gel, transferred onto a nitrocellulose membrane, and immunoblotted with an antibody specific for human IgG
To characterize PK-rS1-Ig cells, intracellular and extracellular expression levels of S1 were examined by immunofluorescence, FACS analysis, and western blotting. As shown in Fig. 2A, specific cell staining was clearly evident when PK-rS1-Ig cells were reacted with the anti-human IgG antibody, confirming a consistent high expression level of the S1 protein. The majority of the cells consistently exhibited specific fluorescent signals, indicating a homogenous population of cells expressing S1 (Fig. 2B). Time-course western blot analysis using culture supernatants revealed that the PK-rS1-Ig cells stably express and cumulatively secrete high levels of approximately 160-kDa S1 (Fig. 2C). In addition, the overall growth kinetics of S1-gene-expressing PK-15 cells were found to be similar to those of the parental PK-15 cells, indicating that S1 expression has no effect on cell growth (data not shown). Further, the recombinant S1 protein tagged with the human IgG Fc domain expressed in the supernatants of stable PK-rS1-Ig cells was purified using protein A Sepharose beads. The purified S1 protein was detectable at a high level by Ponceau S staining, and this was confirmed by immunoblotting with anti-human IgG antibody (Fig. 2D). Using our purification and concentration procedures, we were able to obtain approximately 15 μg of the S1 protein from 1 ml culture supernatant of PK-rS1-Ig cells cultivated in one well of a 6-well plate for 72 h.

Constitutive expression of the recombinant S1 protein in PK-rS1-Ig cells. (A) Immunofluorescence assay of the rS1 protein. PK-15 or PK-rS1-Ig cells grown in a 6-well tissue culture plate were fixed with 4 % formaldehyde at 48 h post-seeding and incubated with anti-human IgG antibody (top panels). The cells were then counterstained with DAPI (bottom panels) and examined using a fluorescent microscope at 400× magnification. (B) Intracellular expression of rS1. One million cells were harvested at 48 h post-seeding and incubated with anti-human IgG antibody (red histogram) or an isotype control (white histogram) and analyzed by flow cytometry. (C) Extracellular expression of the rS1 protein. PK-rS1-Ig cells were grown in a 6-well tissue culture plate at 5 × 105 cells/well for 24, 36, and 48 h. Culture supernatants were harvested at the indicated time points and immunoprecipitated with protein A Sepharose beads. The beads were subjected to western blot analysis with anti-human IgG antibody to determine the expression level of the soluble rS1 protein. (D) Purification of the rS1 protein. The recombinant S1 protein was purified from serum-free medium of PK-rS1-Ig grown in a 6-well tissue culture plate. The purified rS1 protein was resolved in a 4–12 % gradient Bis-Tris gel and electrotransferred onto nitrocellulose paper. The membrane was stained with Ponceau S solution (left panel), destained, and blotted with antibody specific for human IgG (right panel) (color figure online)
Antibody response of the recombinant S1 immunogen in rabbits
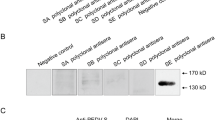
Rabbit antisera were collected before immunization (pre-immune) and at each boost at 2-week intervals. The serum samples at 1:10,000 to 1:100,000 dilutions were examined for binding to the recombinant fusion protein rS1-Ig or the authentic S protein by western blot analysis. The antiserum collected at the second boost reacted with the purified rS1 protein in a concentration-dependent manner (Fig. 3A, top panel), similar to the binding capacity of the anti-IgG secondary antibody, which can directly recognize the protein fused to the Fc region of human IgG1 (middle panel). The antisera were found to retain nearly equivalent reactivity at the third and fourth boosts (data not shown). In contrast, the rabbit antiserum bound weakly to the authentic S protein prepared from cells infected with a PEDV vaccine strain (top panel), whereas an MAb raised against the vaccine strain reacted strongly with the authentic S protein (bottom panel). The rabbit antisera were further tested for their neutralizing activity against the PEDV SM98-1 vaccine strain, which can be grown in Vero cells in the absence of trypsin. As shown in Fig. 3B, the antisera collected at the final boost at 1:16 dilution fully protect Vero cells from SM98-1 infection (left panel), whereas pig hyperimmune sera raised against the vaccine virus were highly effective in inhibiting virus infection with neutralizing antibody titers of >1:256 (right panel). The rabbit sera at the second and third boosts possessed comparable neutralizing activities against the vaccine strain (data not shown). Taken together, our data indicated that the recombinant S1 immunogen elicits potent antibody responses in immunized rabbits. However, the rabbit antisera strongly recognized the homologous S1 protein representing the S protein of field isolates but recognized the heterologous S protein of the PEDV vaccine strain inefficiently, suggesting that there are antigenic differences between the vaccine strain and field PEDVs.

