
Abstract
Key message
Transcriptomic analysis of the relationship between gene expression patterns and flavonoid contents in the flower buds of Lonicera japonica under light-induced conditions, especially the flavonoid pathway genes and transcription factors.
Abstract
Flos Lonicerae Japonicae (FLJ), the flower buds of Lonicera japonica Thunb., has been used to treat some human diseases including severe respiratory syndromes and hand-foot-and-mouth diseases owing to its putative antibacterial, and antiviral effects. Luteoloside is a flavonoid that is used by the Chinese Pharmacopoeia to evaluate the quality of FLJ. Light is an important environmental factor that affects flavonoid biosynthesis in the flower buds of L. japonica. However, how light triggers increases in flavonoid production remains unclear. To enhance our understanding of the mechanism involved in light-regulated flavonoid biosynthesis, we sequenced the transcriptomes of L. japonica exposed to three different light conditions: 100% light intensity (CK), 50% light intensity (LI50), and 25% light intensity (LI25) using an Illumina HiSeq 4000 System. A total of 77,297 unigenes with an average length of 809 bp were obtained. Among them, 43,334 unigenes (56.06%) could be matched to at least one biomolecular database. Additionally, 4188, 1545 and 1023 differentially expressed genes (DEGs) were identified by comparative transcriptomics LI25-vs-CK, LI50-vs-CK, and LI25-vs-LI50, respectively. Of note, genes known to be involved in flavonoid biosynthesis, such as 4-coumarate coenzyme A ligase (4CL), and chalcone synthase (CHS) were up-regulated. In addition, a total of 1649 transcription factors (TFs) were identified and divided into 58 TF families; 98 TFs exhibited highly dynamic changes in response to light intensity. Quantitative real-time PCR (qRT-PCR) was used to test the expression profiles of the RNA sequencing (RNA-Seq) data. This study offers insight into how transcriptional expression pattern is influenced by light in the flower buds of L. japonica, and will enhance the understanding of molecular mechanisms of flavonoid biosynthesis in response to light in L. japonica.
Similar content being viewed by others
Introduction
Lonicera japonica Thunb. (honeysuckle), a member of the Caprifoliaceae family, is a perennial deciduous shrub native to East Asia (He et al. 2010). Its dried flower buds, known as Flos Lonicerae Japonicae (FLJ), are a traditional Chinese medicine that reportedly have anti-inflammatory, antibacterial, antiviral, antitumor, and antidiabetic properties (Liu et al. 2016; Wang et al. 2017a). It has been widely used for preventing and treating upper respiratory tract infections, hyperlipidemia disease, and some epidemic disease such as SARs, H1N1, and hand–foot–mouth disease (Ku et al. 2009). In addition to its medicinal uses, FLJ is also used in healthy foods and cosmetics (Shang et al. 2011; Seo et al. 2012). Due to these reasons, there has been a high demand for FLJ in China. The steady supply of FLJ for traditional Chinese medicine and pharmaceutical companies has been met through increased honeysuckle cultivation.
FLJ contains chemical compounds such as essential oils, flavonoids, iridoid glycosides, and saponins (Shang et al. 2011). In particular, the flavonoid luteoloside is a marker compound used for quality evaluation and standardization of FLJ (Chinese Pharmacopoeia Commission 2015). Increasing the biosynthesis and accumulation of this active compound through metabolic engineering and artificial planting would have practical significance in the improvement of FLJ quality.
Flavonoid biosynthesis and accumulation in plants are related not only to genetics, but also to environmental factors, such as light, temperature, nutritional status, water, soil type, and microbial interactions (Ferdinando et al. 2012). Light not only sustains plant growth and development as an energy source, but also affects the synthesis of multiple secondary metabolites (Zhang et al. 2017). Extensive studies indicate that changes in light intensity can stimulate the synthesis and accumulation of secondary metabolites in medicinal plants (Zoratti et al. 2014; Shao et al. 2014; Yoneda et al. 2017; Ye et al. 2017). Tattini et al. (2004) found that the concentration of quercetin and luteolin derivatives in leaves of L. vulgare increases with light intensity, and that the content of quercetin 3-O-rutinoside and luteolin 7-O glucoside in leaves under 100% natural light is higher than that in plants exposed to only 35% and 6% natural light. Deng et al. (2012) also found that light intensity has a significantly positive effect on the content of total flavonoids, kaempferol, quercetin, and isoquercitrin in leaves of C. paliurus. Xu et al. (2014) reported that full sunlight can promote flavonol biosynthesis, including that of quercetin, kaempferol, and isorhamnetin, in leaves of Gingko biloba.
Flavonoid biosynthesis in plants is closely related to the expression of genes encoding flavonoid biosynthetic enzymes, such as phenylalanine ammonia-lyase (PAL), CHS, flavanone 3-hydroxylase (F3H), and flavonol synthase (FLS) (Winkel 2001). For instance, bagging of Pyrus pyrifolia inhibits the accumulation of anthocyanins as well as the expression of PpCHSs, chalcone isomerase (PpCHIs), PpF3Hs, flavonoid 3′-hydroxylase (PpF3′Hs), dihydroflavonol 4-reductase (PpDFRs), and anthocyanidin synthase (PpANSs). At the same time, the expression of these genes is up-regulated 6 h after the removal of the bag (Bai et al. 2017). Xu et al. (2014) found that full sunlight treatment in Gingko biloba leads to a significant increase in flavonol content in leaves, and that it promotes the expression of PAL, CHS, F3H, and FLS. Pan et al. (2017) also reported an increase in the relative expression of cinnamic acid 4-hydroxylase (C4H), CHS, CHI, and FLS, together with an enhancement in the content of epimedin B, epimedin C, and icariin in Epimedium pseudowushanense exposed to high light intensity.
Various transcription factors, especially MYB, bHLH, WD40, WRKY, and bZIP, regulate the transcription of flavonoid biosynthetic structural genes (Hichri et al. 2011). In turn, plant development and environmental signals, such as light, salinity, temperature, and hormone concentration, affect the regulatory function of these transcription factors (Lai et al. 2013). For instance, Arabidopsis plants exposed to light display enhanced expression of MYB111, MYB12, and HY5 (bZIP family) as well as higher flavonoid content compared to dark-grown plants (Pandey et al. 2014). Koyama et al. (2012) also found that light induces the transcription of an array of R2R3 MYB transcription factors. These studies partly revealed the molecular mechanism of light-induced flavonoids synthesis. However, they also indicated that light-dependent control of signaling and physiological processes represent a complex regulatory network. Therefore, the molecular mechanism of light-induced flavonoid biosynthesis needs further analysis.
With the rapid development of high-throughput sequencing technology, RNA-seq emerged as an efficient approach for gene discovery and expression analysis at the transcriptome level (Robinson et al. 2010). RNA-seq can detect transcripts of low abundance with sufficient sensitivity and does not require prior genomics information (Cullum et al. 2011). Moreover, RNA-seq serves as a powerful technique to describe transcription of metabolic changes in many medicinal plant species in response to various stresses (Wang et al. 2017b; Pan et al. 2017).
