
Abstract
During the 2009 influenza A pH1N1 pandemics in Brazil, the state that was most affected was Rio Grande do Sul (RS), with over 3,000 confirmed cases, including 298 deaths. While no cases were confirmed in 2010, 103 infections with 14 deaths by pH1N1 were reported in 2011. Genomic analysis of the circulating viruses is fundamental for understanding viral evolution and supporting vaccine development against these pathogens. This study investigated whole genomes of six pH1N1 virus isolates from pandemic and post-pandemic periods in RS, Brazil. Phylogenetic analysis using the concatenated genome segments demonstrated that at least two lineages of the virus co-circulated in RS during the 2009 pandemic period. Moreover, our analysis showed that the post-pandemic pH1N1 virus from 2011 constitutes a distinct clade whose ancestor belongs to clade 7. All six isolates contained amino acid substitutions in their proteins when compared to the archetype strains California/04/2009 and California/07/2009. The 2011 isolates contained more amino acid substitutions, and most of their genes were under purifying selection. Based on the amino acid substitutions in HA epitopes from strains isolated in RS, Brazil, in silico analysis predicted a decrease in vaccine efficacy against post-pandemic strains (median 31.562 %) in relation to pandemic ones (median 39.735 %).
Similar content being viewed by others

Introduction
Influenza A virus is a pathogen that causes thousands of human deaths every year. In April 2009, a new H1N1 influenza A virus strain (pH1N1) emerged in North America and rapidly disseminated around the globe, culminating in the first influenza pandemic of the 21st century [1]. By the end of the pandemic period, the pH1N1 strain was responsible for more than 18,000 deaths worldwide [2]. Comprehensive phylogenetic analysis demonstrated that this virus originated from multiple reassortment events, and although it most likely arose in pigs, it also contained genes from viruses that infect birds and humans [3, 4].
Gradual genetic changes in influenza A viruses culminate in their ability to evade recognition by the immune system and therefore allow their constant circulation among human populations [5]. As demonstrated by Nelson et al. [6], at least seven distinct pH1N1 influenza A virus lineages circulated around the globe in the first months of the pandemic period. Considering its ability to mutate rapidly, the major concern was that the virus would acquire mutations that could lead to greater transmissibility and pathogenicity, and also to drug resistance [7–9]. For monitoring these evolutionary changes, genome sequencing is a valuable and accessible approach. Genomic sequences provide fundamental information about the chronological and geographical distribution of the strains, and these data support appropriate vaccine development.
Rio Grande do Sul (RS) was the state that was most affected during the pandemic period in Brazil, with over 3,000 confirmed pH1N1 cases and almost 300 reported deaths [10, 11]. A vaccination campaign started in March of 2010 as a measure to control influenza virus infections, and no cases of pH1N1 were confirmed in 2010. On the other hand, over 100 cases occurred in 2011, including 14 deaths [10, 11].
Studies concerning the molecular evolution of this strain in Brazil are scarce. This shortcoming is even worse in the current post-pandemic period, since there are no data available concerning the evolution of the established pH1N1 viruses. Consequently, this lack of information can compromise local public-health vaccination policies.
In this study, six pH1N1 influenza A genomes – four pandemic and two post-pandemic isolates from RS, Brazil – were analyzed. Phylogenetic analysis using the concatenated genome segments was performed to determine the lineages of these isolates. Also, amino acid substitutions in the viral proteins were mapped, and the efficacy of the vaccine against all isolates was predicted. Finally, the type of natural selection that the isolates were subjected to was evaluated.
Materials and methods
Biological samples, virus identification, and genome sequencing
Nasopharyngeal aspirate samples were collected from patients with acute respiratory infection in the state of Rio Grande do Sul, Southern Brazil, during 2009 and 2011 (Online Resource, Table S1). In order to identify the virus, all samples were analyzed by real-time PCR at the State Central Laboratory (LACEN-RS) as described by Veiga et al. [12]. All ethical issues were approved by the Research Ethics Committee of Universidade Federal de Ciências da Saúde de Porto Alegre (UFCSPA).
Twelve pH1N1 samples with high viral load, eight from the pandemic and four from the post-pandemic period, were sent to the J. Craig Venter Institute. Some of them had an excess of mucus and did not homogenize properly with the viral transport media. Therefore, after RNA extraction, only six samples were viable for amplification by multisegment RT-PCR [13]. Subsequently, whole-genome sequencing was performed on these samples, and the sequences were deposited in GenBank (accession numbers are listed in Table 1).