Antibody responses to the rS1 protein in rabbits. (A) Binding of rabbit antisera directed against rS1. Cell lysates prepared from Vero cells infected with SM98-1 and the purified rS1 proteins were resolved in a 4–12 % gradient Bis-Tris gel using the indicated concentrations and electrotransferred onto a nitrocellulose membrane. The blot was reacted with rabbit serum collected at the second boost (top panel), anti-human IgG antibody (middle panel), or mouse MAb specific for PEDV S (bottom panel). (B) Neutralization of PEDV by rabbit antisera raised against rS1. Rabbit sera collected before starting immunization and at each boost and pig hyperimmune serum directed against PEDV SM98-1 were subjected to the virus neutralization assay against SM98-1 and visualized using crystal violet staining. Only one neutralization assay result for rabbit antiserum collected at necropsy is shown in the left panel. The names of the respective sera are labeled at the top, and antibody titers are indicated on the left
Experimental infection of pigs with the recombinant S1 immunogen
To determine the immunogenicity of the S1-based subunit vaccine, pregnant sows assigned to four groups were immunized intramuscularly, as outlined in Table 1. Serum samples were collected at 6 weeks and 3 weeks prior to farrowing and at delivery and were subjected to a serum neutralization test against the PEDV vaccine strain. Non-immunized sows showed only minimal neutralizing antibody titers, whereas immunized sows exhibited gradually increased neutralizing antibody titers, which increased considerably after the final vaccination (Fig. 4). Briefly, sows immunized with two doses of PEDV live and inactivated vaccines at 2-week intervals (group 1) had neutralizing titers of >1:256 at delivery. In contrast, sows immunized with two doses of PEDV live and S1 protein vaccines (group 2) or three doses of S1 protein vaccine (group 3) at 2-week intervals produced relatively lower neutralizing antibody titers of 1:16 to 1:32 compared to those in group 1. In addition, colostrum samples from each group were found to have neutralizing antibody titers comparable to those in the corresponding serum samples (Fig. 4B). These results were coincident with the rabbit study described above, since the rabbit antisera generated by immunization with the recombinant S1 protein also contained low levels of neutralizing antibodies to the heterologous PEDV vaccine strain.

Virus-neutralizing antibody kinetics in sera from sows. Blood (A) and colostrum (B) samples were collected at the indicated number of weeks prior to farrowing or at farrowing, and at delivery, respectively, and subjected to the virus neutralization assay. Neutralizing antibody titers for groups were plotted as a log2 scale. Values are representative of the mean from three independent experiments carried out in duplicate, and error bars denote standard deviations
Lastly, to assess the efficacy of immunization with the S1 protein, eight 4- to 5-day-old neonatal piglets from each sow were arbitrarily selected and challenged orally with wild-type PEDV. Clinical observations of death, diarrhea, and virus shedding in challenged piglets are summarized in Table 2. In the control group, one piglet died and the other piglet experienced severe watery diarrhea post-challenge. Although none of the piglets from any of the groups of immunized sows died during the challenge experiment, the number of piglets exhibiting diarrhea after challenge varied depending on the group. All piglets delivered from group 1 sows showed mild-to-severe diarrhea lasting for the entire experiment at 2 days post-challenge. In contrast, piglets from sows in groups 2 and 3 experienced only mild diarrhea for 1 or 2 days after challenge or throughout the challenge experiment. Likewise, all piglets from immunized sows (groups 1, 2, and 3) exhibited mild intestinal lesions, and viral antigens were detected only in their small intestines, whereas the majority of enterocytes over the entire villi in the control piglets (group 4) were affected by PEDV, showing moderate-to-severe villous atrophy (Fig. 5). Altogether, all immunization methods used in this study were capable of protecting passively immunized neonatal piglets against mortality and severe disease after challenge. However, immunizations involving at least one dose of PEDV S1 protein vaccine (groups 2 and 3) were more efficacious than immunization of sows with two doses of PEDV live and killed vaccines (group 1) in reducing the overall degree of diarrhea, in terms of the duration and severity, in the suckling piglets.