A previous study found that full light increases the biosynthesis and accumulation of luteoloside in L. japonica. However, the underlying molecular mechanism remains unclear. Here, we analyzed transcriptomes of the flower buds of L. japonica exposed to different artificial light intensities. Our study aimed to construct the transcriptome of L. japonica in relation to light, and annotate the functions of generated unigenes. Besides, we wanted to identify and characterize differentially expressed genes (DEGs) under different light intensity treatments. Moreover, we analyzed and identified transcription factors responding to highly dynamic changes in light intensity. The results not only elucidate the potential light-dependent transcriptional mechanism controlling the accumulation of flavonoids in L. japonica, but also lay a foundation for quality improvement and molecular breeding of this species through genetic engineering.
Materials and methods
Materials and treatments
An experiment was established in an experimental field of the Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China (32º 06′ N, 118º 28′ E). One-year-old healthy and homogenous L. japonica seedlings (ground diameter 5.2~5.3 mm; seedling height 30.0~32.0 cm) were transferred to plastic pots (30.0 cm × 30.0 cm for inner-diameter and height, and one seedling per pot) filled with 2.5 kg of a substrate mixture of vermiculite: peat: soil (3:3:4, v/v/v), and then placed in the greenhouse with a 16:8 light/dark cycle and 65 ± 5% relative humidity at 23 ± 1 °C on February 20, 2017. Six weeks later, the plants were randomly divided into six groups (n = 10 per group), and each light intensity treatment consisted of two groups. Three light intensity levels were created by blocking light penetration using a black cloth stretched over a rigid frame. The light intensity groups were 100% light intensity (non-shade, CK-1, CK-2), 50% light intensity (50% shaded, LI50-1, LI50-2), 25% light intensity (75% shaded, LI25-1, LI25-2) (the fluorescent lights as the light resource). The light intensity was measured with a digital light meter (TES-1330A, Taiwan). The average light intensity passing through in each treatment was about 1300, 640, and 310 μmol m−2s−1 of photosynthetically active radiation (PAR) at noon time (May 2017) for CK, LI50 and LI25. Except for the light intensity, the other culture conditions were the same for each group. After treatment for 30 days, one hundred flower buds from the control and each treated group were harvested at 7:30 a.m. Parts of the collected sample were flash-frozen using liquid nitrogen and stored at − 80 °C until RNA extraction, and the other parts were dried at 45 °C for chemical analysis.
Determination of luteoloside and total flavonoids
The flower buds of each treatment group were collected at the same time with RNA sequencing. All samples were oven-dried at 50 °C and crushed into powder before extraction. The content of luteoloside was determined using HPLC following the current PPRC’s protocol (Chinese Pharmacopoeia Commission 2015). The total amount of flavonoid compounds of each sample was determined according to the method of Hsu et al. (2016).
RNA Extraction, cDNA library construction and Illumina sequencing
Total RNA was extracted from flower buds using RNAiso Plus reagent (Takara Bio, Dalian, China) according to the manufacturer’s instructions. Genomic DNA contamination was eliminated from the extracted RNA by RNase-free DNaseI (TaKaRa Biotech Co., Ltd., Dalian, China). RNA integrity and concentration were then examined by agarose gel electrophoresis (1.2%, w/v) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). The high-quality RNA samples were sent to Gene Denovo Technologies Corporation (Guangzhou, China) for cDNA library construction and sequencing. Poly (A)-containing mRNA was purified from total RNA using poly-T oligo-attached magnetic beads, and mRNAs were fragmented into short sequences (~ 200nt) using fragmentation buffer. These short sequences were transcribed into first-strand cDNA using random hexamer primers and m-muLV Reverse Transcriptase (RNase H-). The second-strand cDNA was synthesized using RNase H and DNA polymerase I. The cDNA fragments were purified using a QiaQuick PCR extraction kit (Qiagen, Inc., Hilden, Germany), and then subjected to end repair and single dATP addition. Thereafter, these short fragments were connected with adapters, and the suitable fragments were screened as templates for PCR amplification. The transcriptome libraries were sequenced using an Illumina HiSeq™ 4000 (Illumina, Inc., San Diego, CA, USA) at the Gene Denovo Institute (Guangzhou, China).
Transcriptome annotation and functional classification
All of the unigenes were searched against annotated public databases, including NR (Non-Redundant Protein Sequence Database), Swiss-Prot, KOG (Clusters of eukaryotic Orthologous Group), GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) using BLASTX analysis with an E-value cut-off of 1.0E-05. Based upon the BLAST results, unique transcripts were assigned to gene ontology categories including cellular component, molecular function, and biological process terms, were annotated using the annotation program BLAST2GO (Conesa et al. 2005). GO functional classification and distribution of gene functions of each assembly unigene were performed using WEGO software at the macroscopic level (Ye et al. 2006).
Identification and analysis of DEGs
To identify DEGs induced by light intensity, the clean reads were mapped to the reference sequence. The relative expression of unigenes was calculated by the RPKM (Reads Per kb per Million reads) method (Mortazavi et al. 2008). The p value was adjusted using the q-value (Anders and Huber 2010). Unigenes with q value < 0.05 and log2 Ratio ≥ 1 were considered to be statistically significant DEGs. All DEGs were mapped to GO and KEGG database for functional and pathway enrichment analysis using the Blast2GO software (http://www.blast2go.org/) and KO-Based Annotation System (KOBAS) software (Mao et al. 2005).
Co-expression network construction
A co-expression network analysis was employed between flavonoid biosynthesis pathway genes and transcription factors in the flower buds of L. japonica. The Pearson correlation evaluation was conducted by SPSS v20 software. The Pearson correlation coefficient of two genes ≥ 0.8 with p value ≤ 0.05 were considered to be co-expressed genes (Fu and Xue 2010). The co-expression networks were visualized with the Cytoscape software version 3.2.0 (Shannon et al. 2003).
qRT-PCR validation
Quantitative reverse transcription-PCR (qRT-PCR) was utilized to validate the patterns of gene expression in the flower buds of L. japonica under light intensity treatments. Total RNA was isolated using the same method mentioned above, and then the first-strand cDNA was synthesized using the Superscript® III First Strand cDNA Synthesis System (Invitrogen, USA) following the manufacturer’s protocol. Actin was used as a reference for calculating the gene expression normalization with the \(2^{{\text{-}\Delta \Delta C_{\text{t}} }}\) method (Livak and Schmittgen 2001). The gene-specific primers were designed with the software Primer Premier 5.0 and are shown in Table S1. The qRT-PCR was performed using SYBR Premix Ex Taq™ II kit (Takara) as per manufacturer’s instructions, and conducted in qTOWER 2.2 Real-Time PCR System (Analytik Jena AG, Jena, Germany) under the following parameters: 94 °C for 2 min, followed by 40 cycles of 94 °C for 20 s, and 60 °C for 34 s. All reactions were performed in three biological replicates and three technical replicate, and the results were reported as mean ± standard error.
Results
Effect of light intensity on the content of luteoloside and total flavonoids
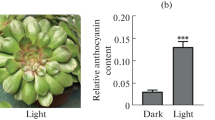
After exposing the plant to different light intensity, we observed the highest luteoloside and total flavonoids contents in CK treatment, followed by LI50 and LI25 (Fig. 1). Therefore, we concluded that high light intensity enhanced luteoloside and total flavonoids production in flower buds of L. japonica.