Sequence alignment and concatenation
Sequences of influenza A virus strains were retrieved from the Influenza Research Database (http://www.fludb.org/), and their accession numbers are listed in Table 1. The genomic segments and genes were aligned using MUSCLE [14], embedded in MEGA 5 software [15]. Amino acid substitutions were visually inspected using the sequences of archetype strains California/04/2009 and California/07/2009 as references. Aligned segment sequences were concatenated using Sequence Matrix (http://code.google.com/p/sequencematrix/).
Phylogenetic analysis
For phylogenetic analyses, the aligned sequences were first evaluated using the FindModel software (http://www.hiv.lanl.gov/content/sequence/findmodel/findmodel.html) in order to identify the evolutionary model that best fit the sequence dataset. Subsequently, phylogenetic analysis was performed on the Phylogeny.fr platform (www.phylogeny.fr) [16]. Phylogenetic trees were reconstructed using the maximum-likelihood method implemented in the PhyML program (v3.0 aLRT). The GTR (general time reversible) substitution model was selected assuming an estimated proportion of invariant sites and four gamma-distributed rate categories to account for rate heterogeneity across sites. The gamma shape parameter was estimated directly from the data. The reliability of internal branches was assessed using the aLRT test (SH-Like).
Detection of adaptive evolution
A codon-based Z-test for detection of positive and purifying selection within viral sequences using the Nei-Gojobori method was conducted in MEGA 5 software [15]. The numbers of synonymous (dS) and nonsynonymous substitutions (dN) per site of each gene from RS influenza A virus pH1N1 isolates in relation to its counterparts from the archetype strains California\04\2009 and California\07\2009 were assessed. The difference between the dS and dN of each gene was computed, indicating the type of adaptive evolution. dN>dS indicates positive selection, and dS>dN indicates purifying selection. The variance of the difference was computed using the bootstrap method (1000 replicates).
Measure of antigenic distance
In this analysis, all HA sequences from pH1N1 strains isolated in RS, Brazil, from 2009 and 2011 deposited in the Influenza Research Database (http://www.fludb.org/) were utilized. The additional HA sequences included in the analysis are from the following strains (accession numbers are in parentheses): Rio Grande do Sul/4509/2009 (CY052048), Rio Grande do Sul/4772/2009 (CY052049), Rio Grande do Sul/4782/2009 (CY052050), Rio Grande do Sul/5395/2009 (CY052347), Rio Grande do Sul/7019/2009 (CY052348), Rio Grande do Sul/7108/2009 (CY054282), Rio Grande do Sul/277/2011 (CY099996), Rio Grande do Sul/278/2011 (CY099997), Rio Grande do Sul/279/2011 (CY099998), Rio Grande do Sul/359/2011 (CY099999), Rio Grande do Sul/360/2011 (CY100001) and Rio Grande do Sul/361/2011 (CY100002).
The antigenic distances of the five epitopes of hemagglutinin of the samples in relation to those of the vaccine strain California/07/2009 were evaluated as described by Deem and Pan [17]. The p-distance is defined as the proportion of different amino acids for each epitope between two strains, and it was measured using MEGA software. The largest of the p-distance values is defined as pepitope and can be used to estimate vaccine efficacy (E) by the equation E = 0.47 − 2.47 × pepitope [17]. The difference of the predicted vaccine efficacies between the pandemic (2009) and the post-pandemic (2011) periods in RS, Brazil, was evaluated by Mann-Whitney test using PAST software [18]. A P-value less than 0.05 was considered significant.
Results
Identification of amino acid substitutions
Six influenza A virus pH1N1 strains, four from the pandemic period (2009) and two from the post-pandemic period (2011), isolated in RS, Brazil were sequenced. Predicted proteins from the isolates were compared with those from the archetype strains California/07/2009 and California/04/2009, and the amino acid alterations are shown in Tables 2, 3, 4 and 5.
The 2011 isolates showed a higher number of amino acid substitutions than the 2009 isolates in relation to the archetype strains, notably in the proteins HA and NA (Tables 2 and 3 and Online Resource, Figures S1 and S2). The HA protein from the 2011 isolates had five modifications in antigenic sites, while that from the 2009 isolates had two modifications (Table 2). The amino acid substitution HA Q310H, which is associated with high mortality rates, was found in the RS samples AVS03 and AVS04 (2009) (Online Resource, Figure S2). However, the alteration D239N/G, which is associated with severe illness [19], was not observed in the isolates. The modification E391K, which is associated with changes in antigenic properties [20], is present in the 2011 isolates (Table 2 and Online Resource, Figure S2).
