
Abstract
Peripheral populations may be crucial for understanding processes underlying adaptive genetic variation. Their evolution and ecology are driven by various genetic and demographic processes, such as selection, gene flow and bottleneck. Peripheral populations often experience a reduction in density resulting in the Allee effect. The presence of interfertile species increases the opportunity for hybridisation, which allows for a rescue from the Allee effect, but at the risk of genetic extinction through introgression. In this article we investigated a peripheral population of Quercus pubescens, a European tree species. The study population is located in Poland, several hundred kilometres northwards from the main species range. Due to geographic separation, the study population exists under strong pressure of introgression from potentially inter-fertile Q. petraea and Q. robur, which are the only common oaks in Poland. The intermediate morphology between typical Q. pubescens and a common oak species found in the study population supports the introgression hypothesis, which could be in line with the earlier studies of this species complex conducted in the main range of Q. pubescens. Alternatively, the intermediate morphology could reflect the founder effect or selection at an ecological extreme. We attempted to verify these hypotheses using microsatellites and a reference of common oak species. The results showed that the study population is genetically distinct from both Q. petraea and Q. robur. Additionally, the population is characterised by a low effective population size and limited gene dispersal. This suggests that the study population reveals strong reproductive isolation from common species, implying alternative sources of atypical morphology.
Similar content being viewed by others
Introduction
Peripheral populations are important for determination of species limits of natural range (Sexton et al. 2009). Because they often occupy marginal habitats compared with a central part of the species range, peripheral populations may be crucial for understanding processes underlying adaptive genetic variation. The knowledge of causes and scopes of adaptive potential of species may then become crucial in the face of climate change. The evolution and ecology of peripheral populations are driven by various genetic and demographic processes. For example, adaptation to local conditions depends greatly on the level of gene flow from the core population (García-Ramos and Kirkpatrick 1997; Lenormand 2002). Although it is generally acknowledged that low gene flow facilitates rapid divergence at a range margin (Felsenstein 1976; Slatkin 1985; García-Ramos and Kirkpatrick 1997), gene flow can play ambiguous roles in adaptation to local conditions, depending on additional factors. If a peripheral population is isolated from the core population, it may never adapt to local conditions because of genetic drift and low mutational input that causes deficiency of beneficial mutations (Kawecki 2000; Bridle and Vines 2006). This may be especially the case when a peripheral population is characterised by low genetic variation, for example due to the founder effect (Pujol and Pannell 2008). On the other hand, gene flow can lead to migration load when the local optimum phenotypic value differs from the average for the core population (García-Ramos and Kirkpatrick 1997; Bolnick and Nosil 2007). As a result, peripheral populations can be subjected to continuous directional selection, being however unable to reach the ecological optimum.
Additional factors influencing populations at range extremes include inverse density dependence (known as the Allee effect) and hybridisation (Bridle and Vines 2006; Goldberg and Lande 2006; Sexton et al. 2009). The former arises because of low population density, which in plants creates opportunity for increased inbreeding and loss of heterozygosity as well as demographic stochasticity, all leading to decreased fitness (Courchamp et al. 1999). In the presence of closely related cross-fertile species, deficiency of conspecifics often leads to inter-specific mating. The process of hybridisation can be recognised as advantageous or disadvantageous, depending on the perspective. Inter-specific crossing potentially allows a rescue from the Allee effect and rapid extinction at deficiency of conspecific mates, because hybridisation increases heterozygosity, reduces inbreeding and introduces novel alleles into a gene pool (Soltis and Soltis 2009). Nonetheless, escaping from the Allee effect may proceed at the cost of introgression and, at the extreme, genetic extinction due to massive invasion of a related species into the focal species’ genome (Petit et al. 2004; Mallet 2005). The latter seems especially likely when marginal (and isolated) populations exist in the presence of commonly occurring related species (because of gene swamping). It should also be stressed that hybridisation may occasionally lead to outbreeding depression in a similar way as in case of crossing between distant populations of the same species (Schierup and Christiansen 1996). Incidentally, hybridisation can drive a process of speciation (Chase et al. 2010).
Oaks are good model organisms for studying evolutionary processes in peripheral populations for several reasons. Oaks are characterised by anemophily, so mating is not vector-constrained but depends entirely on species’ life history traits, such as pollen production, flowering synchrony, plant size and genetic compatibility (Burd and Allen 1988). However, pollen availability in wind-pollinated species can decrease rapidly at low densities, which are likely in peripheral populations, increasing the opportunity for the Allee effect (Davies et al. 2004). Related oak species can hybridise relatively easily, and hybrids are usually fertile (Whittemore and Schaal 1991; Rushton 1993; Tovar-Sánchez and Oyama 2004; Curtu et al. 2007; Peñaloza-Ramírez et al. 2010). Furthermore, related oak species can differ in ecological optima (Whittemore and Schaal 1991; Timbal and Aussenac 1996; Petit et al. 2004; Sánchez de Dios et al. 2006; Gugerli et al. 2007), so that they often occupy different niches and can reveal variable adaptation to local environments, making room for exogenous selection (Barton and Hewitt 1985).
In this study we investigated genetic and demographic aspects of a peripheral population of Downy oak (Quercus pubescens Willd.), which is the northernmost isolated enclave of this species. The population is located in the lower reaches of the Oder River in Bielinek (Poland), far away from the main species range (Fig. 1), but within a natural range of two other oak species (Q. robur and Q. petraea), which are common in Central Europe. The population of Downy oak was first discovered at the beginning of the 20th century (Schalow 1925, after Celiński and Filipek 1958), and its systematic status has been a matter of discussion for a long time (Celiński and Filipek 1958; Staszkiewicz 1977; Danielewicz et al. 2002). Based on leaf morphology, Bielinek was studied first with a reference of French pure Q. pubescens and hybrid Q. pubescens × petraea populations (Staszkiewicz 1977). The results suggested that Q. pubescens from Bielinek corresponds at best to the hybrid population, being however distinct from both Q. petraea and, even more evidently, from Q. robur (see also Danielewicz et al. 2002). The observed phenotypic distribution in the Bielinek population may be explained by introgression between rare and common oak species. Generally, wind pollination is recognised as a mechanism that allows pollen to be transported over long distances without losing viability (over tens or even hundreds of kilometres) (Lindgren et al. 1995; Bohrerova et al. 2009). In the case of white oaks, recent studies showed that pollen can be easily exchanged between populations separated by 30 km (Schueler and Schlüzen 2006). Parentage-based studies of gene flow accord with this finding. A common observation is that a few tens percent of acorns result from pollination with immigrating pollen (e.g. Streiff et al. 1999; Curtu et al. 2009; Lepais and Gerber 2011; Buschbom et al. 2011), and the immigration rate is relatively stable at the stage of established seedlings (Chybicki and Burczyk 2010; Lepais and Gerber 2011). This suggests that the population of Q. pubescens in Bielinek should experience a permanent oak pollen flow from the outside as well. However, because Bielinek is located at least several hundred kilometres from the potential sources of intra-specific pollen, almost certainly all immigrating pollen comes from two other oak species (Q. robur and Q. petraea). Given that the study stand is a mixture of three oak species, it becomes evident that the population of Q. pubescens in Bielinek is being exposed to inter-specific pollen and subjected to a strong pressure of introgression, which seems to be stronger than within previously studied populations of the species complex (e.g. Finkeldey 2001; Curtu et al. 2007, 2009; Salvini et al. 2009; Lepais et al. 2009; Lepais and Gerber 2011). Although the introgression from Q. petraea could be the case, accompanying or alternative processes such as genetic drift and/or selection at the extremes of species ecological spectrum cannot be definitely excluded. Therefore, it remains unclear to what extent the population of Q. pubescens in Bielinek can be considered as a representative of Q. pubescens.