The contents of luteoloside and total flavonoids in L. japonica under different light intensity treatments. The data are expressed as the mean ± SD. Different letters (a, b and c) indicate significant differences among the different light intensity treatments (p ≤ 0.05; n = 10)
Transcriptome sequencing profile of L. japonica under different light intensity treatments
To develop a global overview of the L. japonica transcriptome under different light intensity treatments, we extracted total RNA from the flower buds of CK, LI50, and LI25 plants. To minimize the bias from the Illumina sequencing and transcriptome sampling, we constructed six cDNA libraries: CK-1, CK-2, LI50-1, LI50-2, LI25-1, LI25-2, and sequenced them separately using an Illumina HiSeq™ 4000 genome analyzer.
Each sequenced sample yielded 2 × 150 bp independent reads from either end of a cDNA fragment, with approximately 52–61 million raw reads from each sample. After removing low-quality sequences, we obtained 51–60 million clean reads with 97.96–98.88% Q20 bases, while the GC contents of all samples were 46.23–47.82% (Table 1).
In total, we obtained 77,297 unigenes with lengths ranging from 201 to 15,715 bp using theTrinity program. The average length of these unigenes and N50 were 809 bp and 1281 bp, respectively (Table 2). Figure S1 shows unigenes with a distribution larger than 200 bp.
Sequence annotation and classification
We searched these unigenes against the NR, Swiss-Prot, KOG, GO, and KEGG databases. The results indicated that 42,096 (54.46%) unigenes were annotated in the NR database, 32,190 (41.64%) in the Swiss-Prot database, 27,027 (34.97%) in the KOG database, 16,687 (21.59%) in the KEGG database, and 12,740 (16.48%) in the GO database (Table 3).
For KOG analysis, there were 27,027 unigenes divided into 25 groups for functional prediction and classification (Fig. 2, Table S2). Among them, the “General function prediction only” (9711) contained the largest number of unigenes, followed by “Posttranslational modification, protein turnover, chaperons” (4835), “Signal transduction mechanisms” (4777), “RNA processing and modification” (2647), and “Transcription” (2372).

The classifications of assembled unigenes against euKaryotic Ortholog Groups (KOG). All assembled unigenes were aligned to the KOG database, and each bar represents the number of unigenes classified functionally into the 25 molecular families
In addition, 12,740 unigenes were annotated to one or more GO terms, and classified into 3 categories and 46 terms (Fig. 3, Table S3). For the “Biological Process” (BP) category, the term “Metabolic process” (7290) represented the largest cluster, followed by “Cellular process” (6668), “Single-organism process” (5291), “biological regulation” (2020), and “response to stimulus” (1728). In the “Molecular Function” (MF) category, the terms “Catalytic activity” (7250), “Binding” (5119), “Transporter activity” (609), “nucleic acid binding transcription factor activity” (136), and “structural molecule activity” (123) were among the most frequent. Within the “Cellular Component” (CC) category, the top five terms were “Cell” (4125), “Cell part” (4125), “Organelle” (2984), “Membrane” (2550), and “Membrane part”(1795).

The classfications of assembled unigenes against Gene ontology (GO). The unigenes were systematically classified into three GO sub-categories: biological process, cellular component, and molecular function. Each bar represents the number of unigenes classified under each subcategory
To classify gene function and identify biochemical pathways, we mapped all annotated unigenes onto the KEGG database. 16,687 unigenes were annotated and assigned to 130 KEGG pathways (Fig. 4, Table S4). Among them, the largest category was “Metabolism pathways” containing “Carbohydrate metabolism” (2103), “Global and Overview” (1384), “Amino acid metabolism” (1190), “Lipid metabolism” (1032), “Energy metabolism” (751), “Nucleotide metabolism” (609), “Biosynthesis of other secondary metabolites” (476), “Metabolism of co-factors and vitamin” (432), “Metabolism of other amino acids” (376), “Metabolism of terpenoids and polyketides” (370), and “Glycan biosynthesis and metabolism” (287). Other groups with a high number of members were “Genetic Information Processing” (4106), “Cellular Processes” (829), “Environmental Information Processing” (585), and “Organismal Systems” category (382).

The classifications of annotated unigenes against Kyoto Encyclopedia of Genes and Genomes (KEGG). Five main KEGG metabolism pathway categories are metabolism (a), genetic information processing (b), environmental information processing (c), cellular processes (d), and organismal systems (e). Each bar represents the numbers of unigenes annotated to each KEGG term
DEGs analysis of assembled L. japonica transcripts under different light intensity treatments
We identified a total of 4188 DEGs, including 249 up-regulated and 3939 down-regulated unigenes in the comparison between LI25 and CK. Comparing LI50 and CK, we found 1545 DEGs, including 213 up-regulated and 1332 down-regulated unigenes. The comparison between LI25 and LI50 indicated that there were 1023 DEGs, including 119 up-regulated and 904 down-regulated unigenes (Fig. 5).

Differentially expressed genes (DEGs) of L.japonica under different light intensity treatments. a Scatter-plot graphs of the differential gene expression patterns in the comparison between LI25 and CK, LI50 and CK, and LI25 and LI50, respectively. Reds pots represent up-regulated DEGs, blue spots indicate down-regulated DEGs, and gray spots indicate unigenes that did not change significantly under light intensity treatments. b Number of DEGs showing up-(red) or down-(blue) regulation in pairwise comparisons of the three libraries. c Venn diagram describing the number of upregulated and downregulated genes revealed by paired comparison
To further analyze the functions of these DEGs, we searched and annotated all DEGs with GO and KEGG databases. GO analysis indicated the presence of 37, 36, and 31 functional groups in the comparisons between LI25 and CK, between LI50 and CK, and between LI25 and LI50, respectively (Figure S2). In these three pairwise comparisons, “Metabolic process” and “Cellular process” were dominant within the “Biological process” category. “Cell” and “Cell part” categories were the most represented category with “Cellular component”. “Catalytic activity” and “Binding” were predominant in the “Molecular function” category.
KEGG pathways analysis categorized 1650, 560, and 307 DEGs in LI25-vs-CK, LI50-vs-CK, and LI25-vs-LI50 comparisons into 121, 110, and 90 KEGG pathways, respectively (Table S5). In the comparison between LI25 and CK, 519 (31.45%) DEGs were significantly enriched in 19 pathways, including “Proteasome” (28), “Biotin metabolism” (12), “Ribosome” (89), “Oxidative phosphorylation” (53), “Biosynthesis of unsaturated fatty acids” (18), “Citrate cycle (TCA cycle)” (25), “Fatty acid metabolism” (30), “Valine, leucine and isoleucine degradation” (20), “Fatty acid degradation” (17), and “Steroid biosynthesis” (15). Among the DEGs identified from the comparison of LI50 and CK, 135 (24.11%) DEGs were significantly enriched in 10 pathways, containing “Ribosome” (58), “Oxidative phosphorylation” (23), “Monoterpenoid biosynthesis” (3), “Biosynthesis of unsaturated fatty acids” (7), “Betalain biosynthesis” (1), “Carbapenem biosynthesis” (1), “Cyanoamino acid metabolism” (7), “Phenylpropanoid biosynthesis” (14), “Vitamin B6 metabolism” (3), and “RNA transport” (18). For the comparison between LI25 and LI50, 93 (30.29%) DEGs were significantly enriched in 15 pathways, including “Sesquiterpenoid and triterpenoid biosynthesis” (10), “Monoterpenoid biosynthesis” (3), “Isoquinoline alkaloid biosynthesis” (4), “Cutin, suberine and wax biosynthesis” (4), “Nitrogen metabolism” (4), “Ascorbate and aldarate metabolism” (5), “Fatty acid metabolism” (8), “Starch and sucrose metabolism” (12), “Pentose and glucuronate interconversions” (7), and “Fatty acid biosynthesis” (5).