In the NA protein, there were two substitutions in antigenic sites in the post-pandemic isolates, and only one in the pandemic samples analyzed (Table 3). None of the NA proteins contained the modifications H275Y and S247N (Online Resource Figure S3), which are associated with oseltamivir resistance [21].
The 2011 isolates had more alterations in RNA polymerase complex proteins when compared to the 2009 isolates (Table 4).
Phylogenetic analysis
Preliminary phylogenetic analysis of each segment confirmed that all isolates were phylogenetically closer to pH1N1 than to other influenza A virus strains (Online Resource, Figure S3). Subsequently, whole-genome phylogeny was performed in order to determine the lineages of the isolates. The segments were aligned with those from representative strains of each of the seven clades determined by Nelson et al. [6]. All segment alignments were concatenated and subjected to phylogenetic analysis using maximum likelihood under the GTR model, which had the best performance in the test of nucleotide substitution models (Online Resource, Table S2).
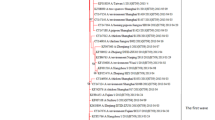
As shown in Figure 1, the Brazilian isolates were distributed between clades 6 and 7, which had high aLRT values (96 and 99, respectively). These isolates exhibited the characteristic amino acid changes that define each clade [6]. Isolates from clade 6 had changes in HA (K2E and Q310H), NP (V100I) and NA (V106I and N248D), and those from clade 7 had substitutions in HA (S220T), NP (V100I), NA (V106I and N248D) and NS1 (I123V) (Tables 2, 3 and 5 and Online Resource, Figures S1 and S2).

Phylogenetic tree of concatenated genomic segments of RS influenza A pH1N1 viruses. The tree was constructed using the maximum-likelihood method. aLRT values greater than 50 % are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The RS H1N1 isolates are in bold
The 2009 RS isolates from clade 6 were phylogenetically the closest to each other. In clade 7, the strain Brazil/AVS/06/2009 was closer to the strain Omsk/02/2009 than to the strain Brazil/AVS/07/2009.
The post-pandemic strains formed a monophyletic clade and shared a most recent common ancestor with clade 7 strains. Also, these strains accumulated more mutations than the pandemic strains, as shown by their longest branches in the phylogenetic tree.
Natural selection analysis
Genes from the pH1N1 isolates were tested for purifying and positive selection. No positive selection was detected in any of the genes of the isolates analyzed (Online Resource, Table S3). Results of the purifying selection test are listed in Table 6. Purifying selection was not found in the M2, NS1 and NEP genes of any of the isolates.
Notably, among the pandemic isolates, the polymerase genes PA, PB1 and PB2 were more frequently affected by purifying selection than other genes. Furthermore, the pattern of purifying selection was not found in any HA genes, while for the NA gene only one isolate (Brazil/AVS03/2009) presented such a pattern of natural selection.
Analysis of the post-pandemic isolates indicated that the HA, NA, M1, NP, PA, PB1 and PB2 genes were under purifying selection.
Vaccine efficacy prediction
The antigenic distances of five epitopes of HA from all isolates in relation to those from the vaccine strain California/07/2009 were computed in order to estimate the efficacy of the vaccine against these isolates using the pepitope method [17], as shown in Table 7. The pepitope of all pandemic strains was 0.029 (dominant epitope E), culminating in a vaccine efficacy estimation of 39.735 %. The dominant epitope varied among the post-pandemic HA1 proteins, and the predicted vaccine efficacy values (median = 31.562 %) were lower than those from the pandemic strains (Mann-Whitney test P-value<0.05) (Table 7). The relative vaccine efficacy medians of the pandemic and post-pandemic strains compared with a perfect-match virus (pepitope = 0) were 84.543 % and 67.154 %, respectively.
Discussion
In this study, genomes of pH1N1 viruses from the pandemic (2009) and post-pandemic periods (2011) isolated in Rio Grande do Sul (RS), Southern Brazil, were investigated.
Whole-genome phylogenetic analysis of the pandemic isolates revealed that clade 6 and clade 7 pH1N1 strains co-circulated in RS in 2009. In a previous study using HA gene sequences, clade 6 strains were isolated in São Paulo (Southeast Brazil), and clade 7 strains in Mato Grosso and Distrito Federal (Center-West region, Brazil), and São Paulo [22]. However, these Brazilian strains were not the most similar to the RS isolates, considering all HA sequences deposited in the Influenza Research Database (http://www.fludb.org/) (data not shown). Therefore, it is likely that in 2009 multiple pH1N1 phylogenetic sublineages could have been introduced in Brazil. The assumption is also supported by the fact that pandemic RS isolates from clade 7 were not the most similar to each other.