PEDV detection in small intestine tissues of piglets. Tissue specimens were collected from small intestine of piglets from each group at the time of necropsy. The formalin-fixed and paraffin-embedded tissue sections were deparaffinized and stained with an anti-PEDV-N antibody. The sections were then counterstained with DAPI and examined using a fluorescent microscope at 200× magnification
Discussion
PEDV continues to have a severe economic impact in swine-raising countries in Asia and, more recently, in the United States. Vaccination against PEDV is an important and effective prevention measure. PEDV entry into host cells is mediated by the S glycoprotein on the viral surface, which interacts with the cellular receptor and induces direct membrane fusion. The PEDV S protein is also responsible for inducing neutralizing antibodies in the natural host and hence is a logical target in the development of effective vaccines. Furthermore, the S1 domain is a key functional portion of the S protein, which is associated with viral binding to host-cell receptors and contains neutralizing epitopes [17, 32]. Therefore, the S1 protein of PEDV is considered to be a potential candidate antigen for vaccination attempts. In the present study, the first aim was to stably express the full-length, codon-optimized S1 gene of PEDV in porcine-origin cells and to evaluate the immunogenicity and efficacy of the recombinant S1 protein. Our codon-optimization approach dramatically enhanced the expression level of the gene of interest upon transient transfection using a mammalian expression system. Subsequently, we were able to successfully generate a stable PK cell line continuously producing large amounts of the codon-optimized S1 protein. Following the purification and concentration processes, approximately 15–20 μg of the recombinant S1 protein could be consistently harvested from PK-rS1-Ig cells in each well of a 6-well plate.
Since the antibody response is a critical indicator to assess the effect of a vaccine, we immunized rabbits with the S1-based immunogen prepared from culture supernatants of PK-rS1-Ig cells and determined whether or not they developed humoral immunity. PEDV-specific antibodies were strongly detectable in rabbit sera collected from the second boost, even at the highest dilution (1:100,000), when reacted with the recombinant PEDV-S1 protein purified from S1-expressing PK-rS1-Ig cells. In contrast, the binding capacity of the antiserum was dramatically reduced when the authentic S protein in whole-cell lysates prepared from cells infected with SM98-1 was used as the antigen for western blotting. Moreover, the rabbit antisera could completely block infection of SM98-1 only at a serum dilution of 1:16, whereas the pig sera raised against SM98-1 contained high levels of neutralizing antibody against the homogeneous virus (>1:256). Since the PEDV field isolate propagated in the current culture system is unavailable to us, we were unable to test the neutralizing capacity of those antisera against the field virus in this study. However, it is conceivable that rabbit antisera directed against the S1 protein may be more effective in neutralizing infection with the field virus than with SM98-1. On the other hand, the weak interaction and neutralizing activity of the anti-S1 rabbit serum against the SM98-1 S protein may be attributed to genetic variations between the S proteins of the vaccine strain and field isolates. Indeed, the Korean field isolates, including the challenge strain used in this study, were found to display a high degree (>10 %) of genetic heterogeneity, especially in the S1 domain, compared with the PEDV vaccine strain SM98-1 [16, 18]. Nevertheless, western blot and virus neutralizing assays showed that the recombinant S1 protein efficiently elicits humoral immune responses against PEDV.
In Korea, several management strategies, including vaccination, have been employed to control PEDV in pig farms. The most highly recommended immunization schedule involves two doses of attenuated live and inactivated killed vaccines in gilts at 2- to 3-week intervals before mating and in pregnant sows at 12 and 14 weeks of gestation. In the present study, we compared the efficacy of this common vaccination protocol and S1 protein-based vaccination. Although all of the immunized sows developed neutralizing antibodies against SM98-1, only the sows immunized with live and killed vaccines (group 1) had high neutralizing antibody titers, ranging from 1:32 to 1:1024. However, consistent with the rabbit immunization study, sows in groups 2 and 3, immunized with at least one dose of the S1-protein-based subunit vaccine, developed weak neutralizing responses to the vaccine virus, with titers ranging from 1:8 to 1:32. Successful protection against PEDV is based on the presence of specific neutralizing antibodies in immune sows that are passively transferred to their piglets through colostrum and milk. Our data showed that, regardless of vaccination group, all neonatal piglets from immunized sows survived after challenge with virulent PEDV, suggesting that the S1 subunit vaccine provides effective lactogenic immunity to prevent mortality comparable to whole-virus-based vaccines. However, the S1-based vaccination strategies, which produced relatively low neutralizing antibody responses, exhibited more-efficient protection, with respective to the duration and severity of diarrhea, than the common live and killed vaccination procedure. It is therefore plausible that the actual levels of neutralizing antibodies raised against the S1 subunit vaccine are underestimated by the vaccine-virus-based neutralization test. Thus, improved diagnostic tools are needed to differentiate vaccinal antibodies from those resulting from natural infection with the field virus. For this purpose, the recombinant S1 protein purified from PK-rS1-Ig cells could be further used as the diagnostic antigen in an enzyme-linked immunosorbent assay (ELISA), and this aspect is currently under investigation. Additionally, it is possible that the full-length S protein may induce stronger immune responses than S1 alone because it contains multiple functional domains and neutralizing epitopes. In fact, Meng et al. [20] have recently reported that the full-length PEDV S gene induces a better immune response than the N-terminal half alone, using the recombinant DNA plasmid in a mouse model. However, production of the full-length S protein may be unachievable in our system because its expression may cause cytotoxicity due to the presence of potential fusion activity in the C-terminal portion of the S protein.