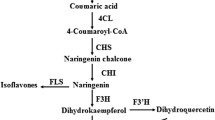
Structural genes involved in luteoloside biosynthesis
Luteoloside is a flavonoid compound, an important group of plant secondary metabolites with various pharmacological activities, whose synthesis is influenced by a large number of enzymes. Here, we identified 23 unique sequences encoding eight enzyme families involved in luteoloside biosynthesis, including PAL, C4H, 4CL, CHS, CHI, Flavonol synthase (FSII), F3´H, and UDP-glucose: flavone 7-O-beta-glucosyltransferase (UFGT) (Fig. 6). The expression levels of the two identified PAL genes, Unigene0038027, and Unigene0005847, increased in response to light intensity. We also identified four C4Hs, among which Unigene0036844 reached the highest expression level in LI25 treatment. The levels of Unigene0036843, Unigene0034399 and Unigene0034398 varied with the increase of light intensity. Five 4CL genes were identified. The expression level of these genes gradually increased with the increase of light intensity. Six unigenes encoding CHS exhibited two expression patterns. Of these genes, the expression levels of Unigene0035685, Unigene0019785, Unigene0035684, Unigene0022607, and Unigene0065625 decreased with the decrease in light intensity. By contrast, the transcription levels of Unigene0008477 reached the highest at LI50. Two identified CHIs showed differential expression pattern between the three treatments. The expression level of Unigene0033496 was highest at LI50. In contrast, the transcription of Unigene0029885 gradually increased as the light intensity was increased. The transcription levels of Unigene 0021760 and Unigene 0027683, which encode FSII and F3´H, respectively, displayed similar expression pattern with 4CL genes. The expression level of Unigene 0041850, which encodes UFGT, gradually decreased with light intensity decreased. Conversely, the expression level of Unigene 0036141 reached the highest at LI25.

Simplified scheme and a heat map of the expression of genes related to luteoloside biosynthesis in L. japonica. Enzyme names, unigene IDs, and expression patterns are indicated at the right side of each step
Transcription factors (TFs)
We identified a total of 1649 TFs and divided them into 58 families aligning the annotated L. japonica unigenes to the Plant TFDB database (Fig. 7). Members of the C2H2, ERF, bHLH, MYB, NAC, MYB-related, bZIP, C3H, WRKY, and FAR1 families, containing more than 58 unigenes, scored in the top 10 classes. The C2H2 and ERF families with 120 members were the largest family. In addition, EIL, LSD, HB-PHD, NF-X1, Whirly, LFY, S1Fa-like, RAV, STAT, VOZ, and NZZ/SPL families contained fewer than 5 members.

Distribution of transcription factor (TF) families
To better reveal the transcriptional regulation mechanism of luteoloside biosynthesis under light, we identified 98 TFs that exhibited highly dynamic changes in response to light intensity (Fig. 8). Among them, C2H2 constituted the largest group (19, 19.38%), followed by WRKY (12, 12.24%), MYB (8, 8.16%), WD40 (7, 7.14%), bZIP (6, 6.12%), ERF (6, 6.12%), NAC (6, 6.12%), GATA (4, 4.08%), bHLH (3, 3.06%), M-type (3, 3.06%), and MYB-related (3, 3.06%).

Heat map showing the expression profiling of DEGs annotated as transcription factors
The expression levels of these C2H2s, except for Unigene0023926, Unigene0006264, and Unigene0063182, decreased with the decrease in light intensity. The MBW transcription complex composed of MYB, bHLH, and WD40 proteins activate structural genes in the flavonoid biosynthesis pathway (Xu et al. 2015). In our study, the expression levels of four MYB TFs (Unigene007630, Unigene0024490, Unigene0026235, and Unigene0036537), two bHLH TFs (Unigene0072300 and Unigene0076621), and seven WD40 TFs (Unigene0002370, Unigene0019012, Unigene0040850, Unigene0064230, Unigene0067152, Unigene0068542 and Unigene0075099) were up-regulated under light intensity treatment. Furthermore, we also identified twelve WRKY TFs, of which five increased in response to the increase in light intensity. Moreover, Unigene0009170, Unigene0012620, Unigene0024130, Unigene0027671, Unigene0041877, Unigene0068250, and Unigene0075386 reached the highest expression at LI50. In addition, six NAC TFs showed a differential expression pattern. The expression levels of the four NAC TFs Unigene0025772, Unigene0034001, Unigene0035139, and Unigene0074792, increased with the increase of light intensity. By contrast, the two other NAC TFs, Unigene0014311 and Unigene0075492 reached the highest expression level at LI50.
Validation of RNA-Seq Data by qRT-PCR
To validate the reliability of our RNA-Seq data, we selected 12 genes, including luteoloside biosynthetic pathway-related structural genes and TFs involved in the regulation of flavonoid biosynthesis, for a qRT-PCR experiment using CK, LI50, and LI25. The expression patterns of these genes (Unigene0028388, Unigene0024490, Unigene0076621, Unigene0035139, Unigene0065625, Unigene0034685, Unigene0026235, Unigene0073241, Unigene0019660, and Unigene0068250) were consistent with our transcriptome sequencing results (Fig. 9), suggesting that our RNA-Seq data were reliable.

Validation of RNA-seq data by qRT-PCR. The data are expressed as the mean ± SD. The actin genes were used as reference genes
Co-expression network analysis between flavonoid biosynthesis pathway genes and transcription factors
Transcriptome co-expression analysis is an excellent tool for predicting regulatory networks of genes involved in linked processes (Yonekura-Sakakibara and Saito 2013). To further understand the relationships between the light-responsive flavonoid biosynthesis genes and TFs in the flower buds of L. japonica, 21 unique sequences encoding 6 flavonoid biosynthesis genes (PAL, C4H, 4CL, CHS, CHI, FSII, and F3′H) and 98 TFs were used to construct a co-expression network and identify those that may regulate flavonoid biosynthesis (Fig. 10). A total of 69 TFs were co-expressed with these flavonoid biosynthesis-related genes. PAL, 4CL, C4H, CHS, and CHI were found to be co-expressed with more TFs, indicating that these genes might have critical roles in light-induced flavonoid biosynthesis. In addition, twelve unigenes including Unigene0064230 (WD40), Unigene0075099 (WD40), Unigene0033261 (WRKY), Unigene0056344 (WRKY), Unigene0072300 (bHLH), Unigene0076621 (bHLH), Unigene0024090 (ERF), Unigene0007630 (MYB), Unigene0036537 (MYB), Unigene0026235 (MYB), Unigene0034001 (NAC), and Unigene0035139 (NAC) are simultaneously co-expressed with the flavonoid biosynthesis genes. These results suggest that the candidate TFs may contribute to the light-induced flavonoid biosynthesis.