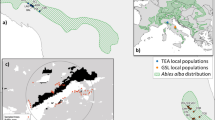
A map of the natural distribution of Quercus pubescens (based on Atlas Florae Europaea) showing large geographical separation of the study population (indicated with the black dot) from both the closest island populations in the Czech Republic and Germany and the main species range
In this article, using nuclear microsatellite markers, we investigated the species status and demographic history of the peripheral population of Downy oak. In particular, we attempted to reveal the importance of the genetic drift, mating system, hybridisation and gene dispersal as mechanisms shaping genetic and morphological variation of this unique population.
Materials and methods
Bielinek reserve is located on the steep slopes (the average grade is 63%) along the postglacial valley of the Oder River (70 m above the river level) about 60 km due south from Szczecin, Poland (14°10′34″ E, 52°55′21″ N, Fig. 1). Due to southern exposition the site is mostly characterised by a relatively dry and warm microclimate, except for shaded and more humid ravines. The soil substrate is composed of glacial clay with a high content of calcium carbonate. Besides Q. pubescens, several other rarities in Polish flora are also noted here, including Dorycium herbaceum Vill., Lithospermum purpurocaeruleum L. and Stipa pulcherrima C. Koch (Celiński and Filipek 1958). The oak population comprises ~2,200 individuals of different ages (the oldest are >250 years old), mostly several decade old natural recruits. Based on exhaustive mapping within the ca. 0.5-ha plot, we estimated the census density of the oak population to be about 1,000 mature individuals/ha (d = 0.1/m2). The earlier surveys of leaf morphology suggested that trees in the study population reveal great variation spanning among three species: Q. pubescens, Q. petraea and Q. robur (Celiński and Filipek 1958; Staszkiewicz 1977; Danielewicz et al. 2002). However, because these species are potentially cross-fertile (Lepais et al. 2009; Salvini et al. 2009; Lepais and Gerber 2011), we assumed that the study population embraces all 2,200 individuals, which however can mate non-randomly due to taxonomic or other barriers (e.g. distance, flowering synchrony). It is worth noting that the Bielinek reserve is a part of a larger forest complex (composed of Scots pine, common oaks and beech). Also, a few-hectare oak stand is located just next to the study population. Thus, oak pollen flow might be considered as a potentially important factor determining the genetic and taxonomic structure of the study population.
The history of the reserve has been well documented since 1957 (when the reserve was established), but the area has been under protection since 1927 (or even earlier). Certainly the reserve has not been used for any commercial purposes for the last 80 years. However, it is thought not to have been influenced much by humans even before this period due to specific conditions (including very steep slopes as well as low humidity). For example, it seems hardly economically justified to use the slopes for wood production, as the slopes are located just next to flat, historically forested areas. Similarly, nothing can be stated with certainty about the population origin. Some authors suggested a human-induced origin (e.g. Schwartz 1937; after Celiński and Filipek 1958), stressing the discontinuity of Q. pubescens distribution in Central Europe. On the other hand, more recently, geobotanists have postulated spontaneous colonisation in the Holocene climate optimum epoch (Czubiński 1950; Celiński and Filipek 1958). They found the specific (though impoverished and influenced by present-day flora) plant association Querceto-Lithospermetum in Bielinek, the presence of which cannot be easily explained unless we assume that it is a relic of the past flora. Although the closest population of Q. pubescens is located 300 km away due southwest on Kunitzberg Mountain near Jena (Germany) (Celiński and Wojterski 1958), numerous populations of the associated herbaceous species are located much closer, suggesting the route of a possible colonisation from Thuringia (Germany) due east and then due north, along the Oder River.
In May 2010, 100 mature individuals were sampled for leaves and mapped using a GPS mapping system (Pathfinder ProXT, Trimble). The samples were taken within a core patch of Q. pubescens (about 1 ha in total), at the highest elevations, at the southwest exposition of the slope. The southeastern edge of the study plot bordered on a ~10-m wide ravine, where the oak population reveals spatial discontinuity (i.e. the census density decreases ca. ten times in the ravine). Two distinguishable elevations were systematically explored, with denivelation reaching up to 15 m. Fresh leaves were used for the extraction of the total genomic DNA following the CTAB protocol (Doyle and Doyle 1990, with minor modifications). As genetic markers we used 14 nuclear microsatellites (Table 1), amplified in three multiplex PCR reactions (according to the procedures described in Chybicki and Burczyk 2010). To strengthen the power of multilocus clustering analysis (see below), we included the loci that appeared to be highly discriminative in the earlier studies on European white oaks (e.g. Curtu et al. 2007; Gugerli et al. 2007; Lepais et al. 2009). PCR products were sized using an ABI 3130XL sequencer (Applied Biosystems, Foster City, CA, USA) and the attached software (Genemapper 4.0).
Genetic structure was described using the observed and effective number of alleles, observed and expected heterozygosity, and the inbreeding coefficient. The latter was used to test against the null hypothesis of no heterozygote deficiency/excess using a permutation procedure. P values were obtained based on 100,000 random permutations of alleles among genotypes, after correction for multiple tests using the Šidak procedure. The computations were conducted with the updated version of INEst software (Chybicki and Burczyk 2009).