In conclusion, to the best of our knowledge, this is the first evaluation of the immunological and protective effects triggered by recombinant S1 protein in rabbit and pig models. The results presented here indicate that the recombinant S1 protein can elicit a specific antibody response and induce neutralizing antibodies, suggesting its excellent immunogenicity in the natural host. Furthermore, challenge experiments revealed that the S1-protein-based vaccine protected passively immunized suckling piglets against field PEDV. Despite the nationwide use of commercial live and killed PEDV vaccines, swine herds in Korea have continued to experience repeated outbreaks, leading swine practitioners and researchers to question their protective efficacy. Based on the present and previous studies, we hypothesize that antigenic and genetic variation between the vaccine virus and field PEDVs responsible for periodic outbreaks in herds may be the cause of the low efficacy or failure of vaccination. Accordingly, current PEDV vaccines manufactured from cell-adapted viruses should be reassessed to determine their efficacy and improved if necessary. Although further studies with a larger number of animals will be needed to better evaluate the efficacy of the S1 protein vaccine and to optimize immunization procedures, the recombinant S1 protein has potential for use in improving or developing effective and safe vaccines for PED prevention.
References
Babcock GJ, Esshaki DJ, Thomas WD Jr, Ambrosino DM (2004) Amino acids 270 to 510 of the severe acute respiratory syndrome coronavirus spike protein are required for interaction with receptor. J Virol 78:4552–4560
Bosch BJ, Van Der Zee R, De Haan CA, Rottier PJ (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77:8801–8811
Chang SH, Bae JL, Kang TJ, Kim J, Chung GH, Lim CW, Laude H, Yang MS, Jang YS (2002) Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells 14:295–299
Chen JF, Sun DB, Wang CB, Shi HY, Cui XC, Liu SW, Qiu HJ, Feng L (2008) Molecular characterization and phylogenetic analysis of membrane protein genes of porcine epidemic diarrhea virus isolates in China. Virus Genes 36:355–364
Chen Q, Li G, Stasko J, Thomas JT, Stensland WR, Pillatzki AE, Gauger PC, Schwartz KJ, Madson D, Yoon KJ, Stevenson GW, Burrough ER, Harmon KM, Main RG, Zhang J (2014) Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the United States. J Clin Microbiol 52:234–243
Debouck P, Pensaert M (1980) Experimental infection of pigs with a new porcine enteric coronavirus, CV777. Am J Vet Res 41:219–223
Duarte M, Laude H (1994) Sequence of the spike protein of porcine epidemic diarrhea virus. J Gen Virol 75:1195–1200
Duarte M, Tobler K, Bridgen A, Rasschaert D, Ackermann M, Laude H (1994) Sequence analysis of the porcine epidemic diarrhea virus genome between the nucleocapsid and spike protein genes reveals a polymorphic ORF. Virology 198:466–476
Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, Gerard C, Sodroski J, Choe H (1999) Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96:667–676
Hofmann M, Wyler R (1988) Propagation of the virus of porcine epidemic diarrhea in cell culture. J Clin Microbiol 26:2235–2239
Jackwood MW, Hilt DA, Callison SA, Lee CW, Plaza H, Wade E (2001) Spike glycoprotein cleavage recognition site analysis of infectious bronchitis virus. Avian Dis 45:366–372
Kim SH, Kim IJ, Pyo HM, Tark DS, Song JY, Hyun BH (2007) Multiplex real-time RT-PCR for the simultaneous detection and quantification of transmissible gastroenteritis virus and porcine epidemic diarrhea virus. J Virol Methods 146:172–177
Kocherhans R, Bridgen A, Ackermann M, Tobler K (2001) Completion of the porcine epidemic diarrhea coronavirus (PEDV) genome sequence. Virus Genes 23:137–144