Co-expression network analysis between flavonoid biosynthesis pathway genes and transcription factors
Discussions
Illumina sequencing of L. japonica
RNA-seq has played an important role in illuminating the molecular interactions underlying specific biological events in non-model plants (Roy et al. 2011; Ward et al. 2012). Traditional Chinese medicine uses L. japonica to treat various diseases due to the anti-bacterial, and anti-viral properties of this plant. Some transcriptome-based reports focused on luteoloside biosynthesis, different tissues and floral developmental stages (Yuan et al. 2012; He et al. 2013; Zhang et al. 2016b; Rai et al. 2017). Here, we found that light could promote the accumulation of luteoloside in L. japonica. Several studies elucidated the molecular mechanisms of light-induced secondary metabolism biosynthesis using Illumina deep sequencing technique (Zhang et al. 2016a; Hao et al. 2017). Therefore, we also attempted to explain the light-induced flavonoid accumulation in L. japonica using this technology creating a transcriptome for different light intensity treatments. In our study, we sequenced six libraries (CK-1, CK-2, LI25-1, LI25-2, LI50-1 and LI50-2) from the flower buds of L. japonica under the different light intensity treatments using the Illumina Hiseq 4000 platform. We obtained 77,297 unigenes with an N50 value of 1281 bp, representing a better result than those obtained in previous works. For example, Zhang et al. (2016b) generated 150,523 unigenes with an N50 value of 947 bp using Illumina HiSeq™. Furthermore, we observed that 43.94% of the unigenes were not found in any database searches. This might be the absence of genome information or the unique genes functions of L. japonica.
We performed differential expression analysis between CK, LI50, and LI20 after transcriptome assembly, sequence annotation, and classification. We identified 4188 DEGs in total, including 249 up-regulated and 3939 down-regulated unigenes between LI25 and CK, 1545 DEGs including 213 up-regulated and 1332 down-regulated unigenes between LI50 and CK, 1023 unigenes with 119 up-regulated and 904 down-regulated unigenes between LI25 and LI50.
Previous works reported that shading affects the biosynthesis of secondary metabolites (Xu et al. (2014). In our study, DEGs were significantly enriched in pathways including “Monoterpenoid biosynthesis”, “Phenylpropanoid biosynthesis”, “Sesquiterpenoid and triterpenoid biosynthesis”, and “Isoquinoline alkaloid biosynthesis”. Previous studies also reported similar results in Litchi chinensis and Chrysanthemum × morifolium ‘Purple Reagan’ (Zhang et al. 2016a; Hong et al. 2015). The findings indicate that these metabolic pathways might be modulated in response to light.
Transcription factors involved in light-regulated flavonoid biosynthesis
Flavonoids are important secondary metabolites in plants, and play multiple roles in plant growth, development, and adaptation to the external environment. In addition, they also have various pharmacological activities, such as antibacterial, anti-inflammatory, antioxidative, and anticancer effects (Duthie et al. 2000). Therefore, promoting the biosynthesis of flavonoids in plants has important practical significance. Extensive studies show that there are a large number of structural gene encoding enzymes, such as PAL, 4CH, CHI, F3H FLS, and UFGT, that directly influence the biosynthesis and accumulation of flavonoids (Winkel 2001). Moreover, the expression levels of these structural genes depend on a series of TFs (Hichri et al. 2010). In recent years, many studies have indicated that manipulation of TFs is a means to enhance flavonoid biosynthesis in plants (Pandey et al. 2014; Dalman et al. 2017). In our study, we identified a total of 1649 unigenes encoding for TFs. Among these, we found 98 DEGs of TFs responding to changes in light intensity, and divided them into 26 subfamilies (Figs. 7, 8). Light enhanced the expression of most of these genes in our experiment, suggesting that these DEGs may participate in flavonoid biosynthesis in L. japonica.
The MYB TFs family takes its name from its conserved MYB domain. It is one of the largest TF families in plants, involved in the regulation of diverse biological processes, including flavonoid biosynthesis (Dubos et al. 2010). Recent works identified and characterized many MYB TFs regulating flavonoid biosynthesis in model species (Gonzalez et al. 2008; Gesell et al. 2014). For instance, in A. thaliana, AtMYB12 activates the expression of AtFLS and AtCHS (Mehrtens et al. 2005). MYBs not only can regulate the expression of structural genes directly, but also can modulate interactions with protein partners such as bHLH or WD40 (MBW transcription complex) (Xu et al. 2015). Abiotic stresses, such as light, soil salinity, moisture, and temperature, affect the expression of TFs. In Litchi chinensis, light exposure promotes the expression of LcMYB1 and anthocyanin accumulation relative to bagging in the pericarp (Lai et al. 2014). In Vitis vinifera, light controls the co-expression of MYB, bHLH, WRKY, and MADS-box, and in turn that of the structural genes in flavonoid biosynthesis (Sun et al. 2017). In this study, the expression of four MYB TFs, two bHLH TFs, and seven WD40 TFs were mostly up-regulated with light intensity enhanced, suggesting that MBW might participate in light-induced flavonoid biosynthesis.
In addition, AP2/ERF (AP2/EREBP) is a large family of transcription factors unique to plants whose members contain the AP2/ERF structural domain composed of 60–70 amino acids (Licausi et al. 2013). They have important functions in plant growth and development, including hormonal signal transduction, response to biotic and abiotic stresses, and regulation of metabolism (Nakano et al. 2006). In L. erythrorhizon, light significantly inhibits the transcription of LeERF-1, required in the biosynthesis of shikonin (Zhang et al. 2011). Moreover, Jiao et al. (1987) found that light reduces ethylene formation. Yuk et al. (2016) suggested that ethylene activates a set of structural genes involved in flavonoid biosynthesis. Here, we identified a total of 120 ERF TFs, representing the largest TF family in our transcriptome data. Among them, we detected six DEGs codifying for ERF TFs, of which three were up-regulated with the increase of light intensity, indicating that these ERFs might be involved in the interaction between light and ethylene.
NACs are another important plant-specific TF family, also involved in regulating flavonoid biosynthesis (Xiong et al. 2005). ANAC078 positively regulates flavonoid biosynthesis in responses to high light intensity in A. thaliana (Morishita et al. 2009). This suggests that NAC TFs may play an important role in light stress (Nakashima et al. 2012). In our experiment, 6 NAC TFs exhibited differential expression, and most of these were up-regulated except for Unigene0014311 and Unigene0075492 in response to high light. Therefore, we hypothesize that NAC TFs might directly or indirectly involve in flavonoid biosynthesis in response to light. As for the NAC family, WRKY is also a large family involved in the metabolic regulation, abiotic stress and physiological processes (Schluttenhofer and Yuan 2015). We found 12 WRKY TFs showing differential expression between three different treatments, including five unigenes up-regulated in response to the increase of light intensity, and seven other unigenes reached the highest expression at LI50, which suggests that these seven genes might also play a regulatory role in responses to light. Previous research demonstrated that the regulation of WRKY TFs in flavonoid production might relate to the MYB–bHLH–WD40 TF complex, indicating an interaction between WRKY TFs and other transcription factors in regulating specialized metabolism (Ishida et al. 2007; Amato et al. 2016). In conclusion, we hypothesize that WRKY TFs may regulate the flavonoid biosynthesis in combination with multiple TFs and hormone signaling.