To assess social structure in the study population, we used a simulated annealing algorithm for pedigree reconstruction implemented in Colony 2 (Jones and Wang 2009). Briefly, the method seeks the most likely full-sib, half-sib and unrelated pairs among a cohort of genotyped individuals. It has a number of advantages, including accounting for non-random mating and genotyping errors (Wang and Santure 2009). The full-likelihood-based analysis (medium likelihood precision and medium length of run) was performed under the assumption of polygamy and monoecy with a possibility of self-fertilisation. Rates for allelic dropout were substituted with locus-specific estimates of null allele frequencies provided with INEst software (Chybicki and Burczyk 2009). Rates of other error types were arbitrarily fixed at 0.01. Allele frequencies were updated during optimisation. No pair of individuals was a priori excluded as a possible full-sib or half-sib. Also, sampled individuals were assumed to form a single generation; therefore parent-offspring relations were not allowed. The analysis was initiated without a prior distribution for the paternal and maternal sib-ship sizes of the offspring.
Based on the reconstructed pedigree we estimated the spatial kinship structure (using spatial correlogram) and effective population size (N e(SA)) according to the method proposed by Wang (2009). Additionally, we measured effective population size (N e(LD)) based on a linkage disequilibrium method (Waples 2006; Russel and Fewster 2009). The N e(LD) was computed after omitting alleles with frequency lower than 0.02. The 95% confidence interval around N e(LD) was approximated using 1,000 bootstrap samples over pairs of loci. Although some loci belong to the same linkage group (the smallest map distance of 15 cM is between ssrQrZAG11 and ssrQrZAG96; Saintagne et al. 2004), we did not notice any meaningful differences between estimates based on the full data set and after omitting linked pairs. Therefore, we reported the results based on the full data set.
In addition to the analysis based on the real data set, we conducted a limited simulation study to test the performance of Colony software. Briefly, to generate a single data set of 100 individuals genotyped at 14 markers (as in the real data), first we generated parental genotypes assuming Hardy-Weinberg conditions (allele frequencies at 14 unlinked markers equalled the observed allele frequencies in the study population). Then, multilocus genotypes of progeny were drawn randomly assuming Mendelian transmission, given known pedigrees equal to the results for the real data set (see “Results”). Finally, we introduced genotyping errors into progeny genotypes at rates equal to the rates assumed for the real data set (see above). Progeny genotypes were then analysed with Colony 2, using the same settings as for the real data. To evaluate possible behaviour of Colony 2 software we repeated the procedure ten times (due to slow convergence). Thus, the above procedure was equivalent to the parametric bootstrap, except for the limited number of repetitions. Artificial data sets were generated using an ad hoc Windows-based computer program written in Delphi 7 (available from IJC upon request).
Using MSVAR software (Beaumont 1999) we verified whether the study population exhibits signatures of a historical demographic decline (or expansion). Briefly, based on the simple demographic model of a single isolated population, the method allows estimating the set of parameters (r = N 0/N 1, t f = T a/N 0 and θ = 2N 0 μ, where N 0 the current effective population size, N 1 the ancestral effective population size before demographic change, T a the time in generations at which the change occurred and μ mutation rate) using the Markov chain Monte Carlo approach. Given parameter estimates, the inference can be made if a population has been characterised by a negative (r < 1) or positive growth rate (r > 1) within a timeframe of last T a generations. We used the basic method as implemented in version 0.4.2 of the software (available at http://www.rubic.rdg.ac.uk/~mab/stuff/), which assumes common demographic parameters for all loci. Although data exploitation might be suboptimal in this way (Storz and Beaumont 2002; Girod et al. 2011), the basic method has an advantage as it requires priors on the scaled demographic parameters (i.e. r and t f), which in our case can be proposed much more easily than priors for the natural demographic parameters (i.e. N 0, N 1 and T a; as in version 1.3) (see “Discussion” in Girod et al. 2011). The analysis was conducted assuming the exponential growth model, with rectangular priors for log(r), log(t f) and log(θ) bounded on −5 and 5 (as in Storz and Beaumont 2002). The Markov chain was run for 3 × 109 steps (keeping every 30,000 update), disregarding the first 3 × 108 updates to avoid dependence of final estimates on initial settings. Convergence was assessed using the potential scale reduction factor (PSRF; Gelman and Rubin 1992) based on four independent chains run with different initial values.
Under assumption of the isolation-by-distance we estimated levels of gene dispersal using indirect methods implemented in SPAGeDi (Hardy and Vekemans 2002). Because the iterative procedure in SPAGeDi requires a priori known effective population density (d e), we used the estimate d e = d × h(N e(SA), N e(LD))/N c, where d = 0.1 is the actual density of the population and h() denotes a harmonic mean.
Finally, we assessed a level of genetic separation of the study population (i.e. putative Q. pubescens) from two other oak species (Q. robur and Q. petraea), which are naturally widespread in this part of Europe. For this purpose we added a reference of 231 trees having a priori assigned species status of Q. robur (147 trees) or Q. petraea (84 trees). The selected reference groups consisted of certified elite oak trees (plus trees) for which the species status was carefully tested by means of morphology and genetic markers (Burczyk J et al., unpublished data). The genetic clustering analysis was conducted based on a common subset of nine nSSR loci (excluding ssrQrAG65, ssrQrAG87, ssrQrAG90, ssrQrAG101, ssrQrAG102) using Geneland package version 3.3 (Guillot et al. 2008). Because Geneland intrinsically estimates the most likely number of clusters (K), no additional inference procedures were required. The analysis was run under the assumption of the F-model for allele frequencies and the spatial-free model for genetic structure. The estimates were obtained in three replicates after 500,000 iterations, keeping every 100th update. Every time the analysis was initiated at K = 1, with K being allowed to change between 1 and 10. Once assignment probabilities were estimated, spatial arrangement of putative taxons was assessed using spatial autocorrelation (Moran’s I) with assignment probability as a variable. The analysis was performed using PASSaGE 2 (Rosenberg and Anderson 2011).
Results
The number of alleles per locus ranged from 8 to 20 with an average of 13.0. Due to generally uneven distribution of allele frequencies, the effective number of alleles was only a half of the observed number. On average, the study population showed a slight deficiency of heterozygotes as compared with Hardy-Weinberg proportions (mean F IS = 0.052). The permutation procedure revealed that F IS deviated significantly from zero at four loci, with three of them showing deficiency and one showing an excess of heterozygotes (Table 1).