On August 2010, the World Health Organization (WHO) declared that the pH1N1 pandemic had ceased and predicted that this virus type would circulate for the years to come, but causing minor problems [2]. In 2010, no cases of pH1N1 infections were detected in RS, Brazil, which was attributed to the intensive vaccination campaign adopted that year, when almost 45 % of the RS population was vaccinated [10]. However, in 2011, 103 cases (14 deaths) of pH1N1 were reported in RS [11]. Phylogenetic analysis of the post-pandemic 2011 RS isolates demonstrated that they belong to a monophyletic group that is nested in clade 7. During the 2009 pandemic, clade 7 strains were the most common in Latin America [22, 23] and in other countries such as the USA [24], India [25] and Canada [26]. The founder effect hypothesis would explain the global dominance of clade 7, since the initial foothold of this lineage occurred in New York State, which could have facilitated its rapid global spread via New York City’s high international interconnectivity [24]. Further studies should be performed to evaluate if most of the currently circulating pH1N1 strains in Brazil were derived from the clade 7 lineage, since the WHO recommendation for influenza vaccine composition is to utilize the clade 1 strain California/07/2009, whose antigenic properties could be distinct from the former ones.
Evolution at high mutation rates is a genetic variation mechanism that ensures that RNA viruses survive [27].
As demonstrated previously, nonepitopic sites tend to accumulate fewer mutations than epitopic ones [28]. This phenomenon is also observed in internal viral proteins, such as matrix (M1), polymerase (PA, PB1, PB2), nucleoprotein (NP), and nonstructural proteins NS, since they are hidden from antibodies and are thus under less selective pressure to change. Furthermore, these proteins have a constrained structure, and mutations that impair their functionality are therefore promptly removed by purifying selection [29]. For instance, polymerase genes have the lowest dN/dS ratio among the virus genes, indicating that intense purifying selection acted on these genes [30].
Among the RS pH1N1 isolates, purifying selection was detected in most genes of the 2011 post-pandemic viruses. However, it was observed that some genes from 2009 pandemic isolates were already under purifying selection, even with the short time of divergence from the common ancestor shared with the archetype strains. Nevertheless, although purifying selection is an important driving force of pH1N1 virus evolution, it is worth noting that RS post-pandemic and pandemic isolates accumulated amino acid mutations relative to the vaccine strain. This observation is in agreement with the findings of Plotkin et al. [29], who suggested that influenza viruses are subject to an intragenomic conflict over the mutation rate: certain genes, and specific residues within those genes, experience frequency-dependent selection to change, while other genes experience purifying selection to remain fixed.
Studies have attempted to correlate the presence of the amino acid alterations in the major viral antigenic determinants HA and NA proteins with prognosis of illness, pathogenicity, virulence, and resistance to antiviral drugs. In this respect, for instance, the substitution Q310H in the HA protein was detected in the isolates AVS03 and AVS04. Although a previous study pointed out the association of this mutation with fatal cases [8], this finding was recently challenged [9, 31].
Amino acid changes in NA associated with oseltamivir resistance were not found in the RS isolates. Assessments of oseltamivir resistance frequency among RS strains during the pandemics and the post-pandemic period (2010-2011) revealed that the H275Y mutation is extremely rare in RS [10, 32]. However, because there are limited options for antiviral treatment, the emergence of resistant strains is a public-health concern [33].
Some amino acid substitutions occurred in antigenic sites of the HA and NA, which could affect influenza vaccine efficacy. As expected, the post-pandemic strains had more amino acid changes in the antigenic sites of HA, culminating in a decrease in the predicted vaccine efficacy against the post-pandemic strains in relation to the pandemic ones. A study performed recently in Japan demonstrated that, in post-pandemic strains containing multiple mutations in antigenic sites of HA (2 to 4 mutations), these mutations did not contribute to a change in antigenicity [34]. However, this probably depends on the site where the mutation occurred and also the type of amino acid substitution. For instance, viruses with the double mutation E391K and D142N have been associated with several breakthrough infections [35]. The D142N alteration was not observed in the 2011 isolates. Nevertheless, other changes in epitopic sites, such as R222K and I233V, which are both lacking in the archetype strains, must be evaluated.