Conclusion
In our study, we investigated the transcriptome profiles of L. japonica using Illumina RNA-seq to identify responsive genes and specific secondary metabolism pathways involved in light responses. A total of 43,334 unigenes with lengths ranging from 201 to 15,715 bp were obtained. We identified 4188, 1545, 1023 differential genes in the comparison between LI25 and CK, LI50 and CK, LI25 and LI50, respectively. In addition, structural genes involved in the luteoloside biosynthesis pathway were identified in L. japonica, and the response mechanism of luteoloside biosynthetic genes to light signaling needs to be studied, especially 4CL and CHI. 98 differentially expressed TFs belonged to 26 subfamilies were identified, and the large number of up-regulated WRKY, MYB, NAC, WD40 and ERF TFs in L. japonica suggests that they may play important roles in light-responsive transcriptional regulation of flavonoid metabolism. We believe that the transcriptome data and DEGs presented in this study will provide valuable information and gene sequences for understanding the regulatory mechanism of light-induced secondary metabolism pathways of L. japonica and other medicinal plants.
Author contribution statement
HL F and XW Q designed the project, performed the experiments and wrote the manuscript. YM L, DB X, X Y, and X L analyzed the data and wrote the manuscript. CY L and WL L revised the manuscript. All authors have read and approved the final manuscript.
Abbreviations
- FLJ:
-
Flos Lonicerae Japonicae
- DEGs:
-
Differentially expressed genes
- 4CL:
-
4-coumarate coenzyme A ligase
- CHS:
-
Chalcone synthase
- TF:
-
Transcription factor
- qRT-PCR:
-
Quantitative real-time PCR
- RNA-seq:
-
RNA sequencing
- PAL:
-
Phenylalanine ammonia-lyase
- F3H:
-
Flavanone 3-hydroxylase
- FLS:
-
Flavonol synthase
- CHI:
-
Chalcone isomerase
- F3′H:
-
Flavonoid 3′-hydroxylase
- DFR:
-
Dihydroflavonol 4-reductase
- ANS:
-
Anthocyanidin synthase
- C4H:
-
Cinnamic acid 4-hydroxylase
- NR:
-
Non-redundant protein sequence database
- KOG:
-
Clusters of eukaryotic orthologous Group
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- FSII:
-
Flavonol synthase
- UFGT:
-
UDP-glucose: flavone 7-O-beta-glucosyltransferase
References
Amato A, Cavallini E, Zenoni S, Finezzo L, Begheldo M, Ruperti B, Tornielli GB (2016) A grapevine ttg2-like wrky transcription factor is involved in regulating vacuolar transport and flavonoid biosynthesis. Front Plant Sci 7:1979. https://doi.org/10.3389/fpls.2016.01979
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11(10):R106. https://doi.org/10.1186/gb-2010-11-10-r106
Bai SL, Sun YW, Qian MJ, Yang FX, Ni JB, Tao RY, Li L, Shu Q, Zhang D, Teng YW (2017) Transcriptome analysis of bagging-treated red Chinese sand pear peels reveals light-responsive pathway functions in anthocyanin accumulation. Sci Rep 7(1):63. https://doi.org/10.1038/s41598-017-00069-z
Chinese Pharmacopoeia Commission (2015) The Pharmacopoeia of the People’s Republic of China. China Medical Science Press, Beijing, p 221
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21(18):3674–3676. https://doi.org/10.1093/bioinformatics/bti610
Cullum R, Alder O, Hoodless PA (2011) The next generation: using new sequencing technologies to analyse gene regulation. Respirology 16(2):210–222. https://doi.org/10.1111/j.1440-1843.2010.01899.x
Dalman K, Wind JJ, Nemesio-Gorriz M, Hammerbacher A, Lundén K, Ezcurra I, Elfstrand M (2017) Overexpression of PaNAC03, a stress induced NAC gene family transcription factor in Norway spruce leads to reduced flavonol biosynthesis and aberrant embryo development. BMC Plant Biol 17(1):6. https://doi.org/10.1186/s12870-016-0952-8
Deng B, Shang XL, Fang SZ, Li QQ, Fu XX, Su J (2012) Integrated effects of light intensity and fertilization on growth and flavonoid accumulation in Cyclocarya paliurus. J Agr Food Chem 60(25):6286–6292. https://doi.org/10.1021/jf301525s
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L (2010) MYB transcription factors in Arabidopsis. Trends Plant Sci 15(10):573–581. https://doi.org/10.1016/j.tplants.2010.06.005
Duthie GG, Duthie SJ, Kyle JAM (2000) Plant polyphenols in cancer and heart disease: implications as nutritional antioxidants. Nutr Res Rev 13(1):79–106. https://doi.org/10.1079/095442200108729016
Ferdinando MD, Brunetti C, Fini A, Tattini M (2012) Flavonoids as antioxidants in plants under abiotic stresses. In: Ahmad P and Prasad MNV (eds) Abiotic stress responses in plants: metabolism, productivity and sustainability. Springer, New York, USA, pp 159–179
Fu FF, Xue HW (2010) Coexpression analysis identifies Rice Starch Regulator 1, a rice AP2/EREBP family transcription factor, as a novel rice starch biosynthesis regulator. Plant Physiol 154(2):927–938. https://doi.org/10.1104/pp.110.159517
Gesell A, Yoshida K, Lan TT, Constabel CP (2014) Characterization of an apple TT2-type R2R3 MYB transcription factor functionally similar to the poplar proanthocyanidin regulator PtMYB134. Planta 240(3):497–511. https://doi.org/10.1007/s00425-014-2098-y
Gonzalez A, Zhao M, Leavitt JM, Lloyd AM (2008) Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLHh/Myb transcriptional complex in Arabidopsis seedlings. Plant J 53(5):814–827. https://doi.org/10.1111/j.1365-313X.2007.03373.x
Hao XL, Zhong YJ, Fu XQ, Lv ZY, Shen Q, Yan TX, Shi P, Ma YA, Chen MH, Lv XY, Wu ZKY, Zhao JY, Sun XF, Li L, Tang KX (2017) Transcriptome analysis of genes associated with the artemisinin biosynthesis by jasmonic acid treatment under the light in Artemisia annua. Front Plant Sci 8:971. https://doi.org/10.3389/fpls.2017.00971
He SQ, Hu QF, Yang GY (2010) Research of honeysuckle. Yunnan Chem Technol 37:72–75. https://doi.org/10.3969/j.issn.1004-275X.2010.03.020
He L, Xu XL, Li Y, Li CF, Zhu YJ, Yan HX, Sun ZY, Sun C, Song JY, Bi YA, Shen J, Cheng RY, Wang ZZ, Xiao W, Chen SL (2013) Transcriptome analysis of buds and leaves using 454 pyrosequencing to discover genes associated with the biosynthesis of active ingredients in Lonicera japonica Thunb. PLoS One 8(4): e62922. https://doi.org/10.1371/journal.pone.0062922
Hichri I, Heppel SC, Pillet J, Léon C, Czemmel S, Delrot S, Virginie L, Jochen B (2010) The basic helix-loop-helix transcription factor MYC1 is involved in the regulation of the flavonoid biosynthesis pathway in Grapevine. Mol Plant 3(3):509–523. https://doi.org/10.1093/mp/ssp118