Using Colony we estimated that the most likely parental population consisted of 56 diploid genotypes. The contribution of reconstructed parents was apparently non-uniform: 3 of the most fecund parents contributed 44 (22%) gametes, while 39 parents contributed no more than 3 gametes. The distribution of the total reproductive success (i.e. jointly through female and male function) fitted well to the log-normal distribution as revealed by the Kolmogorov-Smirnov test (p value >0.05). Two (2%) individuals in the study plot were most likely a result of self-fertilisation. They did not share a mother tree, but in both cases the mothers had estimated relatively high overall reproductive success of eight and nine gametes in total. Out of 4,950 pairs of individuals in total, 38 and 485 fell into full-sib (FS) and half-sib (HS) categories, respectively. A simple estimate of average kinship (i.e., ignoring more distant cryptic relationships) in the study population equalled (0.25 × 38 + 0.125 × 485)/4,950 = 0.014. However, kinship was not randomly distributed in the space (Fig. 2). Individuals separated by up to 20 m were more than twice as related as any two randomly chosen individuals. On the other hand, individuals separated by more than 60 m appeared less related than any two random individuals. Mean distance (standard deviation in parentheses) between FS and HS pairs was 28.5 m (22.1) and 39.9 m (32.0). For comparison, unrelated individuals (within the study plot) were separated by 66.0 m (38.8) on average. Regardless of some differences in the mean distance, distributions of a pairwise distance for FS and HS appeared not significantly different as revealed by the Kolmogorov-Smirnov test (p value >0.1).

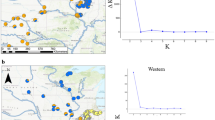
Spatial kinship structure. Frequency of full-sib (FS), half-sib (HS) and unrelated (UR) pairs (lower panel) and corresponding interval-averaged kinship (upper panel) at a given distance interval. Solid and dashed lines in the upper panel show the results of the randomisation procedure (based on 100,000 permutations) and refer to median kinship (solid) and 95% confidence limits (dashed) around a median under a null hypothesis of random kinship distribution
Analyses of simulated data sets revealed that Colony tends to underestimate the number of FS pairs and to overestimate the number of HS pairs, given our marker set. The average number of FS equalled 25.9 (from 24 to 28), while of HS equalled 490.6 (from 448 to 524). In consequence, the results for the real data set might suffer from a systematic bias, influencing both N e(SA) and spatial structure estimates.
The reconstructed pedigree allowed for the estimation of effective population size (N e(SA)). Under the assumption of random mating N e(SA) = 36 with 95% CI between 24 and 59 individuals. Under non-random mating, N e(SA) = 33 with 95% CI between 22 and 55. A supplementary estimate of effective population size based on linkage disequilibrium equalled N e(LD) = 29.8 with 95% CI between 26.7 and 33.3. MSVAR output was first analysed for convergence. The diagnostic measures PSRF for log(r) and log(t f) equalled 1.014 and 1.001, respectively, allowing us to conclude that chains resulting from MCMC simulation reached convergence. Using MSVAR we found strong support for a past population decline, because all the stored updates of log(r) < 0. Estimated average log(r) was −2.323 (95% CI: −2.686, −2.035), while the parallel estimate of log(t f) was −0.287 (95% CI: −0.386, −0.198).
Indirect estimates of gene dispersal were obtained assuming d e = 0.1 × 33/2,200 = 0.0015 (individuals per m2; see “Materials and methods”). However, it is worth mentioning that the iterative procedure yielded almost the same value of the neighbourhood size using the actual census density (d) instead of d e. The analysis based on d e yielded an estimate of the neighbourhood size of 31.0 individuals with 95% CI between 18.3 and 43.6. The corresponding estimate of the root mean squared parent-offspring distance was σ = 40.5 m (95% CI: 32.4–48.6).
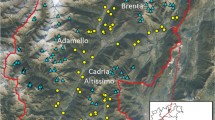
Three replicates of the genetic clustering analysis with Geneland were convergent and all gave strong support for a number of genetic clusters K = 3. We observed that the three independent runs apparently differed in convergence rates. Thus, to obtain estimates unaffected by the initial settings we rejected the first 200,000 updates, leaving only those that corresponded to a stabilised log-likelihood value (Fig. 2b). Based on the truncated series of parameter updates a posterior probability for K = 3 equalled ~1.0 for all three runs. The three genetic clusters corresponded very well to the three putative oak species (Fig. 2a). As expected (based on the earlier unpublished results), the reference samples of Q. robur and Q. petraea showed almost no admixture signatures. Only one individual in the ‘petraea’ reference group showed 0.37 probability of having ancestry in the ‘robur’ group. The average rate of assigning of an individual from the ‘robur’ to the ‘petraea’ reference group was 0.002, whereas the probability of assigning in the opposite direction was 0.007. It is worth noting that neither of the two reference samples showed signatures of the ‘pubescens’ ancestry. The analyses revealed that the study population was clearly distinct from the reference samples. The average assignation probability of an individual from the study population to the ‘pubescens’ genetic cluster was 0.956. However, three individuals were assigned to the ‘petraea’ genetic cluster with a >0.95 probability. Additionally, three individuals clearly had a mixed ancestry either of ‘pubescens’ and ‘petraea’ (two individuals) or ‘pubescens’ and ‘robur’ (one individual). The overall contribution of the ‘robur’ and ‘petraea’ gene pools to the study population equalled 0.2 and 4.3%, respectively. Plotting a distribution map of admixed and pure ‘petraea’ individuals showed that ‘petraea’-like individuals are present mostly at the eastern margin of the study plot (Fig. 3). The spatial autocorrelation test revealed significant autocorrelation of assignment rates between close neighbours (up to 16.8 m) (p value = 0.005), as well as significant non-random overall spatial structure with p value for the entire correlogram <0.003.

Results of genetic clustering analysis using Geneland (Guillot et al. 2008): a each individual is represented by a vertical bar showing probabilities for assignment to ‘pubescens’, ‘robur’ and ‘petraea’ cluster; b behaviour of the likelihood function across iterations in three independent runs with the arbitrarily chosen burnin period (up to 200,000 iteration) shown by the vertical dashed line; c a map of distribution of individuals with assignment results
Discussion
Low estimates of introgression (a total of 4.5% admixture from Q. petraea and Q. robur) suggest that the population of Q. pubescens in the Bielinek reserve reveals apparent genetic isolation from two common oak species (Q. robur and Q. petraea), which are the only representatives of white oaks in the northern part of Central and Eastern Europe. In general, sharp genetic delineation between sympatric species is not very new for the European white oak complex. Several authors have recently found strong genetic separation of species in mixed stands across the main range of coexistence of different oak species (Gugerli et al. 2007; Curtu et al. 2007; Lepais et al. 2009). However, our results provide new evidence for strong reproductive barriers and/or selection mechanisms among European oak species that shape their ecology and evolution at the Northern range margin.