It is worth noting that in 2011, there were 103 cases with 14 deaths due to influenza A virus pH1N1 in RS. In 2012, until October, 525 cases with 67 deaths were confirmed, an increase of 409.71 % and 378.57 %, respectively, in relation to 2011 [11]. Therefore, future studies are necessary to evaluate the efficacy of the current vaccine against the circulating pH1N1 strains in Brazil.
Conclusion
Genome sequences provide fundamental data for the understanding of influenza virus evolution. In this study, we investigated whole-genomic sequences of Brazilian pH1N1 viruses and demonstrated that distinct lineages co-circulated in RS, Brazil. Moreover, we also showed that the pH1N1 isolates contained amino acid substitutions that could alter their biological and antigenic properties. At present, it is evident that the pH1N1 viruses persisted and are constantly evolving. Therefore, monitoring the circulating strains is crucial to ensure proper local prevention and control measures against these pathogens.
References
CDC (2009) Swine influenza A (H1N1) infection in two children-Southern California, March–April 2009. Morb. Mortal Wkl Rep 58
Who (2010) Pandemic (H1N1) 2009—update 112. http://www.who.int/csr/don/2010_08_06/en/index.html
Ding N, Wu N, Xu Q, Chen K, Zhang C (2009) Molecular evolution of novel swine-origin A/H1N1 influenza viruses among and before human. Virus Genes 39:293–300. doi:10.1007/s11262-009-0393-7
Smith GJD, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M et al (2009) Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122–1125. doi:10.1038/nature08182
Medina RA, García-Sastre A (2011) Influenza A viruses: new research developments. Nat Rev Microbiol 9:590–603. doi:10.1038/nrmicro2613
Nelson M, Spiro D, Wentworth D, Beck E, Fan J et al. (2009) The early diversification of influenza A/H1N1pdm. PLoS Curr 1. doi: 10.1371/currents.RRN1126
Deyde VM, Sheu TG, Trujillo AA, Okomo-Adhiambo M, Garten R et al (2010) Detection of molecular markers of drug resistance in 2009 pandemic influenza A (H1N1) viruses by pyrosequencing. Antimicrob Agents Chemother 54:1102–1110. doi:10.1128/AAC.01417-09
Glinsky GV (2010) Genomic analysis of pandemic (H1N1) 2009 reveals association of increasing disease severity with emergence of novel hemagglutinin mutations. Cell cycle 9:958–970
Lee R, Santos C, de Paiva TM, Cui L, Sirota F et al (2010) All that glitters is not gold - founder effects complicate associations of flu mutations to disease severity. Virol J 7:297. doi:10.1186/1743-422X-7-297
Cevs (2011) Boletim Epidemiológico. http://www.saude.rs.gov.br/upload/1343328296_Boletim%20Epidemio%20Influenza.13,%20n.3,set.,%202011.pdf
Cevs (2012) Boletim epidemiológico preliminar da Influenza A (H1N1)2009 no RS até 10/10/12 (semana epidemiológica 41). http://www.saude.rs.gov.br/upload/1349960214_Avaliacao_Influenza_A_H1N1_10_10_12.pdf
Veiga ABG, Kretzmann NA, Correa LT, Goshiyama AM, Baccin T et al (2012) Viral load and epidemiological profile of patients infected by pandemic influenza a (H1N1) 2009 and seasonal influenza a virus in Southern Brazil. J Med Virol 84:371–379. doi:10.1002/jmv.23198
Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M et al (2009) Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol 83:10309–10313. doi:10.1128/JVI.01109-09
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi:10.1093/nar/gkh340
Tamura K, Peterson D, Peterson N, Stecher G, Nei M et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S et al (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi:10.1093/nar/gkn180
Deem MW, Pan K (2009) The epitope regions of H1-subtype influenza A, with application to vaccine efficacy. Protein Eng Des Sel 22:543–546. doi:10.1093/protein/gzp027
Hammer Ø, Harper DAT, Ryan P (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4
Kilander A, Rykkvin R, Dudman SG, Hungnes O (2010) Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome, Norway 2009–2010. Euro Surveill 15