Hichri I, Barrieu F, Bogs J, Kappel C, Delrot S, Lauvergeat V (2011) Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J Exp Bot 62(8):2465–2483. https://doi.org/10.1093/jxb/erq442
Hong Y, Tang XJ, Huang H, Zhang Y, Dai SL (2015) Transcriptomic analyses reveal species-specific light-induced anthocyanin biosynthesis in chrysanthemum. BMC Genomics 16(1):202. https://doi.org/10.1186/s12864-015-1428-1
Hsu HF, Hsiao PC, Kuo TC, Chiang ST, Chen SL, Chiou SJ, Ling XH, Liang MT, Cheng WY, Houng JY (2016) Antioxidant and anti-inflammatory activities of Lonicera japonica Thunb. var. sempervillosa Hayata flower bud extracts prepared by ater, ethanol and supercritical fluid extraction techniques. Ind Crop Prod 89:543–549. https://doi.org/10.1016/j.indcrop.2016.05.010
Ishida T, Hattori S, Sano R, Inoue K, Shirano Y, Hayashi H, Shibata D, Sato S, Kato T, Tabata S, Okada K, Wada T (2007) Arabidopsis transparent testa glabra2 is directly regulated by R2R3 MYB transcription factors and is involved in regulation of GLABRA2 transcription in epidermal differentiation. Plant Cell 19(8):2531–2543. https://doi.org/10.1105/tpc.107.052274
Jiao XZ, Yip WK, Yang SF (1987) The effect of light and phytochrome on 1-aminocyclopropane-1-carboxylic acid metabolism in etiolated wheat seedling leaves. Plant Physiol 85(3):643–647. https://doi.org/10.1104/pp.85.3.643
Koyama K, Ikeda H, Poudel PR, Gotoyamamoto N (2012) Light quality affects flavonoid biosynthesis in young berries of cabernet sauvignon grape. Phytochemistry 78(6):54–64. https://doi.org/10.1016/j.phytochem.2012.02.026
Ku SK, Seo BI, Park JH, Park GY, Seo YB, Kim JS, Lee HS, Roh SS (2009) Effect of Lonicerae Flos extracts on reflux esophagitis with antioxidant activity. World J Gastroenterol 15(38):4799–4805. https://doi.org/10.3748/wjg.15.4799
Lai Y, Li HX, Yamagishi M (2013) A review of target gene specificity of flavonoid R2R3-MYB transcription factors and a discussion of factors contributing to the target gene selectivity. Front Biol 8:577–598. https://doi.org/10.1007/s11515-013-1281-z
Lai B, Li XJ, Hu B, Qin YH, Huang XM, Wang HC, Hu GB (2014) LcMYB1 is a key determinant of differential anthocyanin accumulation among genotypes, tissues, developmental phases and ABA and light stimuli in Litchi chinensis. PLoS One 9:e86293. https://doi.org/10.1371/journal.pone.0086293
Licausi F, Ohme-Takagi M, Perata P (2013) Apetala2/ethylene responsive factor (ap2/erf) transcription factors: mediators of stress responses and developmental programs. New Phytol 199(3):639–649. https://doi.org/10.1111/nph.12291
Liu ZX, Cheng ZY, He QJ, Lin B, Gao PY, Li LZ, Liu QB, Song SJ (2016) Secondary metabolites from the flower buds of Lonicera japonica and their in vitro anti-diabetic activities. Fitoterapia 110:44–51. https://doi.org/10.1016/j.fitote.2016.02.011
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21(19):3787–3793. https://doi.org/10.1093/bioinformatics/bti430
Mehrtens F, Kranz H, Bednarek P, Weisshaar B (2005) The arabidopsis transcription factor myb12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol 138(2):1083–1096. https://doi.org/10.1104/pp.104.058032
Morishita T, Kojima Y, Maruta T, Nishizawa-Yokoi A, Yabuta Y, Shigeoka S (2009) Arabidopsis NAC transcription factor, ANAC078, regulates flavonoid biosynthesis under high-light. Plant Cell Physiol 50:2210–2222. https://doi.org/10.1093/pcp/pcp159
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7):621–628. https://doi.org/10.1038/nmeth.1226
Nakano T, Nishiuchi T, Suzuki K, Fujimura T, Shinshi H (2006) Studies on transcriptional regulation of endogenous genes by erf2 transcription factor in tobacco cells. Plant Cell Physiol 47(4):554–558. https://doi.org/10.1093/pcp/pcj017
Nakashima K, Takasaki H, Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) Nac transcription factors in plant abiotic stress responses. Biochim Biophys Acta 1819(2):97–103. https://doi.org/10.1016/j.bbagrm.2011.10.005
Pan JQ, Chen HM, Guo BL, Liu C (2017) Understanding the molecular mechanisms underlying the effects of light intensity on flavonoid production by RNA-seq analysis in Epimedium pseudowushanense B.L.Guo. PLoS One 12(8):e0182348. https://doi.org/10.1371/journal.pone.0182348
Pandey A, Misra P, Bhambhani S, Bhatia C, Trivedi PK (2014) Expression of Arabidopsis MYB transcription factor, AtMYB111, in tobacco requires light to modulate flavonol content. Sci Rep 4(5):5018. https://doi.org/10.1038/srep05018
Rai A, Kamochi H, Suzuki H, Nakamura M, Takahashi H, Hatada T, Saito K, Yamazaki M (2017) De novo transcriptome assembly and characterization of nine tissues of Lonicera japonica to identify potential candidate genes involved in chlorogenic acid, luteolosides, and secoiridoid biosynthesis pathways. J Nat Med 71(1):1–15. https://doi.org/10.1007/s11418-016-1041-x
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. https://doi.org/10.1093/bioinformatics/btp616
Roy NC, Altermann E, Park ZA, McNabb WC (2011) A comparison of analog and Next-Generation transcriptomic tools for mammalian studies. Brief Funct Genomics 10(3):135–150. https://doi.org/10.1093/bfgp/elr005
Schluttenhofer C, Yuan L (2015) Regulation of specialized metabolism by WRKY transcription factors. Plant Physiol 167:295–306. https://doi.org/10.1104/pp.114.251769
Seo ON, Kim GS, Park S, Lee JH, Kim YH, Lee WS, Lee SJ, Kim CY, Jin JS, Choi SK, Shin SC (2012) Determination of polyphenol components of Lonicera japonica Thunb. using liquid chromatography-tandem mass spectrometry: contribution to the overall antioxidant activity. Food Chem 134(1):572–577. https://doi.org/10.1016/j.foodchem.2012.02.124
Shang XF, Pan H, Li MX, Miao XL, Ding H (2011) Lonicera japonica Thunb.: ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J Ethnopharmacol 138(1):1–21. https://doi.org/10.1016/j.jep.2011.08.016
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Shao QS, Wang HZ, Guo HP, Zhou AC, Huang YQ, Sun YL, Li MY (2014) Effects of shade treatments on photosynthetic characteristics, chloroplast ultrastructure, and physiology of Anoectochilus roxburghii. PLoS One 9(2):e85996. https://doi.org/10.1371/journal.pone.0085996