Kweon CH, Kwon BJ, Jung TS, Kee YJ, Hur DH, Hwang EK, Rhee JC, An SH (1993) Isolation of porcine epidemic diarrhea virus (PEDV) in Korea. Korean J Vet Res 33:249–254
Lee C, Hodgins D, Calvert JG, Welch SK, Jolie R, Yoo D (2006) Mutations within the nuclear localization signal of the porcine reproductive and respiratory syndrome virus nucleocapsid protein attenuate virus replication. Virology 346:238–250
Lee DK, Park CK, Kim SH, Lee C (2010) Heterogeneity in spike protein genes of porcine epidemic diarrhea viruses isolated in Korea. Virus Res 149:175–182
Lee DK, Cha SY, Lee C (2011) The N-terminal region of the porcine epidemic diarrhea virus spike protein is important for the receptor binding. Korean J Microbiol Biotechnol 39:140–145
Lee S, Lee C (2014) Outbreak-related porcine epidemic diarrhea virus strains similar to US strains, South Korea, 2013. Emerg Infect Dis 20:1223–1226
Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q (2012) New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis 18:1350–1353
Meng F, Ren Y, Suo S, Sun X, Li X, Li P, Yang W, Li G, Li L, Schwegmann-Wessels C, Herrler G, Ren X (2013) Evaluation on the efficacy and immunogenicity of recombinant DNA plasmids expressing spike genes from porcine transmissible gastroenteritis virus and porcine epidemic diarrhea virus. PLoS One 8:e57468
Mole B (2013) Deadly pig virus slips through US borders. Nature 499:388
Nam E, Lee C (2010) Contribution of the porcine aminopeptidase N (CD13) receptor density to porcine epidemic diarrhea virus infection. Vet Microbiol 144:41–50
Oldham J (1972) Letter to the editor. Pig Farming Oct. suppl:72–73
Park NY, Lee SY (1997) Retrospective study of porcine epidemic diarrhea virus (PEDV) in Korea by in situ hybridization. Korean J Vet Res 37:809–816
Pensaert MB, de Bouck P (1978) A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58:243–247
Pijpers A, van Nieuwstadt AP, Terpstra C, Verheijden JH (1993) Porcine epidemic diarrhea virus as a cause of persistent diarrhoea in a herd of breeding and finishing pigs. Vet Rec 132:129–131
Puranaveja S, Poolperm P, Lertwatcharasarakul P, Kesdaengsakonwut S, Boonsoongnern A, Urairong K, Kitikoon P, Choojai P, Kedkovid R, Teankum K, Thanawongnuwech R (2009) Chinese-like strain of porcine epidemic diarrhea virus, Thailand. Emerg Infect Dis 15:1112–1115
Saif LJ, Pensaert MB, Sestack K, Yeo SG, Jung K (2012) Coronaviruses. In: Straw BE, Zimmerman JJ, Karriker LA, Ramirez A, Schwartz KJ, Stevenson GW (eds) Diseases of swine, 10th edn. Wiley-Blackwell, Ames, pp 501–524
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ (2013) Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Invest 25:649–654
Sturman LS, Holmes KV (1984) Proteolytic cleavage of peplomeric glycoprotein E2 of MHV yields two 90K subunits and activates cell fusion. Adv Exp Med Biol 173:25–35
Sun DB, Feng L, Shi HY, Chen JF, Liu SW, Chen HY, Wang YF (2007) Spike protein region (aa 636789) of porcine epidemic diarrhea virus is essential for induction of neutralizing antibodies. Acta Virol 51:149–156
Takahashi K, Okada K, Ohshima K (1983) An outbreak of swine diarrhea of a new-type associated with coronavirus-like particles in Japan. Jpn J Vet Sci 45:829–832
Acknowledgments
We gratefully thank Hyeryun Choe from Harvard Medical School for providing reagents. This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2010-0002318).
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Oh and K.-W. Lee contributed equally to this work and share co-first authors.
Rights and permissions
About this article

Cite this article
Oh, J., Lee, KW., Choi, HW. et al. Immunogenicity and protective efficacy of recombinant S1 domain of the porcine epidemic diarrhea virus spike protein. Arch Virol 159, 2977–2987 (2014). https://doi.org/10.1007/s00705-014-2163-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-2163-7