Maurer-Stroh S, Lee RTC, Eisenhaber F, Cui L, Phuah SP et al. (2010) A new common mutation in the hemagglutinin of the 2009 (H1N1) influenza A virus. PLoS Curr 2. doi:10.1371/currents.RRN1162
Hurt AC, Lee RT, Leang SK, Cui L, Deng YM et al. (2011) Increased detection in Australia and Singapore of a novel influenza A(H1N1)2009 variant with reduced oseltamivir and zanamivir sensitivity due to a S247N neuraminidase mutation. Euro Surveill 16
Goñi N, Moratorio G, Ramas V, Coppola L, Chiparelli H et al (2011) Phylogenetic analysis of pandemic 2009 influenza A virus circulating in the South American region: genetic relationships and vaccine strain match. Arch virol 156:87–94. doi:10.1007/s00705-010-0825-7
Barrero PR, Viegas M, Valinotto LE, Mistchenko AS (2011) Genetic and phylogenetic analyses of influenza A H1N1pdm virus in Buenos Aires, Argentina. J Virol 85:1058–1066. doi:10.1128/JVI.00936-10
Nelson MI, Tan Y, Ghedin E, Wentworth DE, St.George K et al (2011) Phylogeography of the spring and fall waves of the H1N1/09 pandemic influenza virus in the United States. J Virol 85:828–834. doi:10.1128/JVI.01762-10
Potdar VA, Chadha MS, Jadhav SM, Mullick J, Cherian SS et al (2010) Genetic characterization of the influenza A pandemic (H1N1) 2009 virus isolates from India. PLoS ONE 5:e9693. doi:10.1371/journal.pone.0009693
Graham M, Liang B, Van Domselaar G, Bastien N, Beaudoin C et al (2011) Nationwide molecular surveillance of pandemic H1N1 influenza A virus genomes: Canada, 2009. PLoS ONE 6:e16087. doi:10.1371/journal.pone.0016087
Domingo E, Holland JJ (1997) RNA virus mutations and fitness for survival. Annu Rev Microbiol 51:151–178. doi:10.1146/annurev.micro.51.1.151
Li W, Shi W, Qiao H, Ho S, Luo A et al (2011) Positive selection on hemagglutinin and neuraminidase genes of H1N1 influenza viruses. Virol J 8:183. doi:10.1186/1743-422X-8-183
Plotkin JB, Dushoff J (2003) Codon bias and frequency-dependent selection on the hemagglutinin epitopes of influenza A virus. Proc Natl Acad Sci USA 100:7152–7157. doi:10.1073/pnas.1132114100
Sinha NK, Roy A, Das B, Das S, Basak S (2009) Evolutionary complexities of swine flu H1N1 gene sequences of 2009. Biochem Biophys Res Commun 390:349–351. doi:10.1016/j.bbrc.2009.09.060
Ferreira JL, Borborema SE, Brígido LF, Oliveira MI, Paiva TM et al (2011) Sequence analysis of the 2009 pandemic influenza A H1N1 virus haemagglutinin gene from 2009-2010 Brazilian clinical samples. Mem Inst Oswaldo Cruz 106:613–616
Souza TM, Mesquita M, Resende P, Machado V, Gregianini TS et al (2011) Antiviral resistance surveillance for influenza A virus in Brazil: investigation on 2009 pandemic influenza A (H1N1) resistance to oseltamivir. Diag Microbiol Infec Dis 71:98–99. doi:10.1016/j.diagmicrobio.2011.05.006
Sheu TG, Fry AM, Garten RJ, Deyde VM, Shwe T et al (2011) Dual resistance to adamantanes and oseltamivir among seasonal influenza A(H1N1) viruses: 2008-2010. J Infect Dis 203:13–17. doi:10.1093/infdis/jiq005
Dapat IC, Dapat C, Baranovich T, Suzuki Y, Kondo H et al (2012) Genetic characterization of human influenza viruses in the pandemic (2009-2010) and post-pandemic (2010-2011) periods in Japan. PLoS ONE 7:e36455. doi:10.1371/journal.pone.0036455
Strengell M, Ikonen N, Ziegler T, Julkunen I (2011) Minor changes in the hemagglutinin of influenza A (H1N1) 2009 virus alter its antigenic properties. PLoS ONE 6:e25848. doi:10.1371/journal.pone.0025848
Acknowledgments
This work was supported by grants from CNPq, CAPES (Auxpe- PNPD- 3047/2010) and JCVI/NIH. This project has been funded in whole or in part by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract number HHSN272200900007C.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sant’Anna, F.H., Borges, L.G.A., Fallavena, P.R.V. et al. Genomic analysis of pandemic and post-pandemic influenza A pH1N1 viruses isolated in Rio Grande do Sul, Brazil. Arch Virol 159, 621–630 (2014). https://doi.org/10.1007/s00705-013-1855-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1855-8