The main conclusions about genetic isolation of the study population could be drawn based solely on the apparent genetic separation of Q. pubescens in Bielinek from two common oak species, as revealed by the Bayesian clustering method. Nonetheless, further evidence comes from a small effective population number and strong kinship structure with the corresponding estimate of restricted historical gene dispersal (40.5 m on average). Two independent analyses showed that the study population has a historical N e of about 33 equally-contributing individuals. Such a small N e is rather unusual for wind-pollinated stand-forming tree species (Bucci et al. 1997; Chybicki et al. 2008; Wachowiak et al. 2011), unless a small population is highly isolated (Bucci et al. 1997; Chybicki et al., in review). Interestingly, the so-called Wright neighbourhood size estimated from the spatial genetic structure was almost of the same magnitude. This suggests that the number of individuals involved in the reproduction is very limited. Because a strong spatial genetic structure was found, small N e can be attributed primarily to restricted gene dispersal. In oaks, restricted dispersal of acorns was recognised as a primary mechanism shaping the spatial autocorrelation of genes (Streiff et al. 1998; Grivet et al. 2009; Nakanishi et al. 2009; Chybicki and Burczyk 2010). Based on the results alone, we obviously cannot exclude that a similar phenomenon occurred in the study population. Nonetheless, in the case of Bielinek seed dispersal can be additionally driven by a very steep slope; thus it seems possible that the average gene dispersal distance of 40 m might result from both restricted seed and pollen dispersal. In European white oaks effective pollen dispersal is usually bimodal, as a result of limited pollen dispersal within a population and a large pollen immigration from outside (e.g. Streiff et al. 1999; Salvini et al. 2009; Chybicki and Burczyk 2010; Buschbom et al. 2011; Lepais and Gerber 2011). However, due to very low effective pollen immigration (as our results suggest), in the study population a distribution of mating distances can be unimodal with a peak at short distances. Small pollination distances could explain the fact that pairs of full siblings were on average (although not significantly) closer than half-sibs in the study plot.
Pollen limitation is often found in peripheral populations as an effect of limited number of pollen donors, (Busch 2005; Michalski and Durka 2007; Mimura and Aitken 2007). It results in reduced female fecundity and increased self-fertilisation (reviewed in Knight et al. 2005). Given a small number of reproducing individuals and low effective pollen immigration, the population of Q. pubescens in Bielinek might be subject to pollen limitation syndrome as well. However, we did not find any strong support for this hypothesis. Based on field observations we noted that acorn production is not evidently suppressed. On the contrary, we observed that relatively young individuals start to produce acorns. Also, our results did not support an increased self-fertilisation rate, as our estimate of the contemporary selfing rate equalled 2%, which is comparable with other findings for European white oaks in the main species range (Streiff et al. 1999; Curtu et al. 2009; Chybicki and Burczyk 2010; Lepais and Gerber 2011). It seems that the study population produced enough pollen to maintain cross-pollination, and/or some pre- or post-zygotic mechanisms (Buschbom et al. 2011) prevent more intense inbreeding.
Given the strong introgression pressure suggested above, the question remains how then the apparent separation of the gene pool of Q. pubescens in Bielinek can be explained. Three mutually non-exclusive possibilities could occur: (1) reproductive barriers, (2) microsite selection and (3) assortative mating (Bruschi et al. 2000; Curtu et al. 2007; Gugerli et al. 2007; Varela et al. 2008; Lepais et al. 2009). Controlled pollination experiments revealed that oaks in the studied species complex are potentially interfertile (Steinhoff 1993; Orlik and Kjaer 2007). This potential has been confirmed in real populations. Nonetheless, different studies demonstrated a wide range of hybridisation rates (Curtu et al. 2009; Salvini et al. 2009; Lepais et al. 2009). For example, relatively high hybridisation rates were detected in an Italian mixed oak stand of Q. petraea and Q. pubescens (Salvini et al. 2009), reaching up to 30% of within-population mating events, whereas 8.4 and 9% hybridisation events were reported in Romanian and French mixed stands, respectively (Curtu et al. 2009; Lepais and Gerber 2011). All the studies detected directional hybridisation with one species more often playing the pollinator and a second the receptor. However, it seems that the direction of hybridisation is a population-specific phenomenon and may depend on such characteristics as the species abundance, stand density and spatial arrangement of species. Interestingly, these studies generally agreed that hybrid individuals prefer to backcross with their parental species. Backcrossing can be a very important process that facilitates reconstruction of the pure Q. pubescens in Bielinek after spontaneous hybridisation. However, because of a large supply of pollen from Q. petraea and Q. robur, preferential backcross of hybrids with Q. pubescens would require additional factors, such as selective superiority of Q. pubescens in the study site or assortative mating (Petit et al. 2004). The three oaks differ substantially in their ecological optima, causing usually spatial species separation within mixed oak populations (e.g. Bacilieri et al. 1996; Curtu et al. 2007; Gugerli et al. 2007). Because the study site is characterised by shallow soils, a steep slope and southern exposition, Q. pubescens may outcompete both common oak species due to relatively high tolerance to drought. Interestingly, our results suggest that there could be a non-random species arrangement in the site, with Q. petraea or hybrid individuals located closer to shaded and more humid ravines. Also, visiting the Bielinek reserve in September 2011 we found that a rate of leaf damage by insects was substantially lower in pubescent-like individuals as compared with petraea- and robur-like individuals. Therefore we cannot exclude that leaf morphology or other genetically determined characters (Simchuk 2008) can play an additional role in driving the local superiority of Q. pubescens over two common oak species and hybrids. If hybrid individuals were less competitive, then the observed genetic separation could arise as an effect of the reinforcement (Howard 1993). An additional possibility is non-overlapping flowering periods between different species, which would lead to strong assortative mating (Gömöry and Schmidtová 2007). Nonetheless, further studies are needed to test these hypotheses.