Sun RZ, Cheng G, Li Q, He YN, Wang Y, Lan YB, Li SY, Zhu YR, Song WF, Zhang X, Cui XD, Chen W, Wang J (2017) Light-induced variation in phenolic compounds in cabernet sauvignon grapes (vitis vinifera L.) involves extensive transcriptome reprogramming of biosynthetic enzymes, transcription factors, and phytohormonal regulators. Front Plant Sci 8:547. https://doi.org/10.3389/fpls.2017.00547
Tattini M, Galardi C, Pinelli P, Massai R, Remorini D, Agati G (2004) Differential accumulation of flavonoids and hydroxycinnamates in leaves of Ligustrum vulgare under excess light and drought stress. New Phytol 163(3):547–561. https://doi.org/10.1111/j.1469-8137.2004.01126.x
Wang DY, Zhao XM, Liu YL (2017a) Hypoglycemic and hypolipidemic effects of a polysaccharide from flower buds of Lonicera japonica in streptozotocin-induced diabetic rats. Int J Biol Macromol 102:396–404. https://doi.org/10.1016/j.ijbiomac.2017.04.056
Wang R, Xu S, Wang N, Xia B, Jiang Y, Wang R (2017b) Transcriptome analysis of secondary metabolism pathway, transcription factors, and transporters in response to methyl jasmonate in Lycoris aurea. Front Plant Sci 7:1971. https://doi.org/10.3389/fpls.2016.01971
Ward JA, Ponnala L, Weber CA (2012) Strategies for transcriptome analysis in nonmodel plants. Am J Bot 99(2):267–276. https://doi.org/10.3732/ajb.1100334
Winkel SB (2001) Flavonoid biosynthesis: a colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol 126(2):485–493. https://doi.org/10.1104/pp.126.2.485
Xiong YQ, Liu TY, Tian CG, Sun SH, Li JY, Chen MS (2005) Transcription factors in rice: a genome-wide comparative analysis between monocots and eudicots. Plant Mol Biol 59(1):191–203. https://doi.org/10.1007/s11103-005-6503-6
Xu Y, Wang GB, Cao FL, Zhu CC, Wang GY, El-Kassaby YA (2014) Light intensity affects the growth and flavonol biosynthesis of ginkgo (ginkgo biloba, L.). New Forests 45(6):765–776. https://doi.org/10.1007/s11056-014-9435-7
Xu W, Dubos C, Lepiniec L (2015) Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci 20:176–185. https://doi.org/10.1016/j.tplants.2014.12.001
Ye J, Fang L, Zheng HK, Zhang Y, Chen J, Zhang ZJ, Wang J, Li ST, Li RQ, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:293–297. https://doi.org/10.1093/nar/gkl031
Ye SY, Shao QS, Xu MJ, Li SL, Wu M, Tan X, Su LY (2017) Effects of light quality on morphology, enzyme activities, and bioactive compound contents in Anoectochilus roxburghii. Front Plant Sci 8:857. https://doi.org/10.3389/fpls.2017.00857
Yoneda Y, Shimizu H, Nakashima H, Miyasaka J, Ohdoi K (2017) Effects of light intensity and photoperiod on improving steviol glycosides content in stevia rebaudiana (bertoni) bertoni while conserving light energy consumption. J Appl Res Med Aro Plants 7:64–73. https://doi.org/10.1016/j.jarmap.2017.06.001
Yonekura-Sakakibara K, Saito K (2013) Transcriptome coexpression analysis using ATTED-II for integrated transcriptomic/metabolomic analysis. Methods Mol Biol 1011:317–326. https://doi.org/10.1007/978-1-62703-414-2_25
Yuan Y, Song LP, Li MH, Liu GM, Chu YN, Ma LY, Zhou YY, Wang X, Gao W, Qin SS, Yu J, Wang XM, Huang LQ (2012) Genetic variation and metabolic pathway intricacy govern the active compound content and quality of the Chinese medicinal plant Lonicera japonica thunb. BMC Genomics 13:195. https://doi.org/10.1186/1471-2164-13-195
Yuk HJ, Song YH, Long MJC, Kim DW, Woo SG, Lee YB, Uddin Z, Kim CY, Park KH (2016) Ethylene induced a high accumulation of dietary isoflavones and expression of isoflavonoid biosynthetic genes in soybean (glycine max) leaves. J Agric Food Chem 64(43):8272. https://doi.org/10.1021/acs.jafc.6b02543
Zhang W, Zou A, Miao J, Yin Y, Tian R, Pang YJ, Yang R, Qi JL, Yang YH (2011) LeERF-1, a novel AP2/ERF family gene within the B3 subcluster, is down-regulated by light signals in Lithospermum erythrorhizon. Plant Biol 13(2):343–348. https://doi.org/10.1111/j.1438-8677.2010.00375.x
Zhang HN, Li WC, Wang HC, Shi SY, Shu B, Liu LQ, Wei YZ, Xie JH (2016a) Transcriptome profiling of light-regulated anthocyanin biosynthesis in the pericarp of Litchi. Front Plant Sci 7:963. https://doi.org/10.3389/fpls.2016.00963
Zhang L, Yan L, Fu C, Xiang J, Gan J, Gang W, Jia HB, Yu LJ, Li MT (2016b) Different gene expression patterns between leaves and flowers in Lonicera japonica revealed by transcriptome analysis. Front Plant Sci 7:637. https://doi.org/10.3389/fpls.2016.00637
Zhang RT, Kong ZY, Chen SH, Ran ZS, Ye MW, Xu JL, Zhou CX, Liao K, Cao JY, Yan XJ (2017) The comparative study for physiological and biochemical mechanisms of Thalassiosira pseudonana and Chaetoceros calcitrans in response to different light intensities. Algal Res 27:89–98. https://doi.org/10.1016/j.algal.2017.08.026
Zoratti L, Karppinen K, Escobar AL, Häggman H, Jaakola L (2014) Light-controlled flavonoid biosynthesis in fruits. Front Plant Sci 5(5):534. https://doi.org/10.3389/fpls.2014.00534
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31500249), the Natural Science Foundation of the Jiangsu Province (BK20160603, BK20161381).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No potential conflict of interest was reported by the authors.
Availability of supporting data
We have deposit our data in Sequence Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/sra/), the accession for our submission is: SRP132670.
Additional information
Communicated by Petersen.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
468_2019_1916_MOESM1_ESM.png
Supplementary material 1 (PNG 19 kb). Fig S1 Length distribution of the assembled unigenes with a distribution larger than 200 bp in L. japonica
468_2019_1916_MOESM2_ESM.png
Supplementary material 2 (PNG 101 kb). Fig S2 Gene ontology (GO) classifications of DEGs in pairwise comparisons (LI25-vs-CK, LI50-vs-CK, and LI25-vs-LI50). The DEGs were classified into three main categories: (A) cellular component, (B) molecular function, and (C) biological process. Each bar indicates the number of unigenes classified under each specific category
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Fang, H., Qi, X., Li, Y. et al. De novo transcriptomic analysis of light-induced flavonoid pathway, transcription factors in the flower buds of Lonicera japonica. Trees 34, 267–283 (2020). https://doi.org/10.1007/s00468-019-01916-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00468-019-01916-4