The origin of Downy oak in Bielinek is unsure and mostly speculative. However, the results of MSVAR obtained in this study provide some suggestions about the demographic history of the population. First, let us recall that the effective population size was assessed as for 33 individuals. This estimate can be used as a proxy of the current effective population size N 0 (e.g. given that the LD measure stabilises within three to four generations). Based on the estimate of log(r) we found that the current population size was only 0.47% of the ancestral population (see “Materials and methods”). Hence, the ancestral population size might equal 33/0.005 ≈ 7,000 individuals. Therefore a large ancestral population could be formed within a more continuous (e.g. central) part of the species distribution. Given log(t f) of −0.287, we could also estimate that the scaled time since bottleneck equalled 0.516 N 0. Again, making a substitution for N 0 = 33, we estimated ca. 17 generations since the start-up of population decline. To obtain the approximate time in years we might use an estimate of an average species generation time according to T = a [s/(1−s)], where a is the time to maturity (in years) and s is the adult annual survival rate (Spellman and Klicka 2006). In the studied species a ≈ 50 and s = 0.99 or even 0.995 (as typically for long-lived tree species; Cavender-Bares et al. 2011), so that T = 149 or even 249 years. In consequence, the population decline might have begun ca. 2,500 or 4,200 years ago. Although these estimates are largely a subject of speculation, interestingly the latter estimate matches roughly the end of the so-called Holocene climatic optimum. In this epoch, due to warming in the northern hemisphere, southern European species could temporarily shift their range due north (Ellenberg 2009). Thus, it cannot be excluded that in this period Q. pubescens moved northward as well (Czubiński 1950), providing the possibility for colonisation of areas located far outside its current distribution. This would support the possibility of a natural origin of the population in Bielinek. Unfortunately, so far there is no evidence in terms of plant fossils to confirm this hypothesis. Nonetheless, because our results suggest a long time since the bottleneck, the origin of the population by humans seems a rather unlikely scenario.
Finally, we would like to address the problem of atypical morphology of Q. pubescens in Bielinek. Two independent studies showed that morphologically Q. pubescens in Bielinek is rather a hybrid between Q. pubescens and Q. petraea than a pure species (Staszkiewicz 1977; Danielewicz et al. 2002). However, given that introgression from Q. petraea has little support from genetic markers (this study), the intermediate morphology needs an alternative explanation. Here, we speculate that the specific morphology can be generally related with the peripheral location and history of the population. First, we should emphasize that these two species are generally known for a wide morphological variation, which can overlap, sometimes making the assignment of individuals to any species difficult (Dupeouy and Badeau 1993; Bruschi et al. 2000; Finkeldey 2001; Curtu et al. 2007). However, in this respect northern populations of Q. pubescens (e.g. in Switzerland, Slovakia) seem to have a greater affinity with Q. petraea compared with populations in the Mediterranean area (Finkeldey 2001; Jedinaková-Schmidtová et al. 2004). Various authors emphasised a dominant role of recurrent hybridisation as a mechanism that could shape the observed morphological continuum increasing northwards. Because density of Q. pubescens decreases northwards, a hybridisation with common species could allow a rescue from the Allee effect. On the other hand, multilocus genetic data have not fully supported extensive present-day hybridisation in the north (Gugerli et al. 2007; this study; but see Gömöry and Schmidtová 2007). Moreover, it was shown that morphologically intermediate individuals are not obligatory hybrids in terms of their genetic composition (Gugerli et al. 2007). This suggests that the observed similarity between species can be a consequence of more historical processes. The first hypothesis argues that the present-day variation resulted from shared ancestry (Muir and Schlotterer 2005). However, recently Lepais and Gerber (2011) have provided support for another explanation. They found Q. pubescens to be a very invasive species in terms of inter-specific pollination rates. Hence, they proposed introgressive hybridisation as a mechanism of the postglacial expansion of Q. pubescens northwards. Their reasoning is in line with the presumption that, as a thermophilous species, Q. pubescens started the expansion after both Q. robur and Q. petraea. It is thus possible, that Q. pubescens expanded due north through genome invasion into already established oak populations, and the side effect of this process is an increasing northwards affinity of Q. pubescens with Q. petraea. Another explanation of the atypical morphology in Bielinek could be the founder effect or selection occurring away from ecological optima of the species (García-Ramos and Kirkpatrick 1997), with the former being in line with the signs of bottleneck, as revealed in the analyses. Nonetheless, further studies would be helpful to make final conclusions.
To sum up, our study showed that a peripheral population can maintain reproductive isolation from potentially inter-fertile closely related species even in the case of spatial isolation from conspecific pollen sources. We hypothesised that possible mechanisms include reproductive barriers, microsite selection and assortative mating, which in turn can be a basis for the reinforcement. Nonetheless, despite no clear arguments from our data, we cannot exclude that some forms of hybridisation still take place in Bielinek reserve. Because the study plot was established at the highest elevations of the slope, it is certainly not well representative of the entire habitat. We can expect that some ecological conditions (like moisture or insolation) can gradually change both horizontally and down the slope (Celiński and Filipek 1958). In this way we might omit a true hybrid zone (i.e. an area where hybrids would outperform both parental species), which can be located in an ecotone between preferable habitats of parental species (Sánchez de Dios et al. 2006). Future studies need to address this aspect as well.
References
Bacilieri R, Ducousso A, Petit RJ, Kremer A (1996) Mating system and asymmetric hybridization in a mixed stand of European oaks. Evolution 50:900–908
Barton NH, Hewitt GM (1985) Analysis of hybrid zones. Ann Rev Ecol Syst 16:113–148
Beaumont MA (1999) Detecting population expansion and decline using microsatellites. Genetics 153:2013–2029
Bohrerova Z, Bohrer G, Cho KD, Bolch MA, Linden KG (2009) Determining the viability response of pine pollen to atmospheric conditions during long-distance dispersal. Ecol Appl 19:656–667
Bolnick DI, Nosil P (2007) Natural selection in populations subject to a migration load. Evolution 61:2229–2243
Bridle JR, Vines TH (2006) Limits to evolution at range margins: when and why does adaptation fail? Trends Ecol Evol 22:140–147
Bruschi P, Vendramin GG, Bussotti F, Grossoni P (2000) Morphological and molecular differentiation between Quercus petraea (Matt.) Liebl. and Quercus pubescens Willd. (Fagaceae) in Northern and Central Italy. Ann Bot 85:325–333
Bucci G, Vendramin GG, Lelli L, Vicario F (1997) Assessing the genetic divergence of Pinus leucodermis Ant. endangered populations: use of molecular markers for conservation purposes. Theor Appl Genet 7:1138–1146
Burd M, Allen TFH (1988) Sexual allocation strategy in wind-pollinated plants. Evolution 42:403–407
Busch JW (2005) The evolution of self-compatibility in geographically peripheral populations of Leavenworthia alabamica (Brassicaceae). Am J Bot 92:1503–1512
Buschbom J, Yanbaev Y, Degen B (2011) Efficient long-distance gene flow into an isolated relict oak stand. J Hered 102:464–472
Cavender-Bares J, Gonzalez-Rodriguez A, Pahlich A, Koehler K, Deacon N (2011) Phylogeography and climatic niche evolution in live oaks (Quercus series Virentes) from the tropics to the temperate zone. J Biogeogr 38:962–981
Celiński F, Filipek M (1958) Flora i zespoły roślinne rezerwatu leśno-stepowego w Bielinku nad Odrą [Flora and plant communities in the woodland-steppe reserve in Bielinek on the Odra river]. Bad Fizj Pol Zach 4:7–184 (in Polish, with English summary)
Celiński F, Wojterski T (1958) Naukowa wycieczka botaniczna do NRD [Botanical excursion to GDR]. Wiad Bot 2:149–164 (in Polish)
Chase MW, Paun O, Fay MF (2010) Hybridization and speciation in angiosperms: a role for pollinator shifts? BMC Biol 8:45
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113
Chybicki IJ, Dzialuk A, Trojankiewicz M, Slawski M, Burczyk J (2008) Spatial genetic structure within two contrasting stands of Scots pine (Pinus sylvestris L.). Silvae Genet 57:193–202
Chybicki IJ, Burczyk J (2010) Realised gene flow within mixed stands of Quercus robur L. and Q. petraea (Matt.) L. revealed at the stage of naturally established seedling. Mol Ecol 19:2137–2151
Courchamp F, Clutton-Brock T, Grenfell B (1999) Inverse density dependence and the Allee effect. Trends Ecol Evol 14:405–410
Curtu AL, Gailing O, Finkeldey R (2009) Patterns of contemporary hybridization inferred from paternity analysis in a four-species forest. BMC Evol Biol 9:284
Curtu AL, Gailing O, Finkeldey R (2007) Evidence for hybridization and introgression within a species-rich oak (Quercus spp.) community. BMC Evol Biol 7:218
Czubiński Z (1950) Zagadnienia geobotaniczne Pomorza [Geobaotanical problems in Pomerania] Bad Fizj Pol Zach 2:444–658 (in Polish, with English summary)
Danielewicz W, Bąkowska K, Krzakowa M (2002) Variability of downy oak (Quercus pubescens Willd.) marginal population in Bielinek (north-west Poland) in morphological traits of leaves. Rocznik Dendrologiczny 50:43–48
Davies HG, Taylor CM, Lambrinos JG, Strong DR (2004) Pollen limitation causes an Allee effect in a wind-pollinated invasive grass (Spartina alterniflora). Proc Natl Acad Sci USA 101:13804–13807
Doyle J, Doyle J (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Dupeouy JL, Badeau V (1993) Morphological variability of oaks (Quercus robur L., Quercus petraea (Matt.) Liebl., Quercus pubescens Willd) in northeastern France. Ann Sci For 50 (Suppl 1):35s–40s
Ellenberg HH (2009) Vegetation Ecology of Central Europe, 4th edn. Cambridge University Press, New York
Felsenstein J (1976) The theoretical population genetics of variable selection and migration. Ann Rev Genet 10:253–280
Finkeldey R (2001) Genetics variation of oaks (Quercus spp.) in Switzerland. 1. Allelic diversity and differentiation at isozyme gene loci. For Genet 8:185–195
García-Ramos G, Kirkpatrick M (1997) Genetic models of adaptation and gene flow in peripheral populations. Evolution 51:21–28
Gelman A, Rubin D (1992) Inference from iterative simulation using multiple sequences. Stat Sci 7:457–511
Girod C, Vitalis R, Leblois R, Fréville H (2011) Inferring population decline and expansion from microsatellite data: a simulation-based evaluation of the Msvar method. Genetics 188:165–179
Goldberg EE, Lande R (2006) Ecological and reproductive character displacement on an environmental gradient. Evolution 60:1344–1357
Grivet D, Robledo-Arnuncio JJ, Smouse PE, Sork VL (2009) Relative contribution of contemporary pollen and seed dispersal to the effective parental size of seedling population of California valley oak (Quercus lobata, Née.). Mol Ecol 18:3967–3979
Gömöry D, Schmidtová J (2007) Extent of nuclear genome sharing mong white oak species (Quercus L. subgen. Lepidobalanus (Endl.) Oerst.) in Slovakia estimated by allozymes. Plant Syst Evol 266:253–264
Gugerli F, Walser J-C, Dounvai K, Holderegger R, Finkeldey R (2007) Coincidence of small-scale spatial discontinuities in leaf morphology and nuclear microsatellite variation of Quercus petraea and Q. robur in a mixed forest. Ann Bot 99:713–722
Guillot G, Santos F, Estoup A (2008) Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24:1406–1407
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Res 2:618–620
Howard DJ (1993) Reinforcement: origin, dynamics, and fate of an evolutionary hypothesis. In: Harrison RG (ed) Hybrid Zones and the Evolutionary Process. Oxford University Press, New York, pp 46–69
Jedinaková-Schmidtová J, Paule L, Magic D, Gömöry D (2004) Morphological and genetic differentiation among the Central European white oaks. For Genet 11:263–271
Jones O, Wang J (2009) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Res 10:551–555
Kawecki TJ (2000) Adaptation to marginal habitats: contrasting influence of the dispersal rate on the fate of alleles with small and large effects. Proc R Soc London B 267:1315–1320
Knight TM, Steets JA, Vamosi JC, Mazer SJ, Burd M, Campbell DR, Dudash MR, Johnston MO, Mitchell RJ, Ashman T-L (2005) Pollen limitation of plant reproduction: pattern and process. Annu Rev Ecol Evol Syst 36:467–497
Lenormand T (2002) Gene flow and the limits to natural selection. Trends Ecol Evol 17:183–189
Lepais O, Gerber S (2011) Reproductive patterns shape introgression dynamics and species succession within the European white oak species complex. Evolution 65:156–170
Lepais O, Petit RJ, Guichoux E, Lavabre JE, Alberto F, Kremer A, Gerber S (2009) Species relative abundance and direction of introgression in oaks. Mol Ecol 18:2228–2242
Lindgren D, Paule L, Xihuan S, Yazdani R, Segerström U, Wallin J-E, Lejdebro ML (1995) Can viable pollen carry Scots pine genes over long distances? Grana 34:64–69
Mallet J (2005) Hybridization as an invasion of the genome. Trends Ecol Evol 20:229–237
Michalski SG, Durka W (2007) High selfing and high inbreeding depression in peripheral populations of Juncus atratus. Mol Ecol 16:4715–4727
Mimura M, Aitken SN (2007) Increased selfing and decreased effective pollen donor number in peripheral relative to central populations in Picea sitchensis (Pinaceae). Am J Bot 94:991–998
Muir G, Schlotterer C (2005) Evidence for shared ancestry polymorphism rather than recurrent gene flow at microsatellite loci differentiating two hybridizing oaks (Quercus spp.). Mol Ecol 14:549–561
Nakanishi A, Tomaru N, Yoshimaru H, Manabe T, Yamamoto S (2009) Effects of seed- and pollen-mediated gene dispersal on genetic structure among Quercus salicina saplings. Heredity 102:182–189
Orlik DC, Kjaer ED (2007) The reproductive success of a Quercus petraea × Q. robur F1-hybrid in back-crossing situations. Ann For Sci 64:37–45
Peñaloza-Ramírez JM, Gonzalez-Rodriguez A, Mendoza-Cuenca L, Caron H, Kremer A, Oyama K (2010) Interspecific gene flow in a multilocus oak hybrid zone in the Sierra Tarahumara of Mexico. Ann Bot 105:389–399
Petit RJ, Bodénès C, Ducousso A, Roussel G, Kremer A (2004) Hybridization as a mechanism of invasion in oaks. New Phyt 161:151–164
Pujol B, Pannell JR (2008) Reduced responses to selection after species range expansion. Science 321:96
Rosenberg MS, Anderson CD (2011) PASSaGE: pattern analysis, spatial statistics and geographic exegesis. Version 2. Methods Ecol Evol 2:229–232
Rushton BS (1993) Natural hybridization within the genus Quercus L. Ann Sci For 50(Suppl 1):73s–90s
Russel JC, Fewster RM (2009) Evaluation of the linkage disequilibrium method for estimating effective population size. In: Thomson DL et al. (eds) Modeling demographic processes in marked populations, environmental and ecological statistics, vol 3, pp 291–320
Saintagne C, Bodénès C, Barreneche T, Pot D, Plomion C, Kremer A (2004) Distribution of genomic regions differentiating oak species assessed by QTL detection. Heredity 92:20–30
Salvini D, Bruschi P, Fineschi S, Grossoni P, Kjær ED, Vendramin GG (2009) Natural hybridisation between Quercus peteaea (Matt.) Liebl. and Quercus pubescens Willd. within an Italian stand as revealed by microsatellite fingerprinting. Plant Biol 11:758–765
Sánchez de Dios R, Benito-Garzón M, Sainz-Ollero H (2006) Hybrid zones between two European oaks: a plant community approach. Plant Ecol 187:109–125
Schierup MH, Christiansen FB (1996) Inbreeding depression and outbreeding depression in plants. Heredity 77:461–468
Schueler S, Schlüzen KH (2006) Modeling of oak pollen dispersal on the landscape level with a mesoscale atmospheric model. Environ Model Assess 11:179–194
Sexton JP, McIntyre PJ, Angert AL, Rice KJ (2009) Evolution and ecology of species range limits. Ann Rev Ecol Evol Syst 40:415–436
Simchuk AP (2008) The effect of fodder plant genotype on the variation of larval fitness traits in genotype classes of green oak leafroller moth. Rus J Genet 44:481–524
Slatkin M (1985) Gene flow in natural populations. Ann Rev Ecol Syst 16:393–430
Soltis PS, Soltis DE (2009) The role of hybridization in plant speciation. Annu Rev Plant Biol 60:561–588
Spellman GM, Klicka J (2006) Testing hypotheses of Pleistocene population history using coalescent simulations: phylogeography of the pygmy nuthatch (Sitta pygmaea). Proc R Soc B 273:3057–3063
Staszkiewicz J (1977) Pozycja systematyczna dębu omszonego (Quercus pubescens Willd.) z rezerwatu leśno-stepowego w Bielinku nad Odrą w oparciu o analizę biometryczną liści [Systematical position of downy oak (Quercus pubescens Willd.) from the woodland-steppe reserve in Bielinek on the Oder based on the biometric analysis of leaves]. Fragmenta Florist Geobot 23:259–273 (in Polish)
Steinhoff S (1993) Results of species hybridization with Quercus robur L. and Quercus petraea (Matt.) Liebl. Ann Sci For 50(Suppl 1):s137–s143
Storz JF, Beaumont MA (2002) Testing for genetic evidence of population expansion and contraction: an empirical analysis of microsatellite DNA variation using a hierarchical Bayesian model. Evolution 56:154–166
Streiff R, Labbé T, Bacilieri R, Steinkellner H, Glössl J, Kremer A (1998) Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol 7:317–328
Streiff R, Ducousso A, Lexer C, Steinkellner H, Gloessl J, Kremer A (1999) Pollen dispersal inferred from paternity analysis in a mixed oak stand of Quercus robur L. and Q. petraea (Matt.) Liebl. Mol Ecol 8:831–841
Timbal J, Aussenac G (1996) An overview of ecology and silviculture of indigenous oaks in France. Ann Sci For 53:649–661
Tovar-Sánchez E, Oyama K (2004) Natural hybridization and hybrid zones between Quercus crassifolia and Quercus crassipes (Fagaceae) in Mexico: morphological and molecular evidence. Am J Bot 91:1352–1363
Varela MC, Brás R, Barros IR, Oliveira P, Meierrose C (2008) Opportunity for hybridization between two oak species in mixed stands as monitored by the timing and intensity of pollen production. Forest Ecol Manage 256:1546–1551
Wachowiak W, Palme AE, Savolainen O (2011) Speciation history of three closely related pines Pinus mugo (T.), P. uliginosa (N.) and P. sylvestris (L.). Mol Ecol 20:1729–1743
Wang J (2009) A new method for estimating effective population sizes from a single sample of multilocus genotypes. Mol Ecol 18:2148–2164
Wang J, Santure AW (2009) Parentage and sibship inference from multilocus genotype data under polygamy. Genetics 181:1579–1594
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Whittemore AT, Schaal BA (1991) Interspecific gene flow in sympatric oaks. Proc Natl Acad Sci USA 88:2540–2544
Acknowledgments
This study was supported by a research grant from the Polish National Science Centre (NN304 307540) and a research grant from Kazimierz Wielki University (both to IJC). The authors would like to thank two anonymous reviewers for their helpful comments on the earlier draft.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chybicki, I.J., Oleksa, A., Kowalkowska, K. et al. Genetic evidence of reproductive isolation in a remote enclave of Quercus pubescens in the presence of cross-fertile species. Plant Syst Evol 298, 1045–1056 (2012). https://doi.org/10.1007/s00606-012-0614-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-012-0614-8